Pharmacology & Pharmacy
Vol.5 No.7(2014), Article
ID:46794,16
pages
DOI:10.4236/pp.2014.57072
Application of D-Optimal Study Design with Contour Surface Response for Designing Sustained Release Gliclazide Matrix Tablets
Chien Nguyen1, John Mark Christensen2*, Thuy Nguyen1
1National Institute of Pharmaceutical Technology, Hanoi University of Pharmacy, Hanoi, Vietnam
2Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, Corvallis, USA
Email: nguyenngochien@yahoo.com, *jmark.christensen@oregonstate.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 April 2014; revised 17 May 2014; accepted 4 June 2014
ABSTRACT
An optimized formulation of a sustained release tablet of Gliclazide was developed. The use of Doptimal design with a polynomial statistical model to analyze dissolution data reduced the number of laboratory tests required to obtain an optimal dosage form. The final formulation contained 22 mg of Methocel®E15LV, 16.5 mg Methocel®E15 and 10.0 mg of Dibasic Calcium Phosphate per 30 mg Gliclazide sustained release tablet. Dissolution studies performed on tablets from 5000 tablet test batches released greater than 90 percent of loaded drug in eight hours. Drug release from the optimized tablets followed a pattern more closely similar to zero-order than other mechanisms of drug release tested. Storage of tablets in accelerated and ambient conditions for 6 and 12 months respectively did not alter any of the physico-chemical properties, drug release or the drug release rate compared to initial observations and dissolution data of the prepared tablets. The addition of potassium phosphate and monosodium phosphate to the tablet reduced the effect pH has on Gliclazide dissolution compared to the commercially available product.
Keywords:Gliclazide, Sustained Release, D-Optimal, Zero-Order, Response Surface, Modified Release

1. Introduction
Gliclazide 1-(3-azabicyclo (3.3.0)oct-3-yl)-3-p-tolylsulphonylurea is an oral hypoglycemic agent used to treat non-insulin-dependent diabetes mellitus. Dosage forms include immediate and sustained release preparations. Gliclazide plasma half-life is approximately 12 hours [1] . After a single 80 mg dose of gliclazide (Diamicron®) immediate release tablet to 16 healthy volunteers, gliclazide peak plasma concentrations of 5.40 ± 1.03 µg/l were attained in about 3.5 ± 0.9 hours. AUC of immediate release 80 mg gliclazide tablets after 24 hours was 58.02 ± 12.88 µg∙hr/ml [2] . After a single dose of 30 mg gliclazide modified release tablet, Diamicron®MR, to 12 volunteers, mean absolute concentrations of gliclazide ranged between 79% to 110% (average 97%) compared to IV (AUCIV was 17.8 ± 4.8 µg∙hr/ml) showing complete absorption. Tmax of peak gliclazide concentrations from Diamicron®MR 30 mg was between 4 and 8 hours and tlag ranged between 0.5 and 2 hours [3] . Gliclazide plasma concentrations declined monoexponentially from peak concentrations, Cmax and were measurable up to 36 - 72 hours after oral dosing of the modified release gliclazide tablets In addition, no significant differences were observed in AUC, tmax, t1/2, and Cmax after administration of the 30 mg MR gliclazide tablet under fasted and fed conditions. The release of gliclazide over a 24-hour period has been shown to parallel the circadian glycemic profile of type 2 diabetes.
Sustained release formulations of gliclazide have been prepared as matrix tablets, osmotic pump tablet, spheres, and pellets. Among sustained release formulations, hydrophilic matrix tablets have been studied most, probably due to the fact that matrix tablets are easily prepared due to the fact that numerous polymers, such as hydroxylpropylmethylcellulose (HPMC), hydroxypropylcellulose, sodium alginate, Eudragit® EPO, etc., can be used to prepare the hydrophilic polymer matrix system that can easily modulate the drug release kinetic process to a desired drug release rate.
Among studies on sustained release gliclazide tablets, investigators have mainly used HPMC to control drug release. In 2004, Barochez et al. [4] mentioned gliclazide sustained release tablets, Diamicron®MR30 mg MR, in a US patent. The authors used hydroxypropylmethylcellulose (HPMC) combined with glucose syrup, for example, maltodextrin, to make a matrix tablet that provides prolonged release of gliclazide, the release being insensitive to variations in pH of the dissolution media and that ensured continuous, constant and complete release of the active ingredient. Murpani and Madan [5] , 2006, prepared modified release formulations comprising gliclazide and HPMC 4000 cps and HPMC 100 cps. The formulation was composed of an intragranular part and an extragranular part wherein the intragranular part contained gliclazide and dicalcium phosphate (DCP) and was wet-granulated with polyvinyl pyrrolidone to form granules. The extragranular part, including HPMC 4000 cps and HPMC 100 cps, and lubricants, was mixed with intragranular part, and the obtained mixture was compressed to form 12 hours sustained release tablets. The formulations described are said to be bioequivalent with Diamicron®MR. In another invention, Wit et al. [6] , 2010, used the same preparation method and controlled release polymers as that of Murpani’s study but they used a different ratio of polymers and did not use a binder in the intragranular part. The study showed that the formulation achieved a similar sustained-release effect as well as independence of the release profile to variations in pH of the dissolution media. In 2008, Jin et al. [7] developed and optimized the gliclazide extended release formulations by simultaneously using a combination of two hydrophilic polymers: HPMC K15M and sodium alginate to impede drug release. Hydrophilic matrix tablets of gliclazide with HPMC K15M, lactose and sodium alginate were prepared and optimized using a D-optimal experimental design mixed with multiple response optimizations. The final mathematical model was satisfactory for describing the relationships between formulation variables (amounts of HPMC K15M, lactose and alginate) and individual response variables (percentage of drug release at 3, 6, 9 and 12 hours). The results of the optimization-model validation demonstrated the reliability of the assumed model in the preparation of extended-release matrix tablets having zero-order drug release. Raja Rajeswari et al. [8] , 2011, used HPMC in two grades, HPMC 15 cps and Methocel®K4M (HMPC K4M), to make a gliclazide sustained release tablet. All six formulations (where the total weight of the 2 polymers was equal to 40 mg) were developed and evaluated for in vitro drug release up to 16 hours and compared to the dissolution profile of the marketed formulation GMF VI (containing 21 mg Methocel®K4M and 19 mg HPMC 15 cps). The investigators found that the studied formulation and the marketed formulation had similar release patterns and followed zero-order drug release by anomalous diffusion. In the same year, Srivastav et al. [9] , 2011, developed a matrix tablet using water swellable polymers (HPMC K4M and K100LV), and a pH modifier selected from alkaline compounds like sodium hydrogen carbonate, potassium hydrogen carbonate, sodium carbonate, potassium carbonate, magnesium carbonate and calcium carbonate. The results show that the tablets produced were stable for 6 months at 40˚C/75% RH and drug release from tablets was insensitive to variations in the pH of the dissolution medium, and extended release up to 10 - 12 hours. The drug release kinetics of the final formulation followed a zero-order model in which drug release is through swelling and erosion of the tablet.
