Pharmacology & Pharmacy
Vol.5 No.3(2014), Article ID:43744,12 pages DOI:10.4236/pp.2014.53031
Solubility Enhancement of Domperidone Fast Disintegrating Tablet Using Hydroxypropyl-β-Cyclodextrin by Inclusion Complexation Technique
Prakash Thapa1#, Ritu Thapa2#, Uttam Budhathoki1†, Panna Thapa1*†
1Department of Pharmacy, School of Science, Kathmandu University, Dhulikhel, Nepal
2Department of Pharmaceutical Sciences, School of Health and Allied Sciences, Pokhara University, Lekhnath, Nepal
Email: *pannathapa@ku.edu.np
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 21 January 2014; revised 11 February 2014; accepted 28 February 2014
ABSTRACT
Domperidone Maleate (DOM), an antiemetic drug, has been used in treatment of adults and children. It has low aqueous solubility and hence low bioavailability. In present study, an attempt has been made to enhance the solubility of DOM by inclusion complexation with Hydroxypropyl-β- Cyclodextrin (HP-β-CD) using kneading technique and formulation of fast disintegrating tablets by using Sodium Starch Glycolate as superdisintegrant. Solubility analysis of DOM in different concentrations of HP-β-CD was carried out. Design of experiment (DOE) is done by using MINITAB 15.1 software to find out the variable for dissolution and disintegration time. HP-β-CD and SSG were identified as the variable for disintegration time and dissolution. For optimization of the concentration of HP-β-CD and SSG, two factors at two levels design through central composite design (CCD) were used which gave 13 formulations. All formulations are evaluated for characteristics such as weight variation, hardness, friability, disintegration time and dissolution of drug. Solubility of DOM increases linearly with increase in concentration of HP-β-CD. The optimum concentration of HP-β-CD is found to be in 1:2 molar ratios and SSG of 7%. The In-Vitro dissolution studies of optimized formulation and market sample were carried out in USP type II apparatus at different time intervals of 5, 10, 15 and 30 minutes at 50 rpm in 0.1 N HCl. The dissolution and disintegration time of optimized formulation is found better than market sample.
Keywords:Domperidone Maleate; Hydroxypropyl-β-Cyclodextrin; Inclusion Complexes

1. Introduction
Oral route is the most popular route of drug administration for both local and systemic actions. Among various oral dosage formulations, tablet is the most widely used formulation due to the various advantages that it offers in terms of drug manufacture and use [1] . Fast disintegrating/fast dispersible tablet (FDT) is the solid dosage form that disintegrates or dissolves into suspension or solution within 10 seconds to 1 minute in the oral cavity without requirement of water [2] . It is also known as Fast dissolving, Mouth dissolving, Rapid dissolve, Quick disintegrating, Orally disintegrating, Rapid melts, Fast melts, Melt in mouth, Quick dissolving, Porous and Effervescent drug absorption system (EFVDAS) [3] . It is a novel dosage delivery system where rapid disintegration of drug occurs after saliva reaches into the pores of formulation [4] .
It is easy to use, and has palatable taste, manufacturing elegance, improved stability and cost effectiveness [5] . Such formulation improves the therapeutic efficacy and patients compliance especially in paedriatic, geriatric and bedridden patients who have dysphagia [6] . There are various non-patented and patented techniques of manufacturing of such tablets [2] [7] . Oral drug delivery has drawback of low aqueous solubility and permeability through intestine leading to poor dissolution rate, poor absorption which ultimately cause lower oral bioavailability and hence therapeutic failure [8] [9] . Therefore, solubility plays an important role in dissolution of drug. There are various approaches to enhancing the dissolution profile of such drugs viz complexation, size reduction and solid dispersion. Formation of inclusion complexes is one of the precise methods of improving dissolution by complexation [10] . There are various techniques of inclusion complexation [11] . Among all, kneading method is convenient and economic. Inclusion complexation can be done in such formulation by the use of pharmaceutical excipients like Cyclodextrin (CD) where complexation occurs between guest and host molecules and hence, improves the aqueous solubility and which causes increase in oral bioavailability [12] . Chemically modified CD like 2-hydroxypropyl ß-CD (HP-ß-CD) has various applications in the field of drug delivery with improved pharmaceutical properties where the hydrophobic internal cavity of CD forms complex with the guest molecules to increase aqueous solubility [13] [14] . Domperidone is a poorly water soluble dopamine antagonist, antiemetic agent. It is a orally active prokinetic agent. Preparation of FDT by the use of Cyclodextrin would be very beneficial to the elderly and pediatrics with swallowing difficulties due to various physiological and neurological conditions by overcoming the drawbacks of oral drug delivery [15] .
2. Materials and Methods
2.1. Materials
Domperidone Maleate and its reference standard and other excipients; Hydroxypropyl-β-CD, Microcrystalline cellulose (Avicel PH102), Sodium Starch Glycolate, Crospovidone, Croscarmellose Sodium, Sodium Saccharin, Vanilla, Colloidal Silicon Dioxide and Magnesium Stearate were received from Deurali-Janta Pharmaceuticals Pvt. Ltd, Dhapasi, Kathmandu, Nepal as gift samples. A marketed oral disintegrating tablet was purchased from local retail pharmacy and was used as reference product for data analysis.
