Advances in Alzheimer's Disease
Vol.2 No.3(2013), Article ID:37276,16 pages DOI:10.4236/aad.2013.23012
Systems pharmacology modeling in neuroscience: Prediction and outcome of PF-04995274, a 5-HT4 partial agonist, in a clinical scopolamine impairment trial
![]()
1Pfizer Global Research and Development, Groton, USA; *Corresponding Author: timothy.nicholas@pfizer.com
2In Silico Biosciences, Lexington, USA
3ICON, North Wales, USA
Copyright © 2013 Timothy Nicholas et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 May 2013; revised 30 June 2013; accepted 17 July 2013
Keywords: Systems Pharmacology; 5-HT4 Receptor Partial Agonist; Scopolamine-Reversal
ABSTRACT
Background: 5-HT4 receptors in cortex and hippocampus area are considered as a possible target for modulation of cognitive functions in Alzheimer’s disease (AD). A systems pharmacology approach was adopted to evaluate the potential of the 5-HT4 modulation in providing beneficial effects on cognition in AD. Methods: A serotonergic synaptic cleft model was developed by integrating serotonin firing, release, synaptic half-life, drug/tracer properties (affinity and agonism) as inputs and 5-HT4 activity as output. The serotonergic model was calibrated using both in vivo data on free 5-HT levels in preclinical models and human imaging data. The model was further expanded to other neurontransmitter systems and incorporated into a computer-based cortical network model which implemented the physiology of 12 different membrane CNS targets. A biophysically realistic, multi-compartment model of 80 pyramidal cells and 40 interneurons was further calibrated using data reported for working memory tasks in healthy humans and schizophrenia patients. Model output was the duration of the network firing activity in response to an external stimulus. Alzheimer’s disease (AD) pathology, in particular synapse and neuronal cell loss in addition to cholinergic deficits, was calibrated to align with the natural clinical disease progression. The model was used to provide insights into the effect of 5-HT4 activation on working memory and to prospectively simulate the response of PF- 04995274, a 5-HT4 partial agonist, in a scopolamine-reversal trial in healthy human subjects. Results: The model output suggested a beneficial effect of 5-HT4 agonism on working memory. The model also projected no effect or an exacerbation of scopolamine impairment for low intrinsic activity 5-HT4 agonists, which was supported by the subsequent human trial outcome. The clinical prediction of the disease model strongly suggests that 5-HT4 agonists with high intrinsic activity may have a beneficial effect on cognition in AD patients.
1. INTRODUCTION
The use of systems pharmacology modeling in drug development is growing across different disease areas [1, 2]. The concept is derived from the large amount of data and connections (systems biology) that can be generated for physiological systems and the need to understand the quantitative relationship of processes in the disease setting. Where systems biology creates the “map” of a disease, it is the systems pharmacology model that connects the “locations” on the “map” in a quantitative manner. As a picture of the U.S.A. would convey that Berkeley, California is west of Storrs, Connecticut; a quantitative map would allow one to determine not only the direction from one place to another but also quantify the time and speed needed to get there for the most efficient travel route. In the same way, a quantitative systems pharmacology model provides both the magnitude and the direction to have an effect for a given disease area target.
Quantitative drug development is an iterative process in which one collects data, builds quantitative models, generates and tests hypotheses, and then integrates the observations back into the model. Hence taking facts and transforming them into knowledge can be used to simulate and interpret further iterations (Figure 1). This report documents how a systems pharmacology model was used in the development of a novel therapeutic for the treatment of Alzheimer’s disease.
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly and accounts for 50% to 70% of all dementias. AD is clinically characterized by a progresssive memory loss, behavioral disturbances and the inability to perform daily living activities [3].
AD has been histopathologically characterized by amyloid plaque deposition and neurofibrillary tangles (NFT). The pathophysiological progression of AD in the brain has been segregated into six stages [4,5] initiating in the transentorhinal region with mild hippocampal involvement (stages I & II). The pathology increases in severity and neurofibrillary tangles (NFT) in pyramidal neurons [4,6,7] and “ghost tangles” become apparent. By the later stages the majority of the hippocampal region and the isocortex are severely affected [4]. Specific neuronal cell types, characterized by lipofuscin-laden cortical projecttions with long, thin, sparsely myelinated axons were identified as the most vulnerable in the progression of
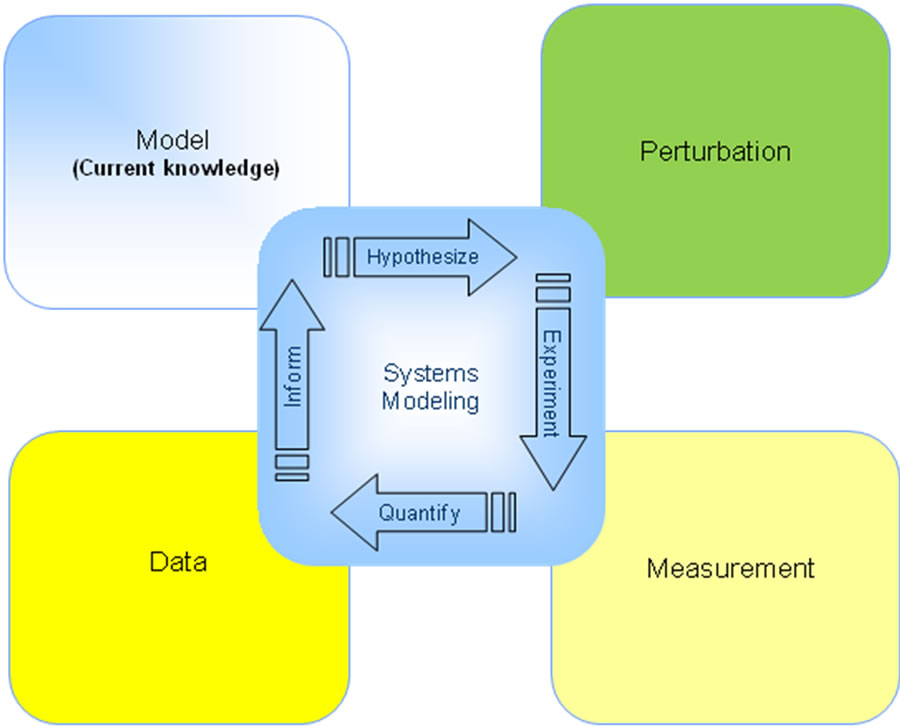
Figure 1. Schematic approach of the Learn-and-Confirm strategy at the heart of quantitative systems modeling. Basically a computer model is developed based on the current knowledge about the neurophysiology; the neuropathology associated with the disease (in this case scopolamine cognitive impairment or Alzheimer’s disease) is added and the clinical effect of a drug intervention is quantitatively simulated. This result is then compared with the actual clinical outcome for the same experiment and feedback allows improvement to the current set of model hypotheses.
AD pathology [8].
The serotonergic neurotransmission system has recently been documented to impact cognition and regeneration Alzheimer’s disease. Recently, a 5-HT6 antagonist SB- 742457 was shown to improve cognitive clinical readouts and mild to moderate AD [9,10]. Studies with amyloid imaging agent PIB-1 suggested that antidepressant use was correlated with lower amyloid load in the brain of Alzheimer’s disease patients [11]. With regard to this paper, the serotonin 5-HT4 (5-hydroxytryptamine 4) receptor is a G-protein receptor that is distributed throughout the body. The distribution of 5-HT4 in the gastrointestinal tract has made this a target for gastro-esophageal reflux disease and other gastro intestinal indications. More recently 5-HT4 receptor agonists have been investigated for possible symptomatic treatment of AD due to their distribution within the brain, primarily in the hippocampus and the cortex. Promnesic activity is thought to be mediated via cyclic adenosine monophosphate (cAMP) secondary messenger system through the inhibition of intra-neuronal calcium and voltage sensitive potassium channels. Additionally, 5-HT4 receptor agonism has been reported to increase acetylcholine release in the cortex and hippocampus [12-15] and to increase the production of soluble amyloid precursor protein alpha (s-APPα) [16- 18].
Previous trials with 5-HT4 modulators for cognition did not show a clear clinical benefit; it is unknown whether this is due to insufficient functional effect or incomplete translation of the biology from rodent to the human. Furthermore the distribution of receptor isoforms is different between rodents and humans [19,20].
One way to assess the impact of this translational disconnect is based upon a quantitative systems pharmacology approach [21,22]. A mechanistic computer simulation of brain circuits relevant for cognitive performance is developed based upon preclinical neurophysiology, human imaging/post-mortem data and calibrated using human clinical outcome data.