Additional investigators have used other hydrophilic polymers besides HPMC. In 2008, Vijayalakshmi et al. [10] utilized a central composite design to develop an extended release tablet formulation of gliclazide with pH dependent matrix forming polymers like Keltone® HVCR (sodium alginate) (X1) and Eudragit® EPO (X2) as independent variables. Five dependent variables were considered: hardness, percent of drug release after 1 hour, percent of drug release after 6 hours, diffusion exponent and the time required for 50% drug release. Response surface methodology and multiple response optimization utilizing a quadratic polynomial equation were used to obtain an optimal formulation. The results indicated that factor X1 (Keltone® HVCR) along with its interaction with factor X2 (Eudragit®-EPO) significantly affected the studied response variables. The release kinetics of gliclazide from optimized formulation followed zero-order release pattern. The dissolution profiles of the optimized formulation before and after stability studies of 6 months in 30˚C/65% RH and 40˚C/75% RH conditions were found to be similar. Jeyaprabha et al. [11] , 2010, prepared a modified release tablet of gliclazide by using different grades of hydroxypropyl cellulose including Klucel® GXF, Klucel® JXF and Klucel® EXF by wet granulation method. Infrared spectroscopy showed that the drug and other excipients were compatible with each other. The authors concluded that the drug release process from the optimized formulation (containing Klucel® GXF 15% and Klucel® EXF 12%) involved erosion and diffusion, and prolonged drug release up to 12 hours. The formulation was stable for three months in the accelerated storage conditions of 75% RH and 40˚C.
In addition to hydrophilic matrix tablets, other dosage forms of gliclazide sustained release formulations have been studied like osmotic pump tablets and microspheres. Li et al. [12] , 2008, compared a conventional PPOP (push-pull osmotic pump) which only had orifice(s) on the side of the drug layer to a novel PPOP that had orifices of the same diameter on both side surfaces of the tablet. The authors concluded that the in vitro drug release rate from both PPOP’s was influenced by coating level and core hardness whereas orifice size did not have much influence on the release of drug. The study also showed that coating level and core hardness could not influence the similarity of the drug release profiles of the two kinds of PPOP. In vivo study also showed that the concentration-time profiles of gliclazide in plasma of the two PPOP were comparable and both of them had good in vitro/in vivo correlation. Kumar M.S. et al. [13] , 2010, made gliclazide microspheres with a coat consisting of alginate and gum kondagogu that were prepared by orifice-ionic gelation method and emulsification gelation technique. The gliclazide microspheres were found to be discrete, more spherical and free flowing with emulsion ionic gelation technique. Drug release from the microspheres was observed to be slow, followed zero order release kinetics with non-Fickian release mechanism where drug release depended on the coat: core ratio and the method employed in the preparation of microspheres. Among the two techniques, the emulsification gelation technique was found to be more suitable for slow and complete drug release over a long time period. These microspheres also showed good mucoadhesive property in an in vitro wash test. In 2007, Devarajan et al. [14] used Eudragit® L100 or Eudragit® RS to produce gliclazide sustained release nanoparticles. The gliclazide release from nanoparticles lasted up to 24 hours andwas affected by pH of the dissolution media. In 2009, Varshosaz et al. [15] applied the Taguchi design to produce gliclazide sustained release chitosan beads using a dispersion technique with tripolyphosphate as the gelation agent. The researchers evaluated the blood glucose lowering effect of beads and the results showed that the optimized formulation decreased blood glucose level in a normal rat for 24 hours. One year later, Barakat et al. [16] also used chitosan to produce gliclazide microparticles by emulsification and ionotropic gelation. Gliclazide chitosan microparticles decreased blood glucose in diabetic rabbits for 18 hours. Wang et al. [17] , 2010, utilized ethyl cellulose, microcrystalline cellulose (MCC) and sodium carboxymethyl starch to make gliclazide loaded matrix pellets by extrusion-spheronization method, then pellets were coated by hot-melt coating, polymer aqueous dispersion film coating, or MCC powder coating to control the initial fast release. The results showed that a MCC powder coating was superior for gliclazide coated pellets studied at 40˚C/75% RH for 6 months. In addition, tableted hot melt drug loaded hydrophobic granules of glyceryl monostearate and stearic acid substantially reduced gliclazide release [18] . Drug release closely followed a square root versus time process. Ficus glomerata mucilage was successfully used as an excipient to sustain release of gliclazide from tablets [19] .
Among the aforementioned studies, several investigators used mathematical statistics and optimization methods in their study designs to minimize the number of trials, delineate the effect of causal factors in the formula by analyzing designed response surfaces and obtain the appropriate formulation within target goals. Response surface methodology (RSM) is a widely applied approach to design an optimized pharmaceutical formulation with an appropriate dissolution rate in a short period of time with minimum laboratory trials. Also, D-optimal design is suitable to investigate and delineate the least number of experiments to perform, analyze the effects that changes in excipient mixture composition produce and aid in the selection of the optimal excipient composition for achieving the optimized formulation [7] . All the response variables are fitted to a quadratic model and regression analysis was preformed to obtain the quantitative relationships between the dependent (i.e., drug release rate) and independent variables such as amount of ingredient needed, tablet hardness, amount of coating, etc. [10] . With the cited advantages, D-optimal design with a polynomial quadratic statistical model instead of a quadratic model was used in this study. The study objectives were to use the D-optimal design with a polynomial quadratic statistical model alongside contour surface response diagrams to assess the effects the independent formulation variables have on drug release to optimize a sustained release gliclazide matrix tablet based on comparison of its dissolution profile to that of a reference product.