2.2. Preparation of Standard Working Solution
60 ml of methanol was added to 20 mg Domperidone maleate weighed accurately and sonicated for 10 minutes. Volume was made up to 100 ml by further addition of methanol. Then absorbance was measured at the range of 200 - 400 nm using UV Visible spectrophotometer (Shimadzu, Japan). λmax was found to be 287 nm.
2.3. Analytical Method Development
All the analytical procedures were validated for Accuracy, Precision, Limit of Detection, Specificity, Limit of Quantitation, Linearity and Range [16] .
2.3.1. Standard Calibration Curve
Absorbance of solutions of concenterations10, 15, 20, 25 and 30 µg/ml was measured using UV visible spectrophotometer at 287 nm. Then, linear equations as well as Correlation Coefficient (R2) were determined using the calibration curve.
2.3.2. Accuracy
It was determined by using the solutions of concentrations 16, 20 and 24 µg/ml of Domperidone Maleate that is ±20% the assay concentration. Recovery of the analytical result indicates the accuracy of method.
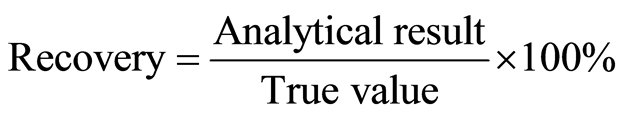
2.3.3. Precision
Assay was done of both the formulated batches and optimized batches. Relative Standard Deviation (RSD) less than 2% indicated the precision.
2.3.4. Specificity
Absorbance was measured of both working standard and excipients at the range of 200 - 400 nm in UV-visible spectrophotometer. No peak at 287 nm indicated specificity.
2.4. Solubility Analysis of Domperidone Maleate
50 mg DOM was mixed with 10 ml of HP-β-CD of 0%, 0.5%, 1%, 2%, 3% and 4%. The solution was then shaken for 24 hours and filtered through Whatsman filter paper No. 42. 1ml filtrate was then further diluted with water and volume made up to 50 ml. Finally, 2 ml of the above dilute solution was measured and volume made up to 20 ml with methanol. Then, absorbance was measured and solubility was determined [17] .
2.5. Design of Experiment (DOE)
MINITAB 15.1 software was used for DOE. Initially, nine excipients (HP-β-CD, SSG, CPVP, CCMC, SS, Vanilla, CSD, Avicel PH 102 and Magnesium Stearate) were chosen to formulate DOM FDT. Then, PlackettBurman Design (PBD) was applied to determine their role in dissolution and disintegration of tablet.
2.5.1. Preparation of Inclusion Complexes and Formulations of Plackett-Burman Design
DOM and HP-β-CD were mixed separately in the molar ratios of 1:0.5 and 1:2 respectively, sieved through sieve number 60 and mixed in mortar and pestle. Then the mixture was kneaded for 45 minutes by the addition of water. It was then dried at 40˚C in hot air oven, and pulverized and passed through 60 mesh sieve and stored in a desiccator [17] . Avicel PH102 and CSD were sieved through mesh size 40, whereas all other excipients were sieved through mesh size 60. The drug complex previously prepared was then mixed with excipients by geometric dilution method, except Magnesium Stearate in a double polythene bag for 15 min then this mixture was lubricated by Magnesium Stearate for 5 minutes. Finally, tablets were prepared by compression in a 10 station compression machine with punch size of 7.95 mm.
2.5.2. In-Vitro Dissolution Studies
Dissolution test was done by using USP dissolution apparatus type II where the DOM tablets were placed in 900 ml of 0.1 N HCL that acts as dissolution medium and maintained at 37˚C ± 0.5˚C and 50 rpm. 10 ml of the aliquot was taken out within 30 minutes and passed through Whatsman filter paper No. 1. Absorbance was measured at 284 nm using UV spectrophotometer [18] .
2.5.3. Surface response Design using Central Composite Design (CCD)
Based on PBD, two factors two levels CCD was applied and 13 experiments were carried out. Inclusion complexes of five different molar ratio of DOM: HP-β-CD was prepared as per experiments and experiments were carried out in same manner as previously described in PBD. Pre-compression parameter like, initial density, tapped density, Carr’s Index, Hausner’s ratio of the formulation were evaluated. Physiomechanical parameters like weight variation, assay, disintegration time, in-vitro dissolution test were performed.
2.5.4. Preparation of Optimized Formulation
On the basis of surface response plot and contour plot obtained from CCD, further optimization of concentration of HP-β-CD and SSG was carried out. DOM and HP-β-CD were taken in 1:2 molar ratios and SSG 7% (w/w). Preparation of inclusion complex and formulation processes were carried out as described in PBD. In-vitro dissolution was carried out by withdrawing 10 ml of aliquot at time intervals 5, 10 15 and 30 minutes, filtering through Whatsman filter paper No. 1 and measuring absorbance at 284 nm [18] . The result of dissolution and disintegration of optimized formulation was then compared with a marketed sample. Similarity and dissimilarity factor between CCD formulations, optimized formulation was compared with marketed formulations using the Equations (1) and (2) respectively.