For instance, increasing evidence suggests that the excitatory-inhibitory balance in cortical and hippocampal networks is fundamentally different between primates and rodents [23]. Monkey basket interneuron cells have a higher input resistance and a lower firing threshold and generate more spikes at near-threshold current intensities. Different interneuron subtypes are found in the primate cortex, with short spike duration, which is not typical for rodent adapting cells [24]. Furthermore, the developmenttal shift in gamma-aminobutyric acid (GABA) (A) receptor alpha subunit expression continues through adolescence in primate cortex, but not in rodents, suggesting species-difference kinetics of GABA neurotransmission [25].
Decreased cholinergic function has been considered one of the abnormalities observed in AD pathology. It is more efficient to study the cholinergic hypothesis by examining in vivo models of cholinergic impairment rather than the AD itself. Similar cognitive deficits as seen in dementia may be generated by the administration of muscarinic receptor antagonists, such as scopolamine [26,27]. Scopolamine competitively inhibits acetylcholine binding to muscarinic receptors and acts as a nonselective muscarinic antagonist. It has been shown to cause cognitive deficits following intravenous (IV) or subcutaneous (SC) administration in healthy volunteers [27-36]. The cognitive effects appear to lag behind the maximal plasma concentrations of scopolamine as described in a PKPD model [31]. The scopolamine model of cognitive impairment has been used in preclinical and clinical settings (normal healthy subjects) to explore the potential of procognitive compounds to reverse the cognitive effects due to cholinergic blockade. Validity of the scopolamine model is based on the similarity between the transient effects of scopolamine in healthy volunteers and the cognitive impairment exhibited in AD patients [27].
PF-04995274 is a new investigative partial 5-HT4 agonist that showed positive results in a preclinical animal model of object recognition during scopolamine-induced deficit (rat Morris water maze study) [37]. The effect of PF-04995274 was simulated using human pharmacology in a humanized environment, a priori, and went on to compare simulations with the clinical response in a human scopolamine-induced deficit paradigm. This is a unique opportunity to test the predictivity of a quantitative systems pharmacology approach. Conversely, feedback from this clinical trial can be used to improve the quantitative systems pharmacology to an improved predictivity level. Indeed learn-and-confirm iterations are at the heart of the quantitative drug development process [38]. This report shows that unlike traditional animal models a quantitative systems pharmacology approach is able to be significantly improved using feedback from actual predictions of clinical trials.
2. METHODS
2.1. Systems Pharmacology Modeling
The quantitative systems pharmacology approach for predicting cognitive effects in an Alzheimer model has been described in detail [39]. Briefly the platform consists of the following elements: 1) a receptor competition model that quantitatively describes the competition between endogenous neurotransmitter (NT), the parent compound and its active metabolite and a radiotracer for proper target engagement in a humanized environment and that is calibrated for serotonin (5-HT) and acetylcholine (ACh); 2) a biophysically realistic computer model of a cortical network that is involved in the maintenance of cognitive traces; 3) implementation of the scopolamine-induced changes and AD pathology related neuropathology and 4) dose-dependent effect of the 5-HT4 modulator on these scopolamine-induced changes and in AD pathology conditions.
2.2. Receptor Competition Model
The receptor competition model (Figure 2) has been described in detail elsewhere [40,41]. This computer model simulates the competition between 4 different agents for the same binding site on the postsynaptic membrane, within the neurophysiology of the realistic central nervous system (CNS) synapse. Presynaptic firings derived from in vivo measurements, while the effect of presynaptic autoreceptor coupling on subsequent neurotransmitter release will be calibrated using both preclinical fast cyclic optometry data and human imaging experiments (see results section). In brief, changes in dynamical binding and unbinding of the different agents to the receptor sites are calculated using ordinary differential equation (ODE) (Eq.1)
 (1)
(1)
with the initial condition that all receptors begin in the free state (subscript and superscript n here refers to the neurotransmitter). Similar equations are used for drug, metabolite (or other compound) and tracer.
The amount of free neurotransmitter depends on two processes, exponential decay and quantal release. Expo-
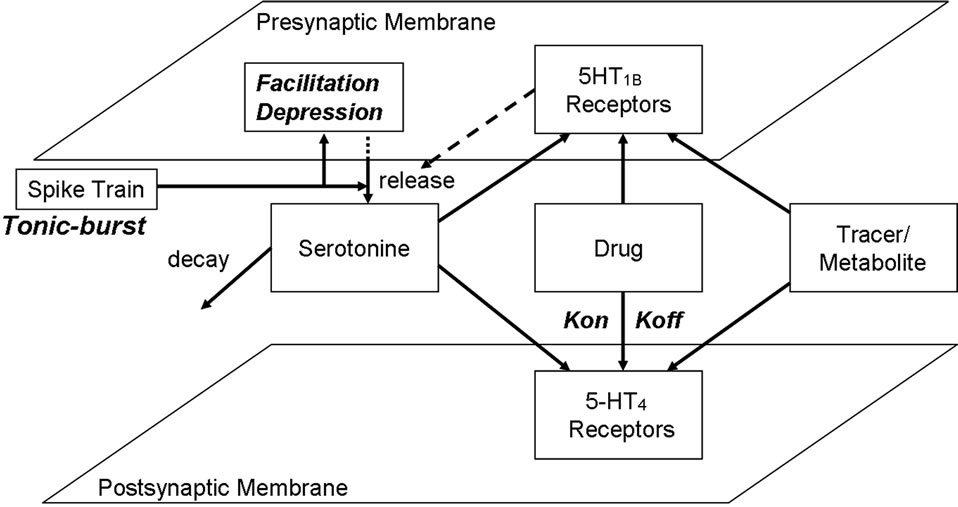
Figure 2. Representation of the generic receptor competition model (see text for more information). The model allows userdefined presynaptic firing patterns for neurotransmitter release and simulates the effect of presynaptic autoreceptor negative feedback on presynaptic neurotransmitter release, facilitation and depression of synaptic release, the decay of serotonin in the cleft due to diffusion, transporters and enzymes, the competition between four agents (neurotransmitter, and up to two drugs and a tracer) and the dynamics of kon/koff binding of each of these agents to their respective receptors using ordinary differential equations (see text), at millisecond time resolution. The output is the time-dependent activation level of preand postsynaptic serotonin receptors, the fraction of each agent bound to these receptors in the low and high affinity state as well as the concentration of free serotonin in the cleft.
nential decay is classically defined as Eq.2
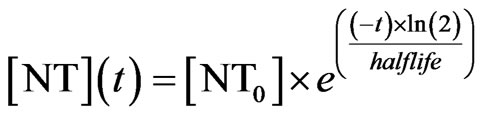 (2)
(2)
where halflife is the half-life of the decay process. At times of release, [NT] is immediately updated by adding the release amount.
The parameters were calibrated so that the coupling of presynaptic 5-HT1B receptor activation to serotonin release reflects actual experimental data in rodents and humans. All differential equations are solved with a fourth-order Runge-Kutta method with a time step of 0.01 msec.
The release can be modulated by a depression or facilitation mechanism [42]. Instead of using internal Ca2+ levels to determine serotonin release, we consider the facilitation and depression of serotonin release based solely on the amount of time elapsed since the previous firing using a phenomenological equation. Thus, the amount of serotonin released is based both on the history of firing and the activation level of the presynaptic 5- HT1B autoreceptors.
This program is coded in Java and visualization as well as manipulation is handled with graphical user interface (GUI) routines.
2.3. Calibration of the Serotonergic Synapse
The serotonergic synapse is calibrated using both in vivo experimental data on free 5-HT levels in preclinical animal models and human imaging data using specific radiotracers, a full detailed description has been published [39]. Basically, the preclinical data measures the free serotonin levels during forced firing frequency of the presynaptic terminals and therefore probe the effect of presynaptic 5-HT1B autoreceptor coupling and facilitation/depression on the release of 5-HT. 5-HT1B is the most important autoreceptor for most projection serotonergic neurons, while 5-HT1A is the major autoreceptor regulating dorsal raphae (DR) firing [43].
Fast cyclic voltametry data in mouse slices of substantia nigra [44] which have been shown to be rich in serotonin innervation was used for validation. Free 5-HT levels are measured after forced firing which ensures that only the effect at the presynaptic 5-HT1B autoreceptor is measured.
An important issue is to quantify the intrasynaptic 5-HT detected by the fast cyclic voltammetry probes. Modeling studies of the glutamate synapse [45] suggest that intrasynaptic levels can be between 1 and 20 times the measured extrasynaptic levels.