2. Methods
2.1. Materials
Gliclazide was a gift from Labochim® (Milan, Italy). Methocel®K15M and Methocel®E15LV were gifts from Colorcon (West Point, PA); disodium hydrogen phosphate, disodium hydrogen phosphate, methanol, and acetonitrile were purchased from Merck (Darmstadt, Germany). Lactose monohydrate, dibasic calcium phosphate, potassium dihydrogen phosphate, polyvinyl pyrrolidone K30 (PVP), talc, and magnesium stearate were obtained from XilongLom (Guangzhou, China). Diamicron®MR 30 mg (Lot No. 021522539, expiration date: 12/2012) was purchased from Les Laboratoires Servier (Paris, France).
2.2. Tablet Formulation Composition and Preparation
Table 1 shows the percentage weight compositions of the different experimental tablet formulations designed by MODDE 8.0 software (Umetrics CO., Sweden).The tablets were prepared by conventional wet granulation method. Gliclazide, Methocel®K15M (K15), Methocel®E15LV (E15), lactose, dibasic calcium phosphate (DCP), potassium dihydrogen phosphate, and disodium hydrogen phosphate dihydrate were weighed for a batch size of 1000 tablets, and blended in a mortar after passing through a 60 mesh sieve. The powder blend was wetted and kneaded with 50 ml of solution of PVP 16% in water. The wet mass obtained was extruded through an 18 mesh sieve, and the granules were dried at 50˚C in an oven until the moisture content decreased to a level of 3% in the tablets. After sieving through an 18 mesh sieve, the dried granules were mixed with talc and magnesium stearate. Finally, the lubricated granules were compressed into tablets using 7 mm flat faced punches in a rotary tablet machine/ZPW21A (Shanghai, China) to a tablet weight of 161 mg, and generating a tablet hardness of between 45 and 75 N.
Three batches of 5000 tablets of the optimal formulation were made to validate the obtained results and to evaluate the stability of the gliclazide tablets using the following processing parameters: blending time of 12 minutes at a speed of 60 rpm, and 20 minutes of wetting the mixture (150 rpm) in Sigma ERWEKA kneader (Heusenstamm, Germany), granulation through 18 mesh sieve using ERWEKA Wet Granulator (Huesenstamm, Germany), 10 minutes for mixing lubricants in ERWEKA Cube mixer (Huesenstamm, Germany) and then, as above, compressing the tablets with the same parameters in a rotary tablet machine/ZPW21A (Shanghai, China).
2.3. Optimization of the Formulation of Tablets
D-optimal design by MODDE 8.0 software along with contour surface response was used to optimize the formulation of gliclazide modified release tablets. The amount of Methocel®K15M (X1); Methocel®E15LV (X2) and DCP (X3) were the three independent factors. The dependent variables were the percentages of drug release after 1 hour (Y1), 2 hours (Y2), 4 hours (Y4), 6 hours (Y6), and 8 hours (Y8). The amount of lactose was adjusted to fix the weight of tablets to 161 mg while keeping the tablet’s other ingredients of gliclazide, KH2PO4, Na2HPO4·2H2O, talc, magnesium stearate and PVP constant.
2.4. Tablet Hardness
Tablets hardness, 10 tablets for each test, was determined as per USP 32 using PTB-511B hardness tester, Pharma Test (Hainburg, Germany).

Table 1. Formulations, drug contents and dissolution data of gliclazide modified tablets.
RP is the reference product (Diamicron®MR 30 mg).
2.5. Drug Content Assay
UV-Vis method (used for freshly prepared formulations): Assay of drug content was performed in triplicate for each gliclazide tablet formulation. An amount of powder equivalent to 26 mg of gliclazide was weighed and transferred to a 50 ml volumetric flask. Methanol (20 ml) and pH 6.8 phosphate buffer solution (30 ml) were used to dissolve the drug under sonication for 15 minutes. Then samples were filtered through a 0.45 µm diameter membrane. Filtered solutions were diluted 50 times with a pH 6.8 phosphate buffer solution and the drug content of the diluted solutions were measured using a UV spectrometer (UV spectrometer model Hitachi U- 1900, Tokyo, Japan) set at a wavelength of 226 nm. The quantity of gliclazide present was determined using a standard curve from drug solutions prepared in pH 6.8 phosphate buffer.
HPLC method: Sample solutions and standard solutions were prepared in the same manner as that of UV-Vis method. The final samples were filtered through a 0.45 µm diameter membrane before injection into HPLC system (Shimadzu, Kyoto, Japan) with the following chromatographic conditions: Mobile phase: acetonitrile: phosphate buffer 0.01 N (60:40) (adjusted to pH 3.0 with phosphoric acid), Phenomenex C8 column (250 × 4.6 mm, 5 µm), mobile phase flow rate of 1.0 ml/minute at 30˚C ± 1˚C, the injection volume of 2.0 µl, with 6 µg glipizide as internal standard, and the UV detector set at a wavelength of λ = 226 nm.
2.6. In Vitro Dissolution Testing
Dissolution studies to determine drug release from each of three or six tablets were performed according to the USP 32, apparatus 2, using 900 ml 0.1 N hydrochloric acid solution for dissolution the first hour and in 900 ml phosphate buffer solution of pH 6.8 over the next 7 hours with a 75 rpm stirring speed. 10 ml samples were collected at predetermined time intervals for the eight hour dissolution study. After collection, the samples were filtered through a 0.45 µm diameter membrane. Filtered drug solution concentrations measured by UV absorbance at 226 nm were compared to gliclazide standard solutions in pH 6.8 phosphate buffer. Once the gliclazide dissolution samples’ concentrations were measured via UV the results were converted to percentage of gliclazide released. Gliclazide dissolution profiles are presented as percent drug release versus time curves.