1) Similarity Factor
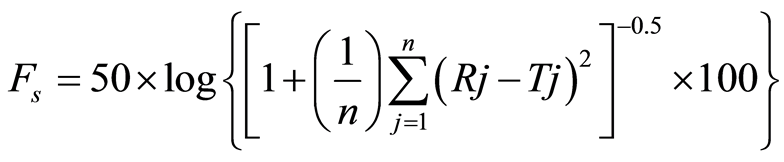 (1)
(1)
where n is the sampling number, Rj and Tj are the % dissolved of reference and the test products at each time points j respectively. fs value higher than 50 and close to 100 show the similarity of the dissolution profiles [19] .
2) Difference Factor
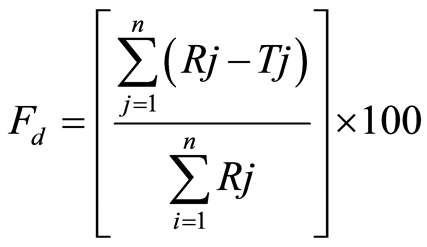 (2)
(2)
The percentage error is zero when the test and drug reference profiles are identical and increase proportionally with the dissimilarity between the two dissolution profiles. Fd values should be close to 0 to be similar. In general, the values lower than 15 or between 0 and 15 show the similarity of the dissolution profiles [19] .
3. Results
3.1. Validation of Analytical Method
3.1.1. Linearity
Absorbance was measured by UV Visible Spectrophotometer at 287 nm of the solutions of 10, 15, 20, 25 and 30 µg. From the working standard solution, the curve was plotted by taking absorbance on y-axis and concentration on x-axis (Figure 1). The graph shows that there is linear relationship between concentration and the absorbance with correlation coefficient (R2) 0.9994 and regression Equation (3).
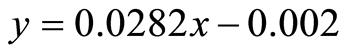 (3)
(3)
3.1.2. Specificity
After scanning the active ingredient and excipients in the range of 200 - 400 nm, in the UV-visible spectrophotometer, a prominent peak was observed by the active ingredient in methanol at 287 nm, while a flat line was

Figure 1. Standard calibration curve of Domperidone Maleate in Methanol.
observed of the placebo at the same range of wavelength.
3.1.3. Accuracy and Precision