Due to the importance of free 5-HT level information in the human situation, the calibration identified the ratio of extravs. intrasynaptic free 5-HT, that has the highest correlation between the output of the synaptic model and actual clinical data in human imaging experiments.
Free 5-HT levels, in human brain, were estimated with the results of PET radiotracer displacement imaging studies using 5-HT receptor specific radio-tracers (WAY 100635 with an affinity of for 0.3 nM 5-HT1A, MPPF with an affinity of 3.1 nM for 5-HT1A and altanserin, setoperone with an affinity of 0.43 and 0.3 nM for 5-HT2A respectively).
Target engagement for the active moiety is calculated as the displacement of the 5-HT4 receptor specific radiotracer SB202644 for which we assume a Ki of 0.18 nM. Using the receptor competition model we then calculate the concentration of the active moiety leading to a specific level of target engagement and then using this concentration to determine the effect on cognitive outcome after scopolamine-induced deficit.
For each of the clinical conditions mentioned above, the displacement of the 5-HT tracers by the appropriate functional brain concentration of the antipsychotic, given its known affinity for the human 5-HT receptor, can be simulated. The amount of tracer displacement will depend upon free 5-HT levels and is a result of complex interactions between tracer, drug and 5-HT. An alignment with the clinical imaging data gives a better idea of the actual 5-HT levels that are driving the 5-HT dynamoics at least in schizophrenia patients. It was assumed that these values can be extrapolated to normal healthy subjects, as schizophrenia is mostly associated with dopamine dysfunction, not 5-HT dysfunction [46]. Indeed, most genetic, clinical and neuroimaging data suggest that schizophrenia is mostly driven by dopamine, glutamate and GABAergic pathologies and that there are no overt serotonergic changes. This doesn’t exclude the presence of subtle serotonergic changes too small to be detected; unfortunately human imaging studies in schizophrenia patients are the only ones that are available for human calibration of the serotonergic dynamics. With these caveats in mind, we assume the 5-HT dynamics and healthy individuals can be calibrated from the imaging studies in schizophrenia.
Ideally one would like to quantify the binding of a specific radio-tracer before and after neuroleptic treatment to correct for any individual baseline variability of the receptor. Although this is possible with our model, it is usually difficult in the clinical setting so that many studies define a binding index (Eq.3) compared to a normal control population
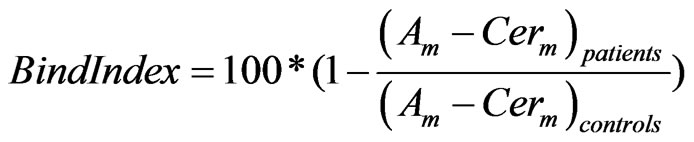 (3)
(3)
Where Am and Cerm are the specific signals in the region of interest (i.e. striatum and cerebellum, respectively).
We defined the apparent receptor occupancy (Eq.4) as
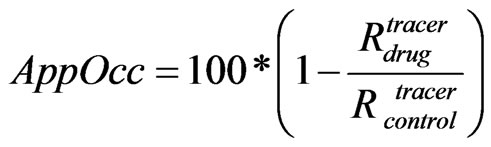 (4)
(4)
where Rdrug and Rcontrol are the receptor tracer occupanies respectively in the presence or the absence of the drug.
Radiotracer displacement is measured functionally and takes into account many confounding issues such as blood-brain barrier transport and free fraction etc. and reflects the actual true functional intra-synaptic concentration of the drug. The clinical imaging experiments include setoperone displacement with 30 mg aripiprazole, altanserin with 300 mg quetiapine, setoperone with 600 mg chlorpromazine, 200 mg clozapine and 10 mg amisulpride and 100 mg loxapine and amoxapine (for a full description see) [39].
Using the functional concentrations of the antipsyhotics derived from the raclopride displacement studies and the appropriate affinities of the schizophrenia drugs against the 5-HT receptor, the displacement of 5-HT2A receptor tracer for the seven clinical cases was simulated and compared the outcomes with the clinically reported data.
2.4. Cortical Network Model
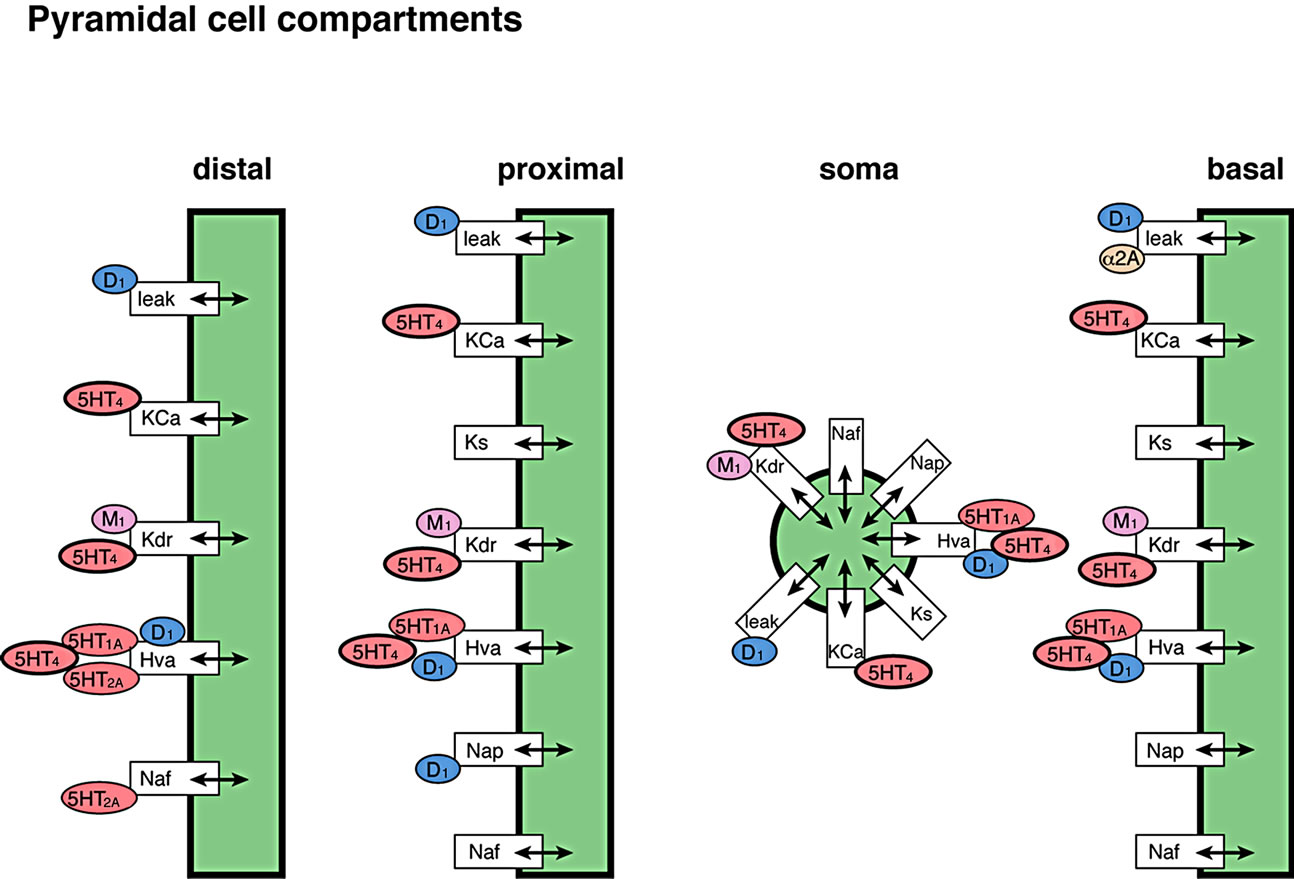
The cortical network model has been described in detail [39]. Basically a biophysically realistic model of a network was extended to comprise of 80 four-compartment pyramidal cells and 40 two-compartment GABA interneurons [47,48] with the receptor physiology of 18 different dopaminergic, serotonergic, noradrenergic, and cholinergic receptors (Figure 3). An mGluR5-dependent delayed after depolarization current that can increase the spiking rate of pyramidal cells for several seconds was implemented as an alpha function in the model with a time constant similar to the observation in [49]. Based on estimates of the relative number of pyramidal cells and interneurons [48,50], 40% of the interneurons synapsed with other GABA interneurons, but not with pyramidal cells.
5-HT4 receptors act to increase the excitability of cortical pyramidal cells, and may improve cognition, learning and memory [51,52]. The effects of 5-HT4 receptor activation in the working memory model was implemented by modulating the delayed rectifier K+ channel, the Ca2+ activated K+ channel and an interneuron-mediated GABA current in pyramidal cells.