Comparison of two drug dissolution release profiles (reference versus test formulations) was performed using the similarity factor, f2, which is calculated as follows (Equation (1)) [20] [21] .
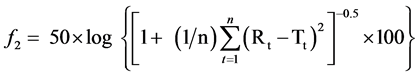 (1)
(1)
where Rt and Tt are the percentages of drug release at time t for the reference and the test formulation, respectively; n is the number of time points. If f2 is equal to or more than 50, the two drug release profiles will be considered to be similar.
2.7. The Dissolution Rate Constants
Dissolution data from gliclazide sustained release tablets were fitted to various mathematical models such as zero-order, Higuchi, First-order, Weibull, Hixson Crowell, Korsmeyer Peppas and Hopfenberg equations. Values for the AIC (Akaike criteria) were used as basis for comparison (Equation (2)) [20] . The models with the smallest AIC were considered to fit the dissolution data best.
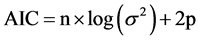 (2)
(2)
where, n is the sample size, σ is the residual standard error which was calculated using S-plus 8.0 statistical software (TIBCO Software Inc., Palo Alto, CA, 94304), and p is the number of parameters in the model.
2.8. Stability Studies
Stability studies were carried out with tablets from the optimal formulation batches of 5000 tablets. The modified release tablets were placed in dark bottles following ICH guidelines at accelerated conditions (40˚C ± 2˚C, RH 75%) in a cooling incubator, Climacell (MMM Medcenter GmbH, Gräfeling, Germany) for six months and ambient room temperature for twelve months. The physicochemical and aesthetic properties of the tablets were evaluated including parameters such as appearance, moisture, hardness, drug content, and in vitro drug release profiles.
2.9. Convolution
Convolution was performed using Kinetica 5.0 (Innaphase, Philadelphia, PA) to simulate plasma concentration time curves of gliclazide after oral administration of Diamicron®MR and the optimal test formulation. Convolution was performed with the following assumptions. The first is that the absorption phase is considered to be that the drug is given as series of IV bolus injections. The second is that gliclazide’s bioavailability is 0.97 and that gliclazide elimination from the body can be described by IV administration data based on the study by Delrat et al., 2002 [3] .
3. Results
A preliminary study was carried out to select the range of input variables that influence the response variables (not shown). Table 1 shows gliclazide formulations based on this preliminary study along with the tablet excipient contents and dissolution data from 19 formulations designed by Modde 8.0. The effects of the independent parameters [X1, X2 and X3 represent the amount of the excipients Methocel®K15M (10 - 20 mg), Methocel® E15LV (10 - 30 mg) and DCP (10 - 50 mg) added, respectively] on response variables (Y1, Y2, Y4, Y6 and Y8 that are the percentages of gliclazide released from tablets at 1 hour, 2 hours, 4 hours, 6 hours, and 8 hours, respectively) were analyzed as follows.
The regression equations of each term were described by the following:
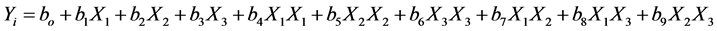 (3)
(3)
where X1, X2 and X3 were amounts of Methocel®K15M, Methocel®E15LV and DCP, respectively; bi was statistically significant when p-value ˂ 0.05; Yi is the percentage of Gliclazide release at time = i hour.
3.1. Effect of Independent Variables on the Response Variables
The regression coefficients for each term in the full regression model are summarized in Table 2. Regression equations show that in the first 2 hours, percentages of drug release (Y-values) were only affected by the amount of Methocel®K15M (variable X1) with all p-values < 0.05 for b1 coefficient at all-time points. The remaining two input variables (amount of Methocel®E15LV and DCP) only had an effect on the formulations at 4 hours (p-values of b8, b9 < 0.05) and 8 hours (p-values of b5, b6, p < 0.05). However, the effects of the ingredients in the formulation are quite complex, requiring each ingredient terms be squared in the equation to fit the observations and the ingredients effects were interdependent.
Table 3 shows several statistical parameters including the multiple correlation coefficients (R2), adjusted multiple correlation coefficients (R2 adjusted), and predicted multiple correlation coefficients (Q2) and the pvalue of the final model of each response variable. The R2 and R2 adjusted values were lower at the first two hours data points (R2 < 0.8 and R2 adjusted < 0.6 at 1 hour and 2 hours). But later time points had higher values of both R2 (R2 > 0.9) and R2 adjusted (R2 adjusted > 0.8). Q2 shows how well the model predicts new data, the higher value of Q2, the better the data prediction. As similar with R2, Q2 also had higher values at the last time points compared with the first 2 hours. In addition, the p-value of the final regression model for each response variable was less than 0.05 and p-values for lack of fit tests (comparing the pure error of the model for each response variable with the full model) were more than 0.05 for all response variables. Thus the final regression equations obtained can be used to describe the relationship between input variables (amount of inert ingredients) and output variables (the percentage of drug release), especially at the time points from 4 to 8 hours.
Table 2.The regression coefficients (bi) for each term in the full regression model.
Table 3. Summary of statistical results from the final regression model and full model.
In addition, response surface analysis also shows the influence of the input variables on the output variables. Figure 1 shows the correlation between input variables and percentage of gliclazide release at 4 hours (Y4), which is near the time 50% of active substance in Diamicron®MR 30 mg, was dissolved. At this point, the effect of Methocel®K15M significantly decreased the percentage of drug release at 4 hour (Y4) with any added amount of Methocel®E15LV as well as DCP (Figure 1(a), Figure 1(c)). Methocel®E15 and DCP also affected the value of Y4, however, these effects depend on the percentage of Methocel®K15M present. When the amount of Methocel® K15M was high, for example equal to 20 mg, increasing Methocel®E15LV also reduced Y4 (Figure 1(a)), while increasing DCP increased Y4 (Figure 1(c)). However this trend became the opposite when the amount of K15M was low. Increasing Methocel®E15LV also increased Y4 (Figure 1(a)), while increasing DCP reduced Y4 (Figure 1(c)). This result is in agreement with the results obtained with regression Equation 3 in Table 2 in which the effects of X1 and X2 on Y4 depend on X3.