To determine the accuracy and precision, 16 mg, 20 mg and 24 mg (each n = 3) of Domperidone Maleate was taken at 287 nm using UV-Visible spectrophotometer. This method was found to be accurate as the recovery values laid within the limit of 98.00% to 102.00% with a lower limit of 100.22% and upper limit of 101.88% while the relative standard deviation (RSD) was calculated and was found to be 0.927%. Thus, the method of analysis was found to be accurate and precise.
3.2. Solubility Analysis of Domperidone Maleate
DOM shows concentration dependent solubility in HP-β-CD as shown in Figure 2. Solubility of DOM increases linearly with increase in concentration of HP-β-CD. This shows that HP-β-CD is useful for enhancing the solubility of drug.
3.3. Plackett-Burman Design
Results of in-vitro dissolution test and disintegration time of PBD formulations were tabulated in Table 1. Among all 12 formulations, F8 shows lowest DT whereas F3 shows highest DT. Likewise, highest dissolution is achieved by F2 and F6 shows lowest dissolution. From the main effect plot Figures 3 and 4, HP-β-CD shows significant role in enhancing the dissolution of drug whereas SSG shows effect in minimizing the disintegration time. Thus both of these excipients were considered as factors for dissolution and DT respectively.

Figure 2. Solubility of DOM in different concentration of HP-β-CD.

Table 1. Design of experiment by Plackett-Burman design along with the parameters.
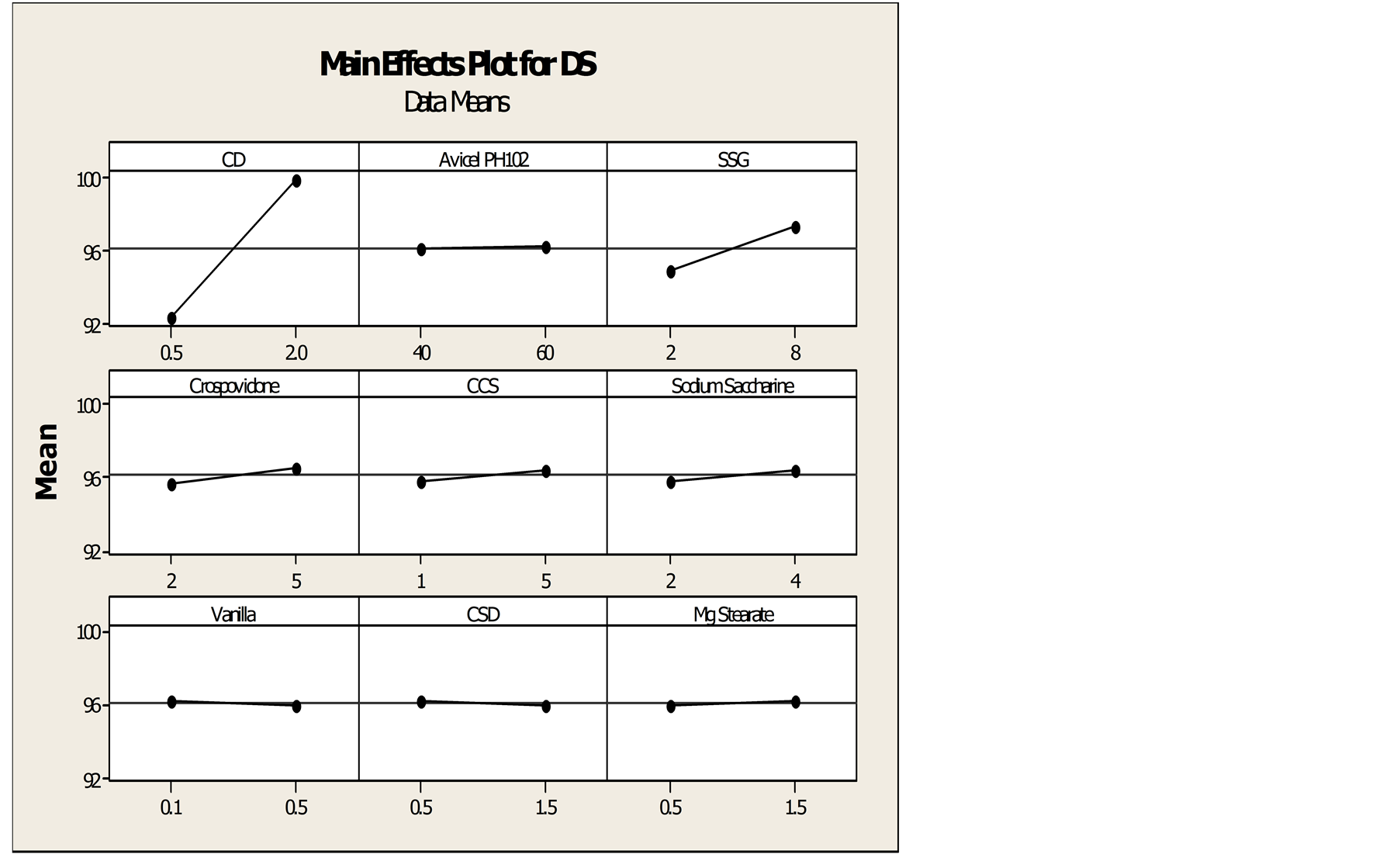
Figure 3. Main effect plots for dissolution (DS).
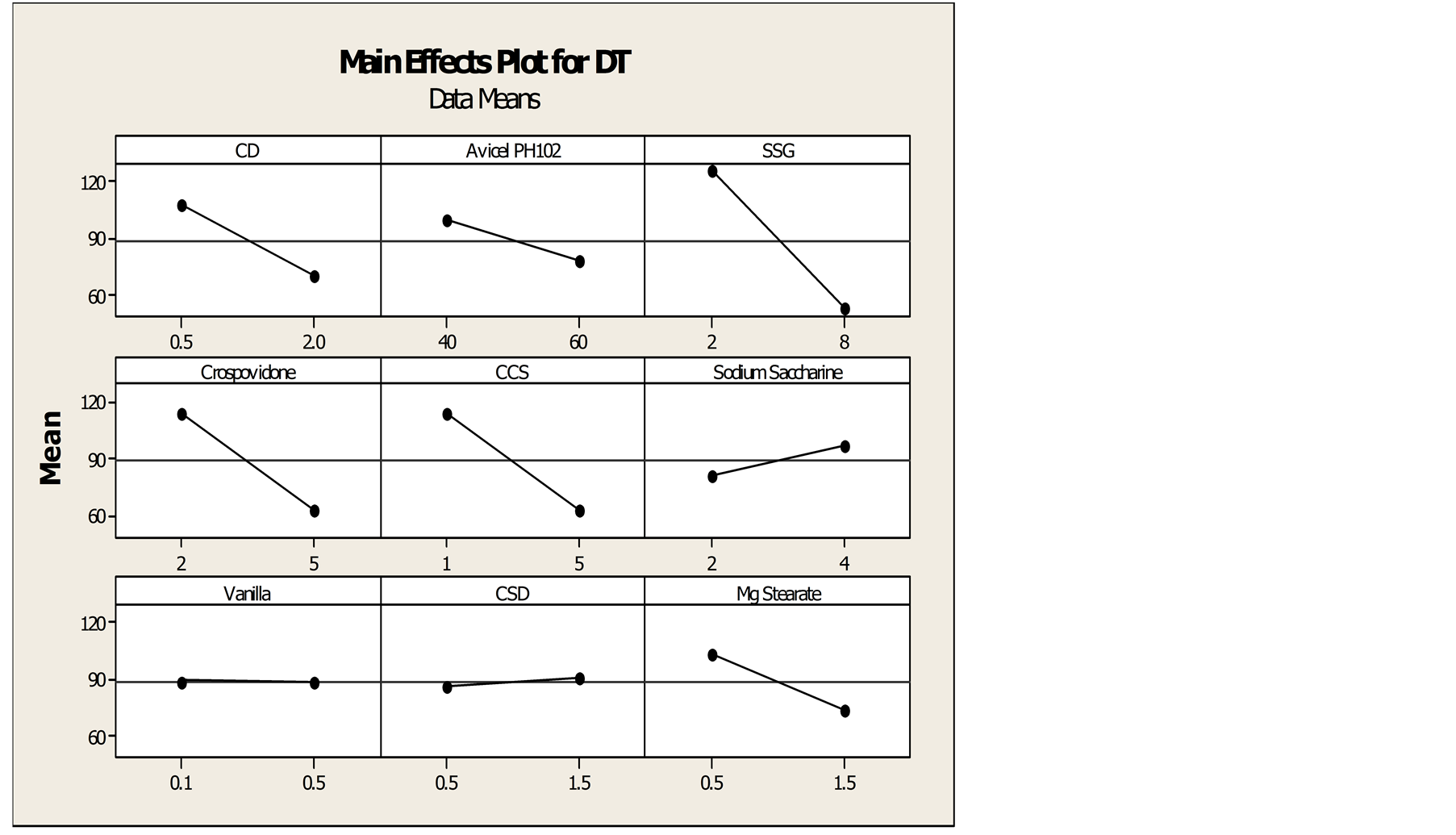
Figure 4. Main effect plot for DT.