Maximum activation of the 5-HT4 receptor reduces the delayed rectifier K-current by 50% [53]. In this implementation (Eq.5), the activation of 5-HT4 receptors reduces the maximum conductance (gKdr) by a linear factor of 0.5 so that
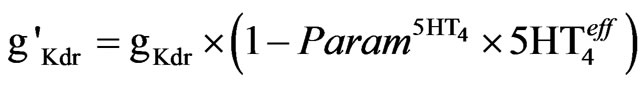 (5)
(5)
where  is the relative effect on 5-HT4 activation levels (see below). It is assumed that the control case is at a balanced level of enzymes so that when we apply the
is the relative effect on 5-HT4 activation levels (see below). It is assumed that the control case is at a balanced level of enzymes so that when we apply the
 (a)
(a)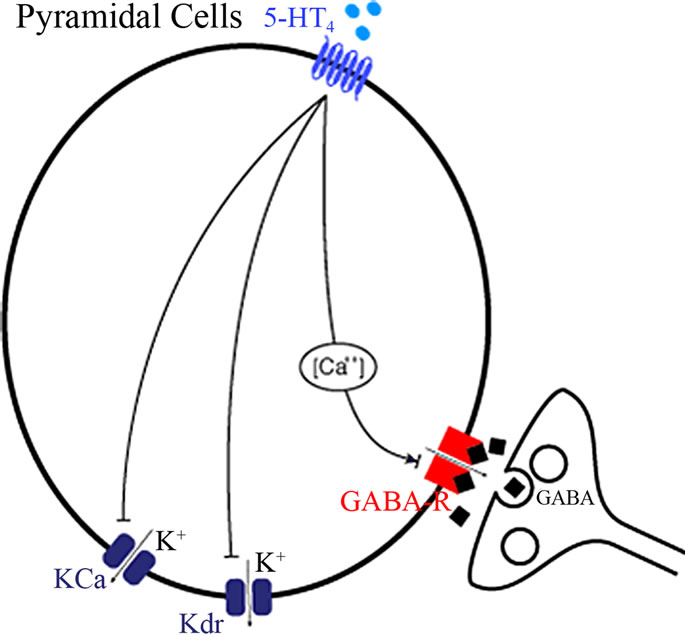 (b)
(b)
Figure 3. Implemented effects of 5-HT4 receptor physiology (see text for more details). (a) In individual neuronal cells, 5-HT4 receptor directly affects the Kdr and the Ca-mediated K channel to alter pyramidal cell membrane excitability in addition to the coupling of GABAAR effects through a Ca-mediated pathway; (b) Localization of the 5-HT4 receptor effect in relation to the other neuromodulatory receptors in the complete cortical network. Note that activation of the 5-HT4 receptor also indirectly increases 5-HT firing through an effect on Dorsal raphe excitability and therefore changes the 5-HT dynamics at 5-HT1A, 5-HT2A, 5-HT3 and 5-HT6 receptors.
equation with the values of the control activation level,  ,
,  is the relative difference from control activation level (Eq.6)
is the relative difference from control activation level (Eq.6)
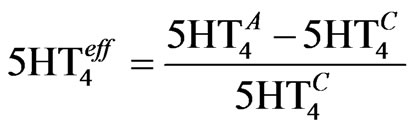 (6)
(6)
where 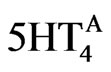 is the actual activation corresponding to the appropriate dose-response. The value for Param5HT4 is determined from the experimental data on the K+ channel [53] and corresponds to 0.2.
is the actual activation corresponding to the appropriate dose-response. The value for Param5HT4 is determined from the experimental data on the K+ channel [53] and corresponds to 0.2.
The 5-HT4 receptor activation reduced the Ca-activated K-currents, responsible for after-hyperpolarization [54], by regulating the release of calcium from internal stores. The model was simplified by directly reducing the maximum conductance (gKCa) of the calcium activated potassium current, as described in Eq.7.
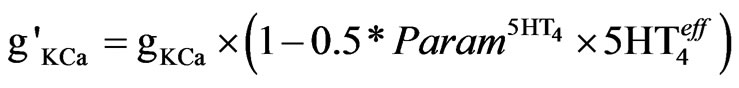 (7)
(7)
As noted above, the value of  is the relative difference from control activation level. The experimental values correspond to a coupling value which is 50% of the coupling for gKdr.
is the relative difference from control activation level. The experimental values correspond to a coupling value which is 50% of the coupling for gKdr.
Agonists of 5-HT4 receptors cause a bi-directional modulation of GABA-A currents in PFC pyramidal neurons [55], depending on the Protein Kinase A (PKA) activation levels in the cell. Internal calcium concentration was used for estimating the PKA activation because of the calcium dependence in PKA’s activation kinetics (Eq.8). A Michaelis-Menten scheme gives a range of ±1 so that
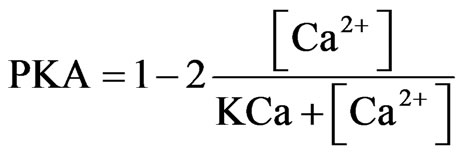 (8)
(8)
where [Ca2+] is the calcium concentration in the model cell and KCa is chosen by observing the calcium concentration in the middle between low and high activity with KCa = 0.0001.
The GABA-A current is modified by multiplying the maximum conductance (gGABA) by the PKA factor and by the %-activation of 5-HT4 receptors (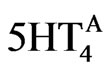 ) as described in Eq.9.
) as described in Eq.9.
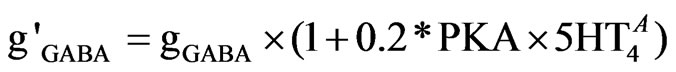 (9)
(9)
The range factor of 0.2 was determined form the physiological range of 5-HT4 receptor effects on GABA current [55].
In addition, 5-HT4 receptor activation increases DR firing, so that in general 5-HT tone is mediated indirectly by 5-HT4 [56,57]. The 5-HT4 receptors are co-localized with 5-HT1A receptors [51] and therefore localized in the same compartments.
2.5. Implementation of Cholinergic Pharmacology
Cholinergic physiology is implemented through the muscarinic acetylcholine receptor (M1 mAChR) and both the α7 and the α4β2 nAChR synapses, although pharmacology at the M2 mAChR can play a role at this presynaptic autoreceptor [39]. Briefly, their interactions are simulated using the cholinergic (muscarinic) receptor competition model.
Nicotinic cholinergic receptors are also included in the model because in AD, many patients are still on cholinomimetics such as acetylcholinesterase inhibitors (AChEI), which significantly reduce the breakdown of ACh and increase the cholinergic tone.
The M1R activation during tonic (4 Hz) and burst firing (20 Hz) was calculated to obtain the appropriate receptor activation levels “M1_Tonic_A” and “M1_Phasic_A”, respectively, as determined from the receptor competition model. A weighted difference, ΔM, was calculated relative to the control levels “_Con” as described in Eq.10.
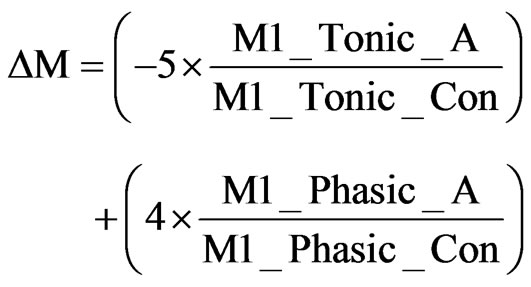 (10)
(10)
The difference in tonic and phasic activation levels led to a change in K+ channel conductance [58]. We incorporate this effect with (Eq.11)
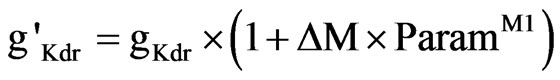 (11)
(11)
where ParamM1 is an adjustable parameter determined from clinical calibrations.
The effect of α7 nAChR physiology was implemented through the modulation of presynaptic glutamate (Glu) release on Glu synapses that connect to pyramidal cells and interneurons [59-61]. This coupling was further calibrated using clinical data on nAChR modulators such as mecamylamine and MEM3454. In addition, α7 nAChR directly affects an inward current on interneurons [62], which was modeled by a decrease in K+ channels on interneuron soma and dendrite.
2.6. Pharmacology of the 5-HT4 Receptor Partial Agonist
The experimental binding affinity data for PF-04995274 are given in Table 1 using radio-active tracer displacement with SB207145, together with the functional doseresponses for the different 5-HT4 receptor isoforms (for further details see [63]).