3.2. Optimization of the Formulation of Gliclazide Modified Release Tablets
Based on the data from the experimental results and dissolution data of Diamicron®MR 30 mg tablets, the range of optimal conditions are presented for dependent variables (Y1, Y2, Y4, Y6 and Y8) in Table 4. Running Modde 8.0 optimization software program using the D-optimal model, the final formulation was extrapolated and is shown in Table 5.
Three batches of 1000 tablets of the optimal formulation were prepared to confirm the predictability of the model. The dissolution profiles of the three batches of optimal formulation tablets were the same as that of Diamicron®MR 30 mg (f2 equal to 67.71, 80.09 and 80.39 for batch 1, 2, and 3, respectively).
Triple batches of 5000 tablets of optimal formulation were also prepared by the same wet granulation method, but using laboratory kneader, wet granulator, cube mixer and rotary tablet press (presented in Materials and Methods). Several properties of the granule (including Carr’s index, and moisture) and tablets (hardness, drug content and dissolution) were evaluated and shown in Table 6.
Table 6 shows that granules from all batches had good flowability (Carr’s index < 16). The assayed contents of drug from all batches were between 95% and 105%. The tablets’ hardness was within the range of 45 - 75 N and the weight variation of tablets complied with the general requirements for tablets. Thus, replication of three batches was shown at the scale of 5000 tablets using laboratory equipment.
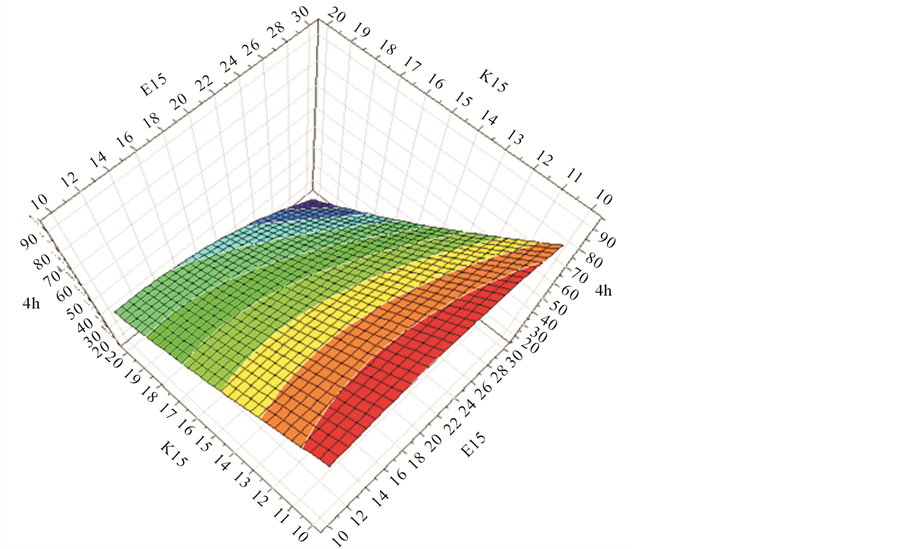 (a)
(a) (b)
(b)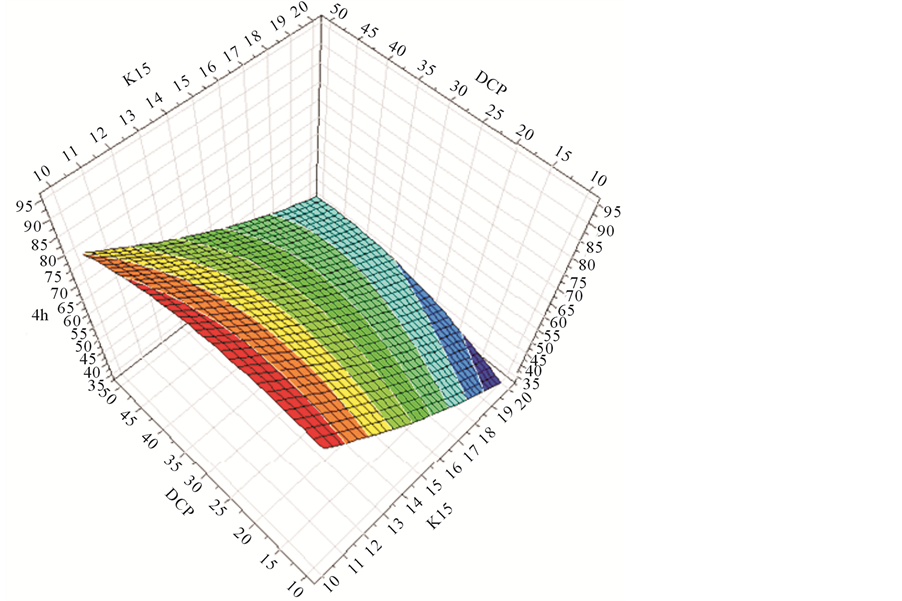 (c)
(c)
Figure 1. Response surface plots of input variables (X1, X2, and X3) versus the percentage of Gliclazide released at 4 hours (Y4).
Table 4. Optimal ranges of dependent variables.