3.4. Formulation and Evaluation of Physicochemical Properties of Surface Response Design Using Central Composite Design (CCD)
Pre-compression parameter like, initial density, tapped density, Carr’s Index, Hausner’s ratio of the formulation were evaluated. Formulation CCD F6 shows the Carr’s Index value 14 which indicates good flow of powder whereas formulation CCD F9 gives highest Carr’s Index value 21 indicates poor flow ability among other formulation. Carr’s Index of the formulated products was found in between 14% - 23% while tapped density ranges from 0.438 - 0.473 g/ml. Details of pre-compression parameters were tabulated in Table 2. Physicomechanical properties were presented in Table 3. Weight variation was in the range of ±5%. The hardness of the formulated products was found in between 2.11 - 4.06 Kg/cm2. The friability was found in the range of 0% - 1.1%. Thickness was found in the range of 1.893 - 1.986 mm. The assay values of the formulated tablets were found in between 95.22% and 103.87%. Disintegration time ranges from 19 - 125 sec. Result of in-vitro dissolution test and DT is shown in Table 4. Formulation CCD F6 shows lowest dissolution whereas highest dissolution is achieved by formulation CCD F1. Similarly, formulation CCD F4 has least disintegration time and formulation CCD F3 shows the highest DT. These results illustrate that HP-β-CD and SSG aid to improve dissolution and DT of DOM FDT. Further optimization of concentration of HP-β-CD and SSG by plotting contour plot and surface plot as shown in Figure 5, Figures 6 and 7 demonstrate that the optimum concentration of DOM:HP-β-CD is 1:2 molar ratio and SSG 7% w/w. Comparison of dissolution profiles of market sample and optimized formulation were shown in Figure 8. The result showed that the dissolution profile of market sample in 30 minutes was 93.06% whereas the optimized formulation had 100.12%.
Table 2. Result of pre-compression parameter of formulated batches by CCD.
Table 3. Physiochemical properties of formulated batches by CCD (weight variation, hardness, thickness, friability and assay).
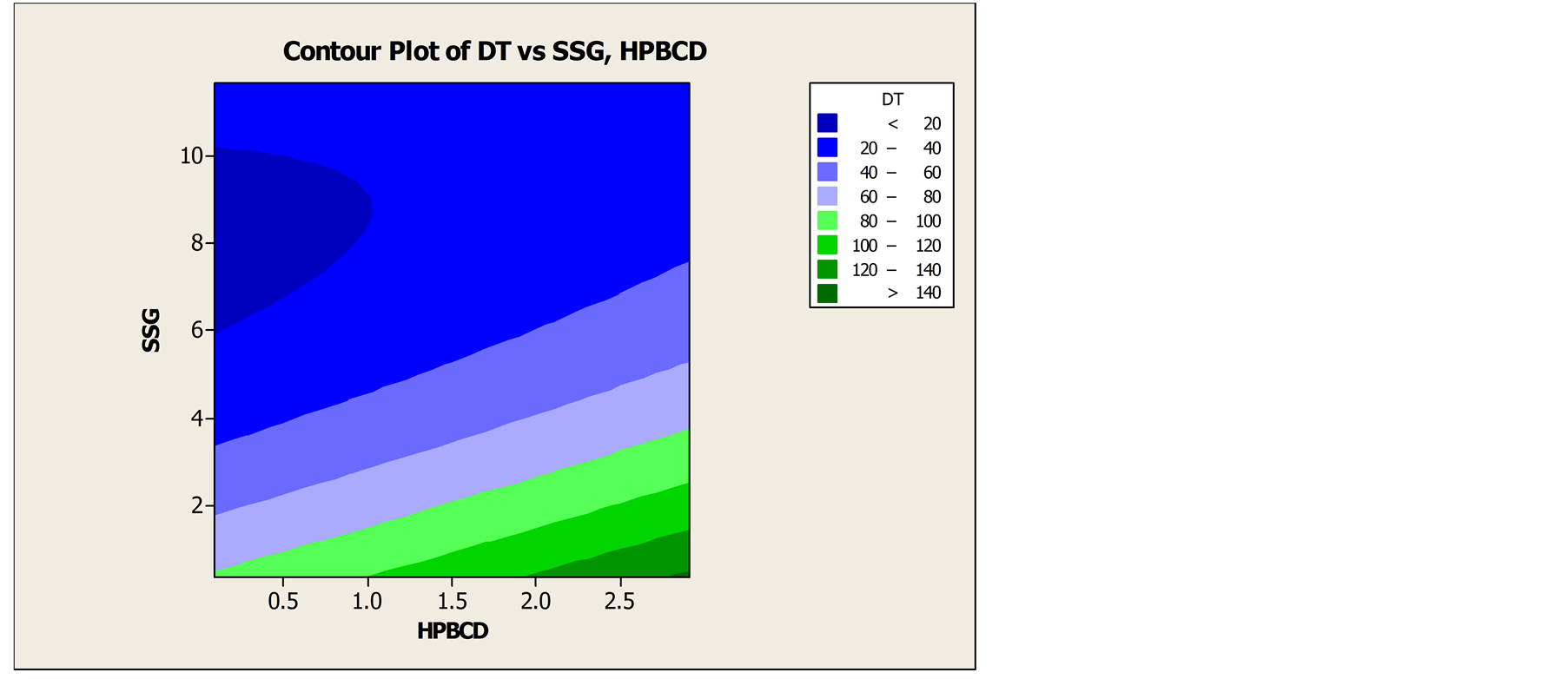
Figure 5. Contour plot of disintegration time vs. SSG and HP-β-CD.