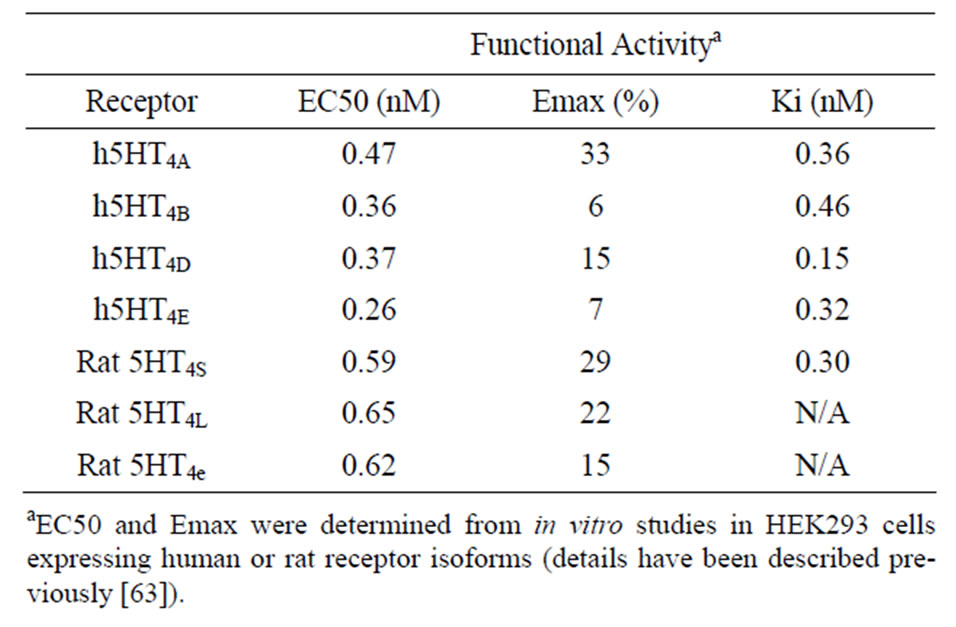
Table 1. Characteristics of the partial 5-HT4 receptor agonist PF-04995274.
2.7. Clinical Study
This randomized, subjectand investigator-blind, sponsor open, placeboand positive-controlled study contained 88 healthy volunteer subjects. The subjects were split into 5 treatment arms that were conducted in parallel. All subjects were administered scopolamine (0.5 mg SC) in addition to donepezil (5 mg or 10 mg PO), PF-04995274 (0.25 mg or 15 mg PO), or placebo (Table 2).
Donepezil was chosen as a positive control as it has been shown to reverse scopolamine induced impairments [34,35]. The 0.25 mg and 15 mg PO doses of PF-4995274 were chosen as these were predicted to cover a receptor occupancy range of 4% - 100%. Both donepezil and PF-04995274 were administered prior to scopolamine such that the maximal plasma concentrations coincided with the time of the maximal cognitive effect from scopolamine (~2 hrs post scopolamine dose).
The change from baseline in the Groton Maze Learning Test (GMLT) at 2 hours post scopolamine dose was used to determine the level of cognitive impairment (or reversal). The GMLT was administered as part of the CogState computerized test battery and was selected based on demonstrated ability to detect scopolamine induced impairments [29,35,64]. The total number of errors made over 5 consecutive trials (lower is better) was quantified as the outcome measure. In an attempt to minimize the learning effects, all subjects were given 2 practice sessions at least 1 day prior to drug administration. A Mixed Model Repeated Measures (MMRM) model was used to determine the maximum likelihood estimates of the log transformed change from baseline (change from baseline was determined on the log transformed values).
3. RESULTS
3.1. Serotonin Synapse Calibration
The 5-HT system calibration is modulated either di-
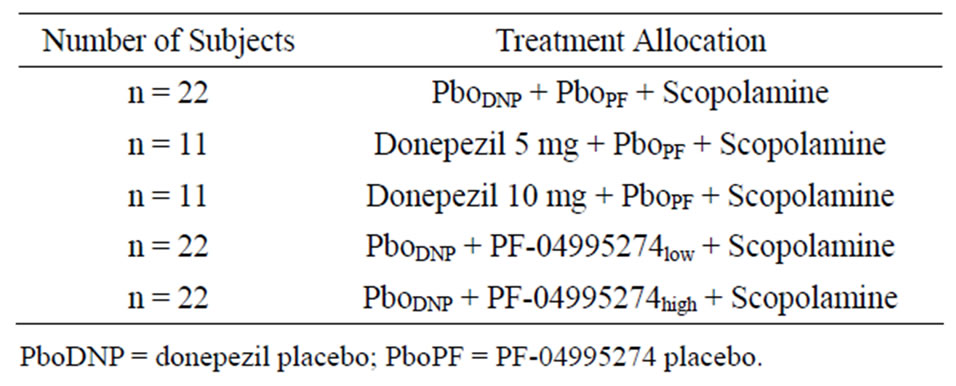
Table 2. Clinical study design and allocation to treatment.
rectly through 5-HT4 receptor activation or indirectly through activation of 5-HT3 and 5-HT6, and is one of the key components of this systems model. The detailed calibration is described elsewhere [39], but briefly recapitulated here. The model shows an excellent correspondence between model output and actual measurements of 5-HT that define the dynamics of coupling between presynaptic 5-HT1B receptor activation and subsequent 5-HT release. Human imaging studies were used to determine which ratio of intraover extrasynaptic 5-HT describes best the clinical outcomes. The results suggest that a two-fold ratio between extra synaptically measured 5-HT and intrasynaptic freely available 5-HT fits the human imaging data.
3.2. Calibration/Validation of the Cholinergic Synapse Model
As the clinical study uses the muscarinic antagonist scopolamine to induce a cognitive deficit, there is a need for proper calibration of the cholinergic synapse. As mentioned before, the full cholinergic synapse model has been calibrated using pharmacology and clinical data on donepezil, rivastigmine and galantamine, [39] an AChE-I with an additional allosteric potentiating ligand effect on nAChR [65]. Basically, a full detailed multi-state receptor model for α4β2 nAChR [66] that simulated the effect of galantamine on the allosteric potentiating ligand site was incorporated into the cholinergic synapse model, starting from a basal ACh release of 500 nM. This computer model included 32 different states with receptor and took into account transitions between activation and desensitization of the ligand-gated ion channel, using actual electrophysiological readouts for calibration, further constrained with microreversibility criteria. The allosteric potentiating effect of galantamine was added using a wide range of electrophysiology data on the amplification of the currents. The full dose-responses for a pure AChE-I and for galantamine are given in Figure 4. Note that for clinically relevant doses of 16 and 24 mg of galantamine, PET imaging of AChE activity using MPT suggest inhibition levels of 20% and 30%, respectively [67].
The model suggests a flat dose-response for galantamine starting at a dose of 16 mg/day; assuming that the
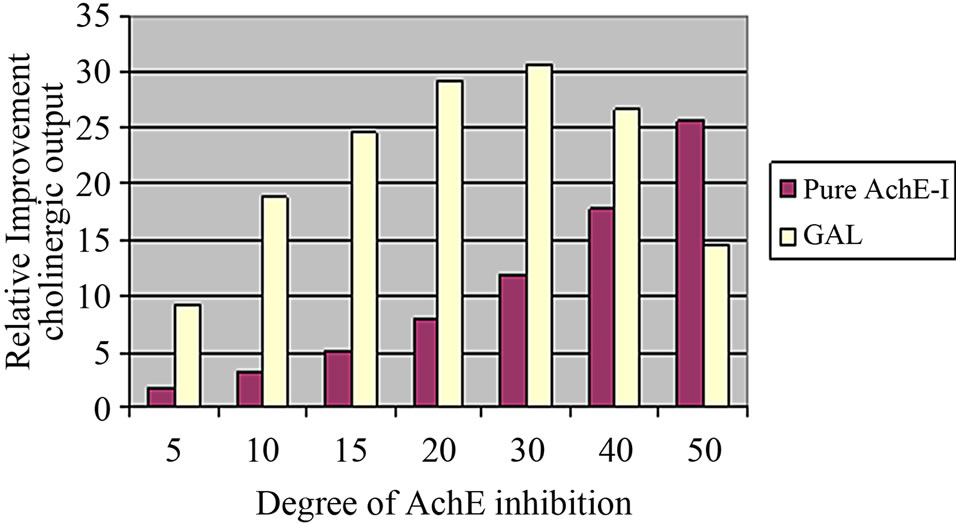
Figure 4. Postsynaptic increase in open α4β2 nAChR with increasing concentrations of galantamine and a pure AChE-I such as donepezil normalized to the degree of inhibition of the enzyme. A daily dose of 16 mg/day corresponds to an inhibition of 20% and a daily dose of 24 mg/day corresponds to 30% AChE inhibition. There is almost no difference in postsynaptic receptor activation at these levels for galantamine, while for donepezil the postsynaptic receptor activation follows a monotonic dose response.
amount of postsynaptic cholinergic receptor activation is proportional to the clinical outcome (this assumes galantamine has no other pharmacological effects); these results correspond with reported clinical data [68,69]. Both in the five-month placebo-controlled study and the 36- month long-term study, patients treated with the 16 and 24 mg daily doses improved essentially to the same degree on the ADAS-Cog (2-point above baseline) and the ADCS-ADL scale (stabilization at baseline values).