Table 5. The optimal formulation of Gliclazide modified release tablets. Table 6. Physico-chemical properties of granules and tablets for three batches of optimal formulation. The dissolution profiles of tablets from three final batches of 5000 tablets are equivalent to dissolution profile of Diamicron®MR 30 mg tablets as shown in Figure 2 (all similarity factors, f2 > 70). Both the optimal test formulation (OF) and Diamicron®MR released gliclazide over eight hours with minimal differences in drug release at all times points. The dissolution results obtained were sufficient to permit analysis of the drug release profiles by the regression using Equation 3 which gave results that was able to describe and predict accurately the drug release profile from gliclazide sustained release tablets. 3.3. Effect of pH of Dissolution Media on Drug Release Figure 3 showed that both reference and test tablets released gliclazide at a greater rate at pH 1.2 compared to the slower rate in phosphate buffer at pH 4.5. The gliclazide tablets’ in vitro dissolution profiles in different media were similar to that of Diamicron®tablets, (similarity factors, f2 were 53.46; 51.67 and 51.13 when comparing dissolution of Diamicron®MR 30 mg and optimal formulation tablets in pH 1.2; phosphate buffer pH 4.5 and pH 6.8 media, respectively). The dissolution profiles of the optimized formulation tablets at pH 1.2 and 6.8 were Figure 2. Gliclazide dissolution profiles of three optimal formulation batches, 5000 tablets (n = 6): B1 = Batch 1; B2 = Batch 2; B3 = Batch 3. Figure 3. Dissolution profilesof Diamicron®MR 30 mg and optimal formulation (OF) in different pH solutions. Where similar (f2 is 61.4). However, the dissolution profile of the optimized formulation tablets at pH 4.5 differs from that of optimized formulation tablets at pH 1.2 and 6.8. 3.4. Mechanism of Gliclazide Release from Sustained Release Tablets Results of fitting of dissolution data to mathematical models are presented in Table 7. Table 7 shows that Korsmeyer-Peppas model best describes the drug release kinetics from gliclazide sustained release tablets (AIC value is the smallest). Korsmeyer-Peppas model is usually used to elucidate the drug release mechanism from a dosage form. If n = 0.89 or above the model indicates case II transport, where drug release does not change over time. The obtained diffusion exponent, n, of 0.832 shows that the mechanism of drug release follows case II transport or zero-order kinetics dominate over square root versus time drug release kinetics. The second choice is Hopfenberg model. Hopfenberg model shows geometry of a dosage form and release mechanism. The value n of Hopfenberg model is 1, 2 and 3 for a slab, cylinder and sphere respectively [20] . In this case, n equaled 1.540 indicating the tablets’ geometry is a cylinder. Zero-order release model is the third choice based on AIC values. Overall, drug release from gliclazide sustained release tablets more closely follows zero-order kinetics up to 8 hours than other mechanisms tested. 3.5. Convolution Using convolution calculations and assuming a linear relationship between dissolution and absorption for both Diamicron®MR and optimal formulation tablets, simulation was performed to predict the drug concentrations versus time curves for these two products in a bioavailability study. Figure 4 shows simulated plasma concentration curves versus time of Diamicron®MR and optimal formulation using dissolution data in two medium testing. Simulated Cmax’s of Diamicron®MR and the optimal formulation are not different by more than 10%. Furthermore, simulated gliclazide plasma concentrations versus time curves of optimal formulation are close to that Diamicron®MR, which implies that an in vivo AUC of the optimal formulation would be close to the AUC of Diamicron®MR. In both cases, bioequivalence is predicted. 3.6. Stability Studies Dissolution and drug contents results from the stability study in accelerated and ambient conditions are summarized in Figure 5 and Table 8. Other specifications of the tablets like tablet hardness and moisture content after 6 months in accelerated storage conditions and 12 months in ambient storage condition are similar to that at the beginning of the storage time (not shown). Figure 5 indicates that during storage, dissolution testing of gliclazide sustained release tablets produced similar release profiles to that of the reference product and itself at time t = 0 in both ambient and accelerated storage conditions (f2 > 50 in all cases). Gliclazide sustained release tablets were stable for 12 months in ambient condition and 6 months in accelerated condition. 4. Discussion Gliclazide is a hydrophobic weak acid, insoluble in water and acidic pH, but soluble in solutions having a pH near or rising towards neutral to alkaline pH. Variation in pH results in inconsistent and irregular release of drug from the commercial dosage form, which is not a desirable feature [9]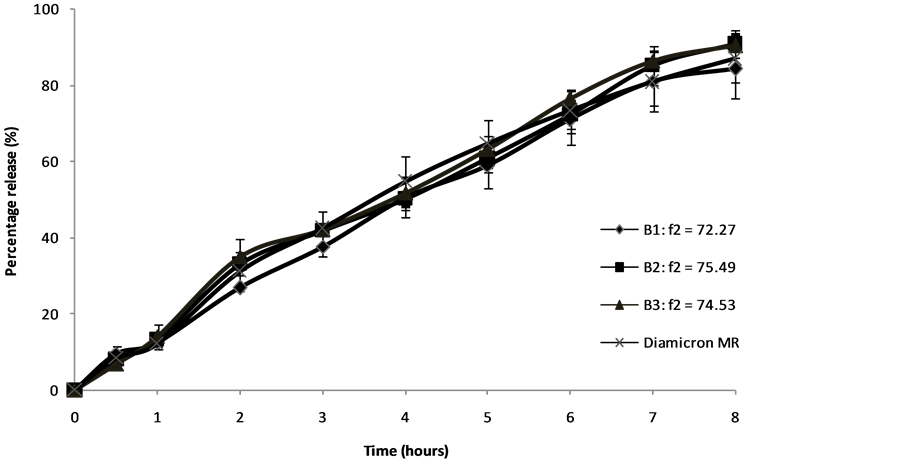

 = OF in pH 1.2,
= OF in pH 1.2,  = OF in pH 4.5,
= OF in pH 4.5,  = OF in pH 6.8;
= OF in pH 6.8;  = Diamicron®MR in pH 1.2;
= Diamicron®MR in pH 1.2;  = Diamicron®MR in pH 4.5,
= Diamicron®MR in pH 4.5,  = Diamicron®MR in pH 6.8.
= Diamicron®MR in pH 6.8.
Table 7. The summary of residual standard errors and AIC for the gliclazide dissolution profile of the optimal formulationfitted by regression to the drug release models.
C and Co is the percentage of drug released at time t and t = 0, respectively; k is the rate constant; σ is residual standard error of each model.