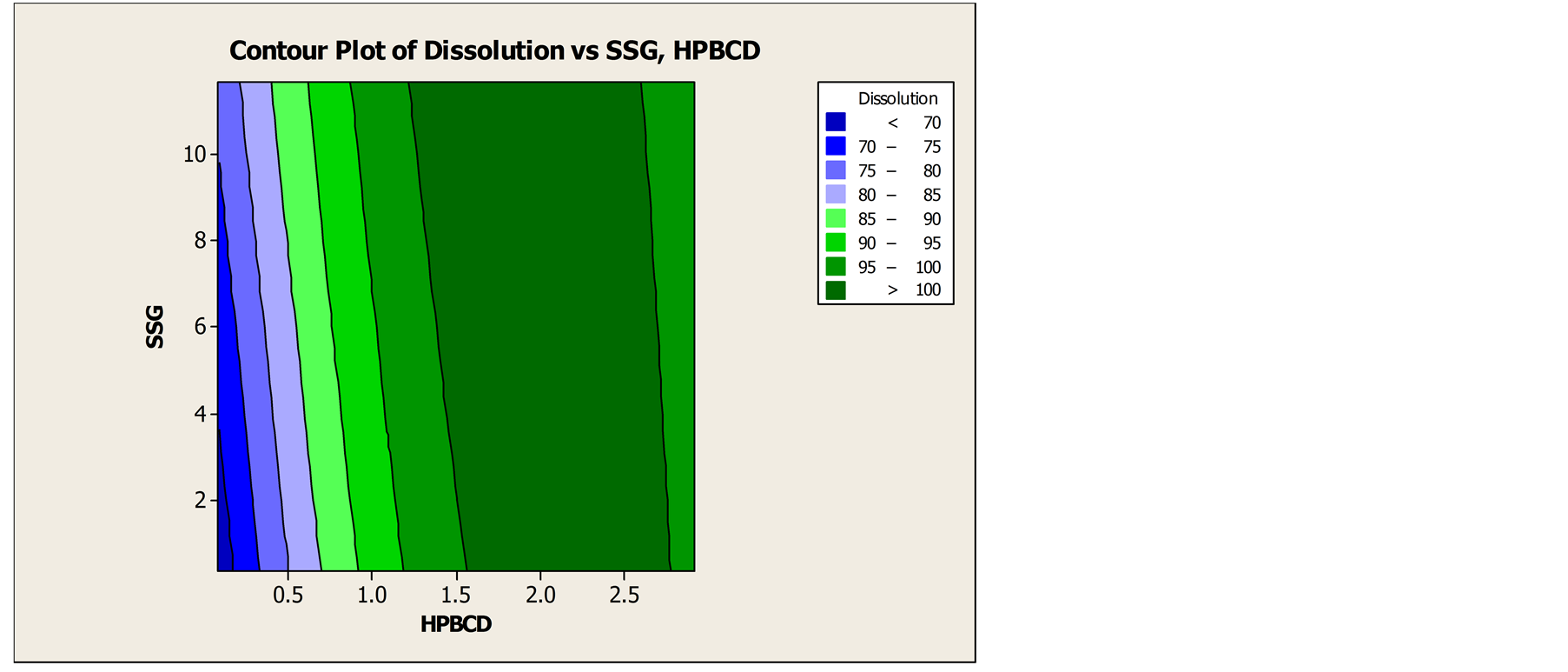
Figure 6. Contour plot of dissolution vs. SSG and HP-β-CD.