The cholinergic synapse was then tested against experimental parameters, derived from work on M2 modulators and M2 KO mice [70]. A two to three-fold increase of free ACh was observed in M2 mAChR KO. Our model simulations yield a 2.7-fold increase, suggesting that relevant coupling parameters are biologically realistic.
Taken together, these results provide an increased confidence of the calibration of the cholinergic cortical synapse that is essential for the working memory performance.
3.3. Dose-Response of 5-HT4 Receptor Activation on Scopolamine-Induced Cognitive Deficit
Scopolamine is a non-selective mAChR inhibitor that affects both postsynaptic M1 (Ki = 1.4 nM) and presynaptic autoreceptor M2 (Ki = 1.2 nM) [71]. Because the affinity of ACh for these two receptors is very different (EC50 of 3.40 nM for M1 mAChR and 340 nM for M2 mAChR), a complex relationship was anticipated. Usually, scopolamine is titrated in a clinical setting until a clear response is observed in human volunteers. No effort was made to determine target engagement, although a clinically approved M2 mAChR specific radiotracer is available [72].
In our simulations, free scopolamine concentrations that yield a substantial deterioration in working memory performance was assumed. This was set in the range of 5 nM, which would result in close to 75% displacement of the radiotracer TZTP.
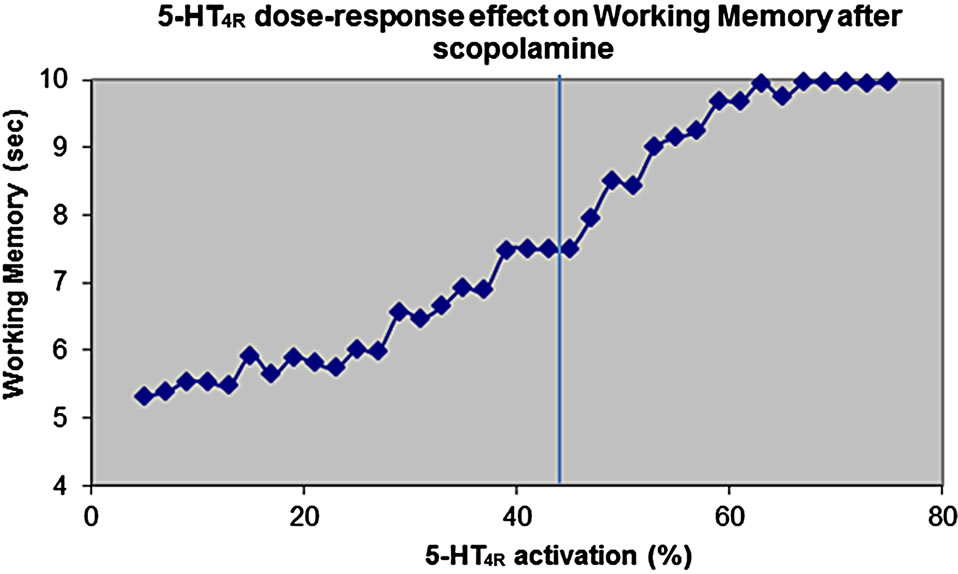
Figure 5(a) shows the effect of pure 5-HT4 receptor activation on cognitive outcome in the cortical network model after scopolamine.
The human brain 5-HT4 receptor isoform is likely to be of either the Aor B-type [19]. Figure 5(b) shows the dose-response of the active moiety of the partial 5-HT4 receptor agonist on cognitive outcome for these two possible relevant 5-HT4 receptor isoforms. These results suggest that at the dose of 0.25 mg of PF, leading to a target engagement of 50% and assuming the 5-HT4 receptor isoform is of the B-type, the compound would reduce the
 (a)
(a)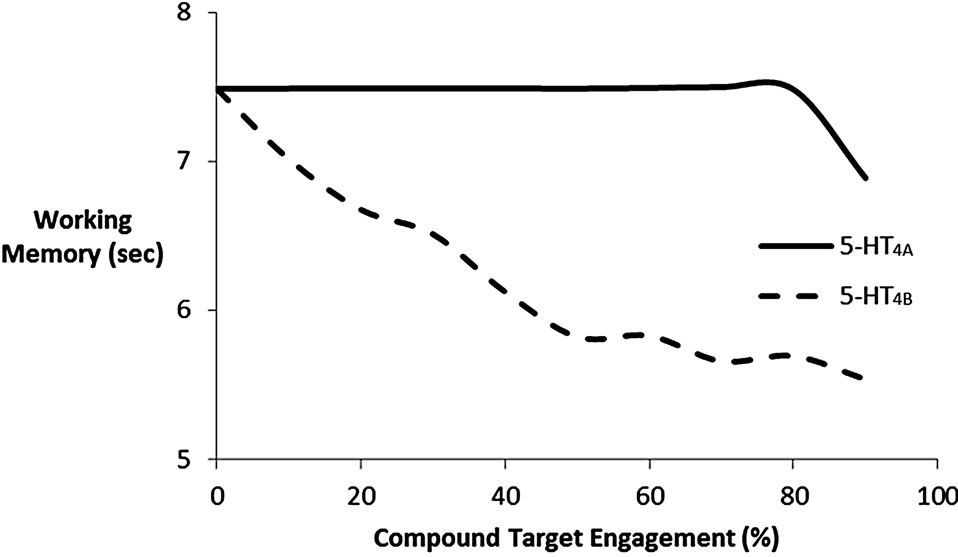 (b)
(b)
Figure 5. (a) Dose-response of pure 5-HT4 receptor modulation on cognitive outcome in the cortical network model after scopolamine-induced deficit. Normal 5-HT4 receptor baseline activity is 45%. It is shown that stimulation of 5-HT4 receptor will improve stability of the memory trace, while reduction of 5-HT4 receptor will decrease cortical model outcome; (b) Doseresponse of the active moiety of the 5-HT4 receptor partial agonist PF-04995274 as a function of target engagement on the working memory readout of the cognitive model in an environment dominated by 5-HT4A or 5-HT4B receptor isoform. The data show that for a 5-HT4B receptor isoform, the compound will actually lead to a decreased cognitive outcome, because it acts as a functional antagonist due to its modest Emax.
outcome by almost 25%. The high dose of 15 mg, corresponding to a target engagement of 95% would reduce the cognitive outcome by almost 30%. In contrast when the 5-HT4 receptor isoform is of the A-isoform type, the low dose would essentially have no effect at all, while the high dose would see a decrease of almost 10%. In the normal case, the average 5-HT4 activation is 45%, but given the Emax of PF-04995274 is 33% and 6% for the A-type and B-type isoforms, respectively, PF-04995274 would act as a functional antagonist. This functional antagonism would be more apparent with the B-type isoform.
3.4. Clinical Outcome of the Scopolamine-Induced Deficit
The demographics of the healthy volunteers are shown in Table 3. Age and weight were comparable across the treatments arms. The mean maximal plasma concentrations (Cmax) of PF-04995274 were 275 pg/mL (56% CV) and 19930 pg/mL (55% CV) for the 0.25 mg and 15 mg PF-04995274 dose, respectively. Donepezil Cmax values were 5.51 ng/mL (32% CV) and 12.4 ng/mL (41% CV) for the 5 mg and 10 mg doses, respectively. Scopolamine Cmax was consistent across treatment arms and ranged from 1.3 ng/mL to 1.6 ng/mL. Exposure of scopolamine, PF-04995274 and donepezil were as expected. No relevant discrepancies were noted based on demographics or drug exposure between the treatment arms.