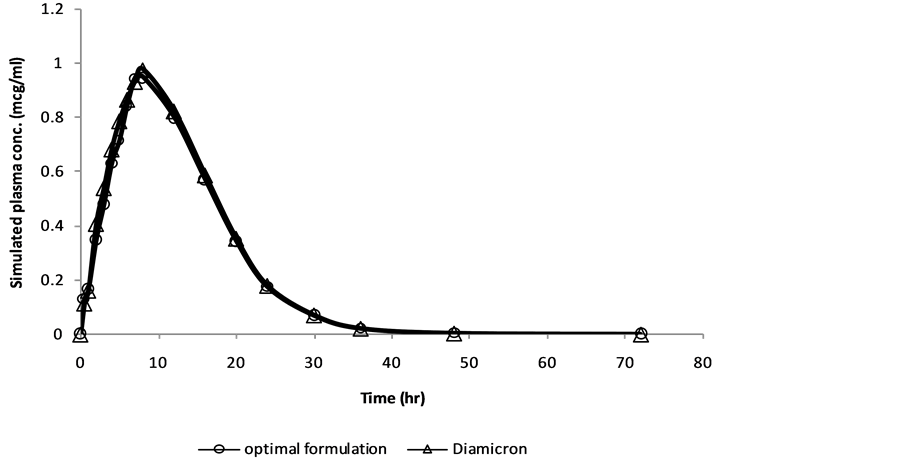
Figure 4. Simulated plasma concentration versus time curves of optimized formulation and Diamicron®MR: o = Optimized formulation; Δ = Diamicron®MR.
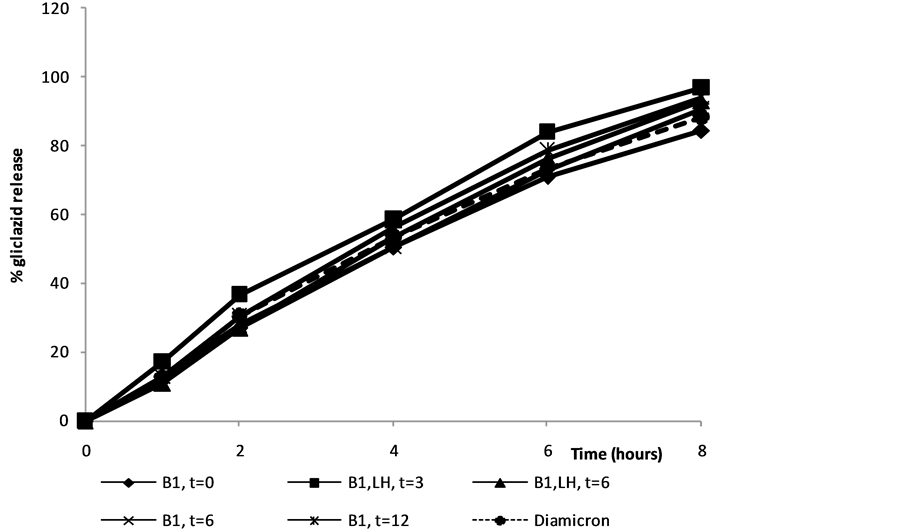
Figure 5. Dissolution profile of OF tablets from batch 1 under different storage conditions. Note:  : OF at time = 0;
: OF at time = 0;  : OF at time = 6 months, ambient condition;
: OF at time = 6 months, ambient condition;  : OF at time= 12 months, ambient condition;
: OF at time= 12 months, ambient condition;  : OF at time = 3 months, accelerated condition;
: OF at time = 3 months, accelerated condition;  : OF at time = 6 months, accelerated condition;
: OF at time = 6 months, accelerated condition;  : Diamicron®MR.
: Diamicron®MR.
and potassium dihydrogen phosphate to obtain an alkaline microenvironment around tablet to minimize the variation of drug release that is characteristic of gliclazide sustained release tablets in different pH dissolution media. However, the results of the dissolution profiles in different pH dissolution media while not exactly identical showed minimal differences in dissolution profiles at pH 1.2 and 6.8 that are acceptable based on f2-value (61.3).
Table 8. Contents and dissolution of gliclazide from optimal formulation in different storage conditions.
Note: a = ambient condition, b = accelerated condition; B2 = Batch 2, 5000 tablet, B3 = Batch 32, 5000 tabletsZero-order release of gliclazide from sustained release tablets was shown for the entire 8 hours of the dissolution test. The results agree with the results of Jin et al. (HPMC K15M, and sodium alginate, 2008) [7] , Raja Rajeswari et al. (HPMC 15 cps and Methocel®K4M, 2011), [8] and Srivastav et al. (HPMC K4M and K100LV) (2011) [9] . In Srivastav’s studies, the author used Zero-order, First-order, Higuchi and Korsmeyer Peppas model to elucidate the release mechanism. In this study, all possible release kinetic models are utilized in order to determine which best fit to the drug’s release. The zero-order release model was in better agreement to the drug dissolution release data of the optimized tablet formulation compared to the other models.
Utilizing MODDE 8.0 software to execute D-optimal mixture design with a polynomial statistical model in the study reduced the number of experiments and the contour surface response diagrams for multiple response optimization allowed rapid optimization of the formulation for the gliclazide modified release tablets. The preparation process for gliclazide modified release tablets was reproducible in the laboratory setting. Gliclazide sustained release tablets were stable for 6 months in accelerated storage conditions. The dissolution profiles remained unchanged throughout for both storage conditions. In particular, the dissolution profiles of gliclazide sustained release tablets were considered equivalent to that of Diamicron®MR 30 mg tablets in vitro.
5. Conclusion
An optimized formulation of a sustained release tablet of Gliclazide was developed. The use of D-optimal design to analyze dissolution data with a polynomial quadratic statistical model reduced the number of laboratory tests required to obtain an optimal dosage form and worked to provide results that rival surface response approaches in predicting an optimal formulation. The final optimized formulation (OF) of the sustained release 30 mg Gliclazide tablet contained 22 mg of Methocel®E15LV, 16.5 mg Methocel®K15M and 10.0 mg of Dibasic Calcium Phosphate. Dissolution studies performed on tablets from 5000 tablet test batches released greater than 90 percent of loaded drug in eight hours. The formulation is readily reproducible as three 5000 tablets batches showed no differences in drug release during dissolution drug testing. Drug release from the optimized tablets followed a pattern similar to zero-order more closely than other mechanisms rested. Storage of tablets in accelerated ambient conditions for 6 and 12 months respectively did not alter any of the physico-chemical properties of the tablet; drug release or the drug release rate compared to initial dissolution or measured data of the prepared tablets. The addition of potassium phosphate and monosodium phosphate dihydrate to the tablet reduced the effect pH has on Gliclazide dissolution compared to the commercially available Diamicron®MR product.