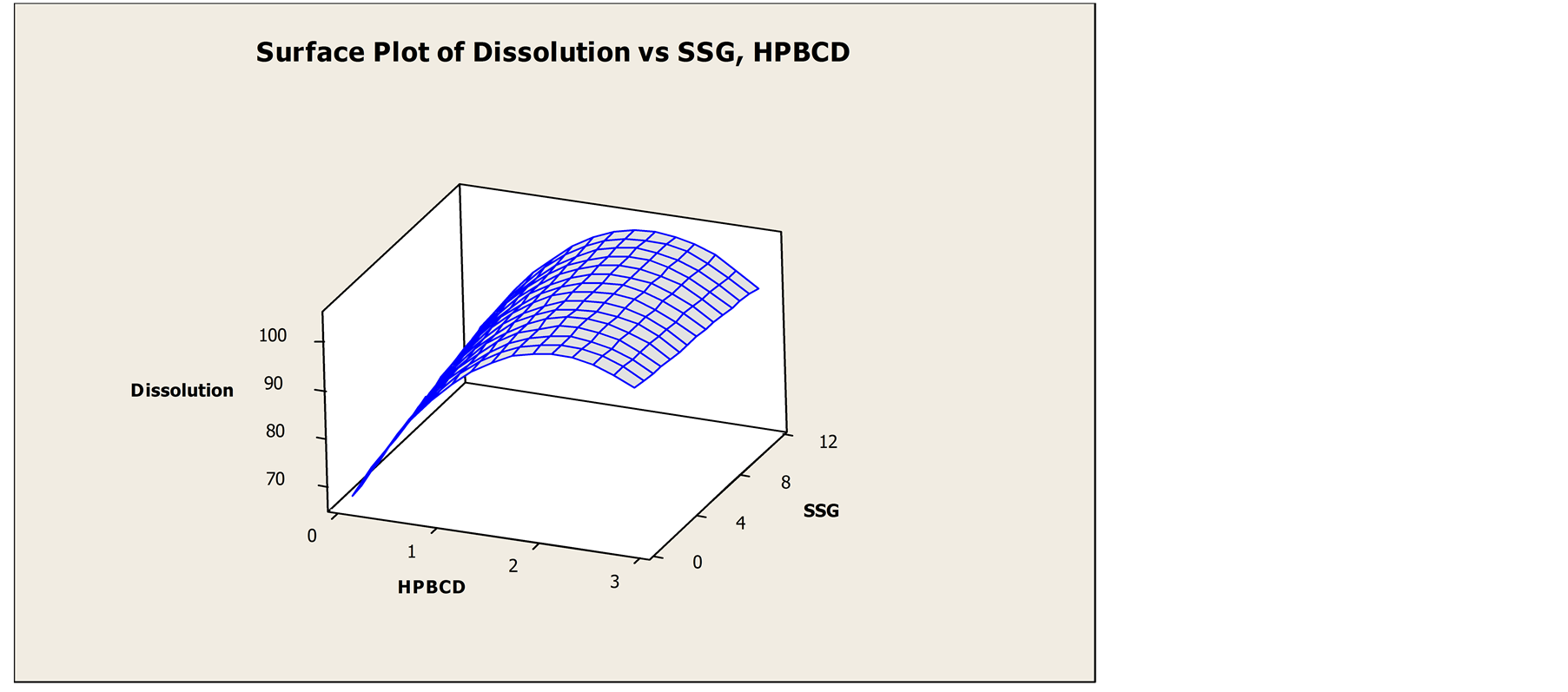
Figure 7. Surface plot of dissolution vs. SSG and HP-β-CD.

Figure 8. Dissolution profiles of optimized formulation and market sample in 0.1 N HCL.

Figure 9. Comparison of disintegration time of market sample and optimized formulation.
Table 4. DT and % drug release in 30 min in 0.1 N HCL.
3.5. Similarity and Dissimilarity Factors
For the more adequate dissolution profile comparison, similarity and dissimilarity factors were applied. The value of similarity and dissimilarity are given in Table 5. CCD Formulations that were similar to the market sample were CCD F1, CCD F2, CCD F4, CCD, F5 CCD F7, CCD F8, CCD F9, CCD F10, CCD F11, CCD F12, CCD F13 and OPT F14. The values in Table 5 clearly indicates that the drug release profile of market sample and these formulation are identical as the range of Fs value is 50 to 100 and the range of Fd is 0 to 15.
4. Discussion
Analytical method validation was done according to ICH guidelines to ensure the reliability of analytical method. Figure 1 shows a linear relationship between the absorbance and concentration of Domperidone Maleate. The λmax was observed at 287 nm in UV-Visible spectrophotometer. The recovery value of DOM was in the range of 98.00% - 102.00% and the RSD was also within the limit i.e. ±2%. All these values are as per the specifications of ICH guidelines. Hence, the procedures carried out were validated methods. The complexation of DOM with HP-β-CD was investigated by Phase Solubility Study. The concentration dependent increment in the solubility as shown by Figure 2 indicates that HP-β-CD is the major factor to enhance dissolution of drug. This can be further illustrated by the main effect plot of Plackett-Burman Design. Disintegration and dissolution time for the formulations made by Plackett-Burman Design as shown in Table 1 was different and hence to find out the reason of such variations, Main effect plot was plotted. After analyzing the results from Main effect plot obtained from Minitab 15.1, it is known that increase in dissolution was due to HP-β-CD and disintegration was due to SSG as shown by Figures 3 and 4. Hence, CCD was applied to find out the optimum concentration of HP-β-CD and SSG. Carr’s Index value 14% - 13% and Hausners ratio 1.22 - 1.29 as shown by Table 2 indicates that the powder has good blending property and hence is suitable for compression while preparing tablets. The tablets so prepared also are in the limits of pharmacopoeial standards. Hardness value 2.11 - 4.06 Kg/cm2 and friability 0% - 1.1% indicates that the tablets can be handled well. Figure 9 shows that disintegration time of optimized formulation is lower than that of marketed formulation. Moreover, Figure 9 shows that dissolution of optimized formulation was higher than marketed formulation. This value of disintegration and dissolution supports higher
Table 5. Similarity and dissimilarity factor.
bioavailability of the optimized formulation. This strongly supports the role of HP-β-CD in enhancing the bioavailability of Domperidone.