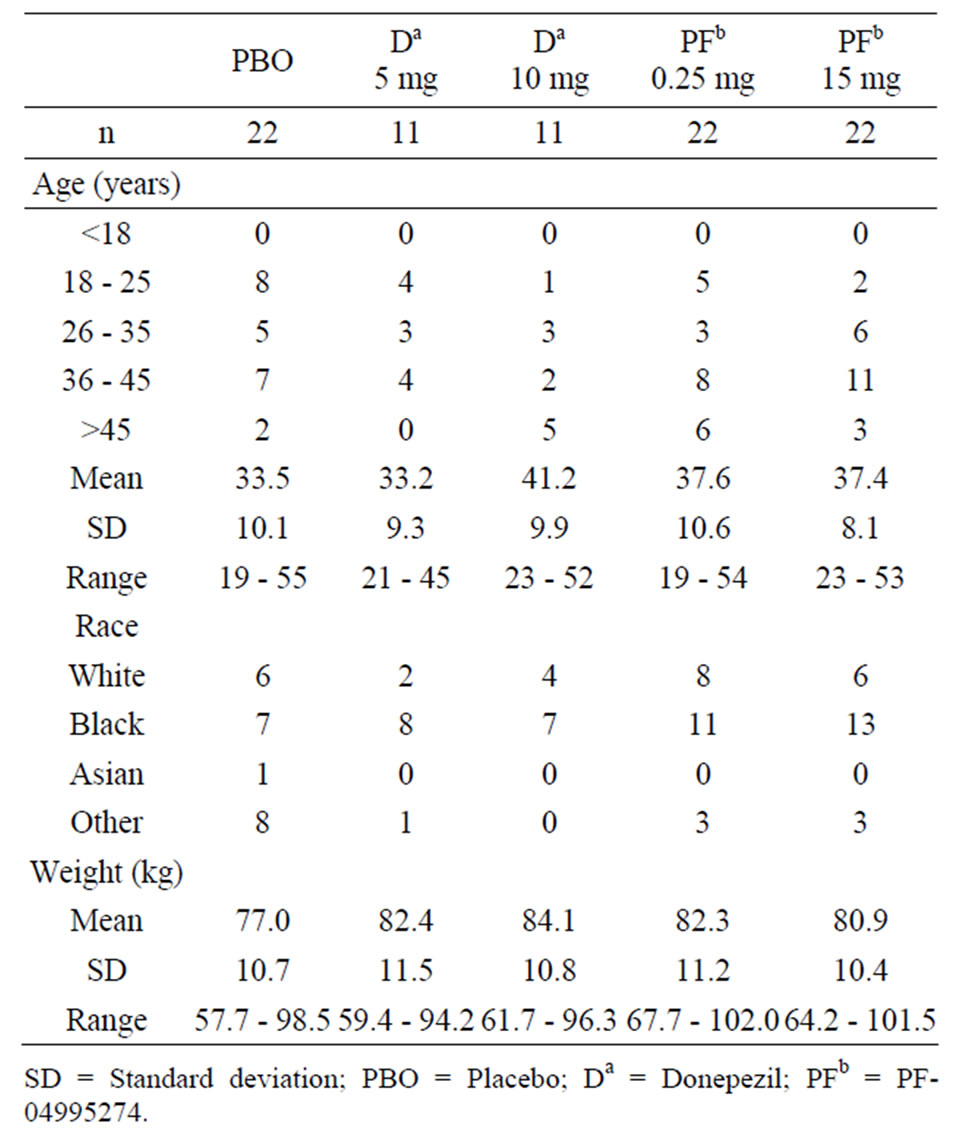
Table 3. Subject demograhic characteristics.
The point estimate for reversal of total number of errors in GMLT for 10 mg donepezil treatment arm was as expected and met the pre-specified decision criteria for a successful scopolamine challenge with donepezil. The 0.25 mg PF-04995274 treatment arm showed an exacerbation of the scopolamine cognitive effects by 26.6% (95% CI: 1.45, 57.91), which was also evident in later time points (data not shown). The higher dose of PF-04995274 (15 mg) had a model based percentage difference of 4.90% (95% CI: -15.72, 30.57) which was not significantly different than the placebo. In the case of the higher PF-04995274 dose the change from baseline number of errors for both the mean (18.9 total errors) and median (14.5 total errors) were qualitatively greater than those from the placebo treated group (18.0 and 10.0 total errors for mean and median, respectively).
The clinical data suggest strongly that the computer model predictions of an exacerbation of scopolamine effect at both doses were qualitatively correct, although the highest dose showed a trend for being slightly better than the lower dose.
3.5. Prediction of 5-HT4 Pharmacology in Alzheimer’s Disease
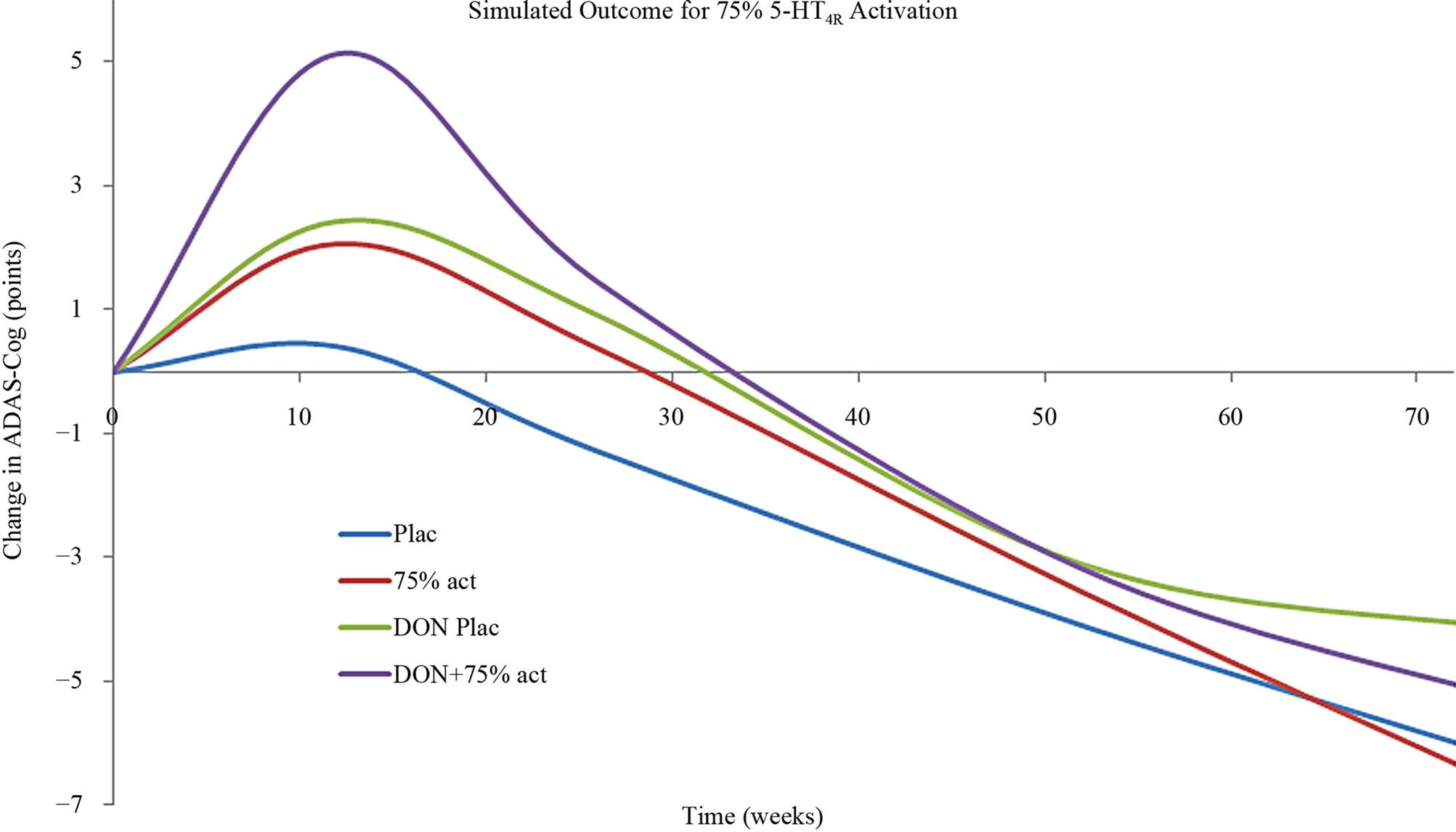
The effect of 5-HT4 pharmacology was then simulated using the calibrated Alzheimer’s disease model [39]. Basically this model takes into account the progressive neurodegeneration of synapse and neuronal cell loss in addition to a constant cholinergic deficit. Figure 6 shows that a strong 5-HT4 receptor agonist at a dose that corresponds to a 75% 5-HT4 receptor activity level clearly improves the symptomatic ADAS-Cog outcome at earlier time-points, but not at later time-points. The effect is additive to cholinergic stimulation by donepezil. Conversely, a weak 5-HT4 receptor agonist (functional antagonist) corresponding to a 5-HT4 receptor activation level of 35% (lower than the baseline value of 45%), as expected worsens the cognition at early time points. This is similar to the observed effect in the clinical trial with scopolamine. An unexpected finding however, was that at later times in the model (corresponding with more progressive neuronal deterioration) a 5-HT4 functional antagonist was predicted to improve cognition.