Acknowledgements
The authors declare no conflict of interest. Deep appreciation is given to the National Institute of Pharmaceutical Technology, Hanoi University of Pharmacy, Hanoi, Vietnam for their support to conduct this research.
References
- Naji, N., Nasir, I., Beshtawi, M., Mohammed, B., Admour, I., Mahmood, S.A., Zaman, Q. and Dham, R. (2002) Bioequivalence Evaluation of Two Brands of Gliclazide 80 mg Tablets (Gliclazide and Diamicron) in Healthy Human Volunteers. Biopharmaceutics and Drug Disposition, 23, 197-202. http://dx.doi.org/10.1002/bdd.310
- Hong, S.S., Lee, S.H., Lee, Y.J., Chung, S.J., Lee, M.H. and Shim, C.K. (1998) Accelerated Oral Absorption of Gliclazide in Human Subjects from a Soft Gelatin Capsule Containing a PEG 400 Suspension of Gliclazide. Journal of Controlled Release, 51, 185-192. http://www.dx.doi.org/10.1016/S01683659(97)00167-3
- Delrat, P., Paraire, M. and Jochemsen, R. (2002) Complete Bioavailability and Lack of Food-Effect on Pharmacokinetics of Gliclazid 30 mg Modified Release in Healthy Volunteers. Biopharmaceutics & Drug Disposition, 23, 151- 157. http://www.dx.doi.org/10.1002/dbb.303
- Barochez, B.H.D., Wuthrich, P. and Martin, L. (2004) Core Tablet for Controlled Release of Gliclazide after Oral Administration. US Patent No: 6733782.
- Murpani, D. and Madan, A. (2006) Modified Release Formulation of Gliclazide. US Patent Application WO 2006/ 123213 A1.
- Wit, J.B. and Doshi, H.A. (2006) Sustained-Release Formulation of Gliclazide. European Patent Application 2181705.
- Jin, X., Zhang, Y., Xaio, L. and Zhao, Z. (2008) Optimization of Extended Zero-Order Release Gliclazide Tablets Using D-Optimal Mixture Design. Yakugaku Zasshi, 128, 1475-1483. http://www.dx.doi.org/10.1248/yakushi.128.1475
- Raja Rajeswari, K. Abbulu, K., Sudhakar, M. and Ravi, N. (2011) Formulation and in Vitro Evaluation of Hydrogel Matrices of Gliclazide Modified Release Tablets. International Journal of Pharmacy, 1, 82-87.
- Srivastav, M., Prabharkar, B. and Omray, A. (2011) Effect of Alkaline Excipients on the Release Profile of Gliclazide Extended Release Tablets. Journal of Pharmaceutical Science and Technology, 3, 462-470.
- Vijayalakshmi, P., Devi, V.K., Narendra, C. and Srinagesh, S. (2008) Development of Extended Zero-Order Release Gliclazide Tablets by Central Composite Design. Drug Development and Industrial Pharmacy, 34, 33-45. http://dx.doi.org/10.1080/0363904071386129
- Jeyaprabha, P., Sudhamani, T., Mahendra, H., Ganesan, V. and Senthil, S.P. (2010) Formulation and Evaluation of Gliclazide Modified Release Tablets Using Hydroxypropyl Cellulose. International Research Journal of Pharmacy, 1, 282-287.
- Li, W., Du, G., Yang, Z., Zhang, Z., Nie, S., Peng, B. and Pan, W. (2008) In Vitro and in Vivo Evaluation of a Novel Push-Pull Osmotic Pump with Orifices on Both Surfaces. Drug Development and Industrial Pharmacy, 34, 1350-1355. http://www.dx.doi.org/10.1080/10673220802122928
- Kumar, M.S., Ramakrishna, R. and Kumar, N.N. (2010) Design and Development of Oral Controlled Release Formulations of Gliclazide Using Natural Polymers. International Research Journal of Pharmacy, 1, 233-242.
- Devarajan, P.V. and Sonavane, G.S. (2007) Preparation and in Intro/in Vivo Evaluation of Gliclazide Loaded Eudragit Nanoparticles as a Sustained Release Carrier. Drug Development and Industrial Pharmacy, 33, 101-111. http://www.dx.doi.org/10.1080/03639040601096695
- Varshosaz, J., Tavakoli, N., Minayian, M. and Rahdari, N. (2009) Applying the Taguchi Design for Optimized Formulation of Sustained Release Gliclazide Chitosan Beads an in Vitro/in Vivo Study. AAPS PharmSciTech, 10, 158-164. http://www.dx.doi.org/10.1208/s12249-009-9191-8
- Barakat, N.S. and Almurshedi, A.S. (2010) Design and Development of Gliclazide-Loaded Chitosan Microparticles for Oral Sustained Drug Delivery: In Vitro/in Vivo Evaluation. Journal of Pharmacy and Pharmacology, 62, 169-178. http://www.dx.doi.10.1111/j.2042-7158.2010.01214.x
- Wang, L., Wang, J., Lin, X. and Tang, X. (2010) Preparation and in Vitro Evaluation of Gliclazide Sustained-Release Matrix Pellets: Formulation and Storage Stability. Drug Development and Industrial Pharmacy, 36, 814-822. http://www.dx.doi.org/10.3109/03639040903520967
- Hussain, T., Saeed, T., Mumtaz, A.M., Javid, Z., Abbas, K., Awais, A. and Idrees, H.A. (2013) Effect of Two Hydrophobic Polymers on the Release of Gliclazide from Their Matrix Tablets. ACTA Poloniae Pharmaceutica-Drug Research, 70, 749-757.
- Kumar, C.S., Reddy-Budidti, K.K., Battula, S.P. and Ayyavala, C.S. (2013) Formulation and Evaluation of Ficusglomerata Mucilage Sustained Release Matrix Tablets. Pakistan Journal of Pharmaceutical Sciences, 24, 399-404.
- Costa, P. and Manuel, J. (2001) Modeling and Comparison of Dissolution Profiles. European Journal of Pharmaceutical Science, 13, 123-133. http://www.dx.doi.org/10.1016/S0928-0987(01)00095-1
NOTES

*Corresponding author.