5. Conclusion
Fast disintegrating tablet of Domperidone was prepared by inclusion complexation using HP-β-CD by kneading method. Solubility of Domperidone was significantly enhanced by the use of HP-β-CD. Likewise, disintegration was enhanced by use of SSG. The optimized formulation meets all the criteria of FDT. Hence, HP-β-CD is effective in enhancing solubility of poorly water soluble drugs of lower molecular drugs like Domperidone.
Acknowledgements
I would like to acknowledge Department of Pharmacy, Kathmandu University for providing its support to conduct the research. I am also very thankful to Deurali-Janta Pharmaceuticals Pvt. Ltd. for providing various raw materials and reagents and granting permission to use the equipments in Research and Development department necessary to conduct the project.
References
- Ansel, H.C., Popovich, N.G. and Allen, L.V. (2005) Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems. 8th Edition, Lippincott Williams & Willkins.
- Zade, P.S., Kabitkwar, P.S. and Sakarkar, D.M. (2009) Formulation, Evaluation and Optimization of Fast Dissolving Tablet Containing Tizanidine Hydrochloride. International Journal of Pharm Tech Research, 1, 34-42.
- Sharma, D. (2013) Formulation Development and Evaluation of Fast Disintegrating Tablets of Salbutamol Sulphate for Respiratory Disorders. ISRN Pharmaceutics, 2013, 1-13. http://dx.doi.org/10.1155/2013/674507
- Chandrasekhar, R., Hassan, Z., Alhusban, F., Smith, A.M. and Mohammed, A.R. (2009) The Role of Formulation Excipients in The Development of Lyophilized Fast-Disintegrating Tablets. European Journal of Pharmaceutics and Biopharmaceutics, 72, 119-129. http://dx.doi.org/10.1016/j.ejpb.2008.11.011
- Amin, A.F. (2006) Emerging Trends in the Development of Orally Disintegrating Tablet Technology.
- Okuda, Y., Irisawa, Y., Okimoto, K., Osawa, T. and Yamashita, S. (2009) A New Formulation for Orally Disintegrating Tablets Using a Suspension Spray-Coating Method. International Journal of Pharmaceutics, 382, 80-87. http://dx.doi.org/10.1016/j.ijpharm.2009.08.010
- Bhowmik, D., Chiranjib, B., Krishnakant, P. and Chandira, R.M. (2009) Fast Dissolving Tablet: An Overview. Journal of Chemical and Pharmaceutical Research, 1, 163-177.
- Salustio, P.J., Feio, G., Figuirinhas, J.L., Pinto, J.F. and Cabral Marques, H.M. (2008) The Influence of the Preparation Methods on the Inclusion of Model Drugs in β-Cyclodextrin Cavity. European Journal of Pharmaceutics and Biopharmaceutics, 71, 377-386. http://dx.doi.org/10.1016/j.ejpb.2008.09.027
- Sachan, R., Khatri, K. and Kasture, S.B. (2010) Self-Emulsifying Drug Delivery System a Novel Approach for Enhancement of Bioavalibility. International Journal of PharmTech Research, 2, 1738-1745.
- Savjani, K.T., Gajjar, A.K. and Savjani, J.K. (2012) Drug Solubility: Importance and Enhancement Techniques. ISRN Pharmaceutics, 2012, 195727.
- Ishiguro, T., Morishita, E., Iohara, D., Hirayama, F., Wada, K., Motoyama, K., Arima, H. and Uekama, K. (2011) Some Pharmaceutical and Inclusion Properties of 2-Hydroxybutyl-ß-cyclodextrin Derivative. International Journal of Pharmaceutics, 419, 161-169. http://dx.doi.org/10.1016/j.ijpharm.2011.07.044
- Osborne, R.J., Slevin, M.L., Hunter, R.W. and Hamer, J. (1985) Cardiotoxicity of Intravenous Domperidone. Lancet, 386, 385. http://dx.doi.org/10.1016/S0140-6736(85)92515-2
- Challa, R., Ahuja, A., Ali, J. and Khar, R.K. (2005) Cyclodextrins in Drug Delivery: An Updated Review. AAPS PharmSciTech, 6, 329-357. http://dx.doi.org/10.1208/pt060243
- Goel, H., Vora, N. and Rana, V. (2008) A Novel Approach to Optimize and Formulate Fast Disintegrating Tablets for Nausea and Vomiting. American Association of Pharmaceutical Scientists, 9, 774-781.
- Shimpi, S., Chauhan, B. and Shimi, P. (2005) Cyclodextrins: Application in Different Routes of Drug Administration. Acta Pharmaceutica, 55, 139-156.
- (1996) ASEAN Guidelines for Validation of Analytical Procedures. ICH Q2B.
- Nalluri, B.N., Chowdary, K.P., Murthy, K.V., Hayman, A.R. and Becket, G. (2003) Physicochemical Characterization and Dissolution Properties of Nimesulide-Cyclodextrin Binary Systems. AAPS PharmSciTech, 4, 6-17. http://dx.doi.org/10.1208/pt040102
- (2007) British Pharmacopoeia, II.
NOTES

*Corresponding author.
#First authors.
†Second authors.