4. DISCUSSION
In this report, we describe a novel quantitative systems pharmacology approach based upon a biophysically realistic model of a cortical neuronal network that is parameterized with human imaging data and is well calibrated using clinical data. We subsequently and prospectively tested the predictions of this model with the actual clinical outcome in a scopolamine-induced cognitive deficit.
Alignment of preclinical experimental voltametry data
 (a)
(a)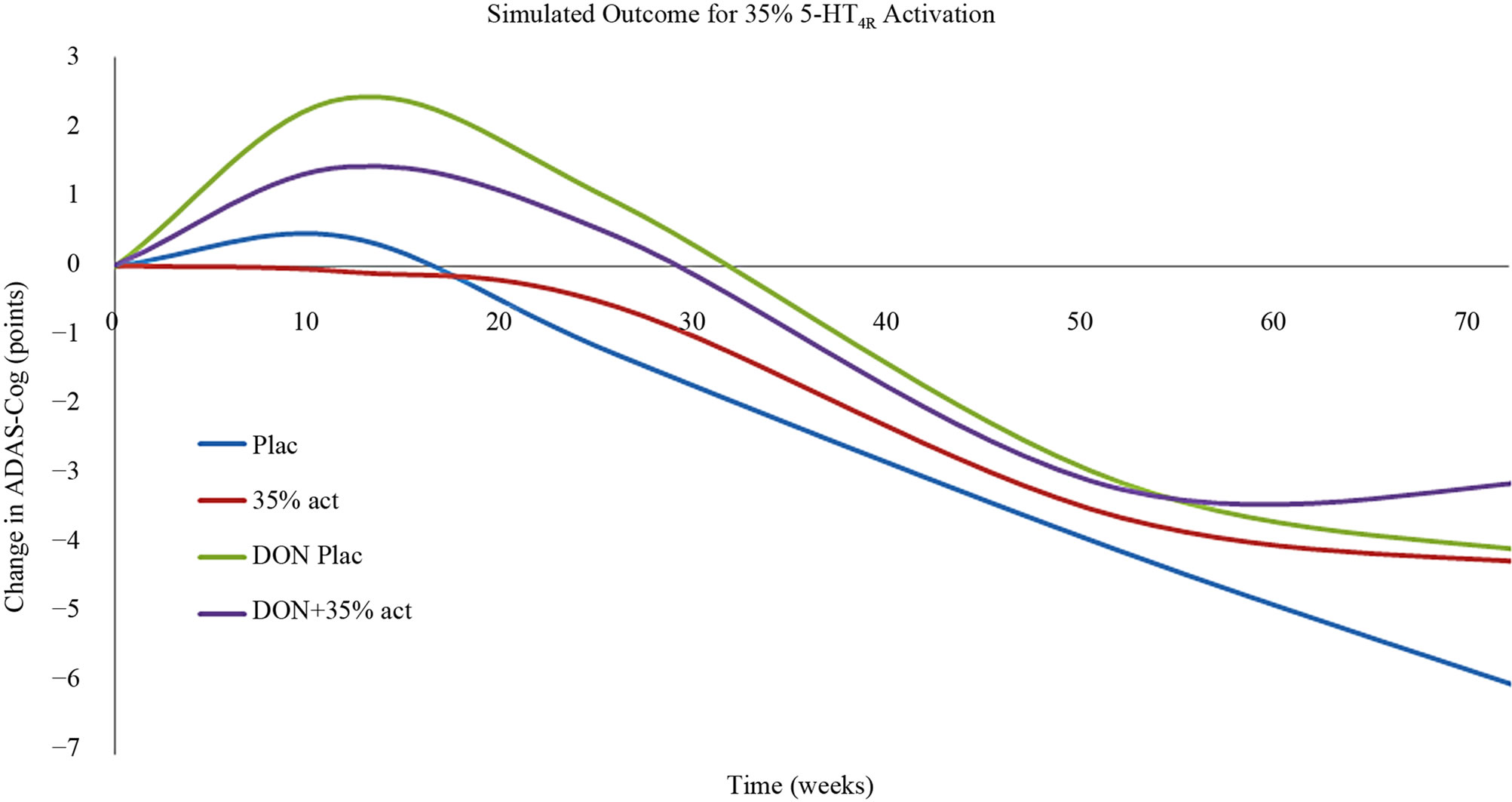 (b)
(b)
Figure 6. (a) Anticipated clinical effect on ADAS-Cog of a strong 5-HT4 receptor partial agonist (corresponding to a situation with 75% 5-HT4 receptor activity) as a function of treatment duration in a mild-to-moderate AD case. Such a compound would be effective at earlier stages of the disease progression and additive to treatment with AChE-I. However with progressive deterioration (i.e. in moderate to severe cases) such a compound would worsen the cognitive outcome; (b) Anticipated clinical effect on ADAS-Cog of a weak 5-HT4 receptor partial agonist (functioning as an antagonist) (corresponding to a situation with 35% 5-HT4 receptor activity) as a function of treatment duration in a mild-to-moderate AD case. Such a compound would worsen the cognitive outcome at earlier stages of the disease progression, similar to the scopolamine effect in healthy volunteers. However with progressive deterioration (i.e. in moderate to severe cases) such a compound would improve the cognitive outcome.
with human brain imaging data enables one to calibrate a serotonergic synapse that is likely to reflect the human cortical serotonergic synapse. Interestingly this leads to a basal activation of about 45% for the 5-HT4 receptor. As a consequence partial agonists with an Emax less than this value will function as antagonists, as they tend to displace the endogenous (full agonist) neurotransmitter from the postsynaptic receptor due to their higher affinity. In this case we had information on full dose-responses of a specific compound PF-04995274 for different receptor isoforms. The maximal activity of the compound against the 5-HT4A and 5-HT4B receptor, likely of importance in the human brain, is relatively modest, leading to a functional antagonism in vivo.
The simulation output from the systems pharmacology model predicted a distinct dependency on the intrinsic activity of the 5-HT4 partial agonist. Low intrinsic activity at the 5-HT4 receptor was predicted to exacerbate the cognitive impairment due to scopolamine, while moderate activity would lead to no effect and a high activity partial agonist would be expected to reverse the effects of scopolamine.
When assuming a 5-HT4B isoform for the human brain, the computer platform would predict a dose-dependent worsening of the cognitive outcome for all doses of PF-04995274. Exacerbation of cognitive outcome beyond scopolamine was indeed unexpectedly observed in the lower PF-04995274 (0.25 mg) treated arm. In contrast, the higher dose of PF-04995274 (15 mg) was not statistically different from the placebo treatment.
The discrepancy between model prediction and clinical outcome may be due to a myriad of possibilities. One possibility might be the variability in the pharmacodynamic effect of scopolamine itself. Another would be the consideration of the relative contribution of the different 5-HT4 receptor isoforms for the clinical measure. Indeed, the model predicted no difference from placebo for the compound assuming a predominant (5-HT4A), where the compound showed a robust 33% Emax; while the exacerbation was predicted for 5-HT4B isoform where the compound had a low 6% maximal activity. The plausible makeup in the healthy volunteer brain would consist of a combination of each of these isoforms leading to a prediction of no change from placebo to a worsening of the cognitive deficit. Even in such a situation, no reversal of impairment was predicted in the computer model, nor observed in the clinical situation. Overall the computer model predictions are congruent to the clinical observations.
It has to be noted that this clinical result was completely unexpected in light of the preclinical findings with this compound. Possible reasons for this translational disconnect include the different pharmacology of the compound for rat vs. human targets in terms of maximal agonist efficacy and possibly the different 5-HT tone in the human brain, leading to different basal activity of the 5-HT4 receptor and the switch from functional agonism to antagonism. Note that nowhere in the model we assume that 5-HT4 receptor would increase levels of ACh and that would not be the reason for reverting scopolamine-induced cognitive deficit. Indeed, experimental data suggest that levels of ACh are increased only at extremely high levels of 5-HT4 activation [73,74]. We suggest that the direct effects of 5-HT4 receptor activation on K+ and GABA-currents might act through the same cAMP-dependent intracellular pathways as for instance M1R. This would explain the beneficial effect of a strong 5-HT4 receptor activation on scopolamine-induced deficit even without an increase in free ACh.
Using feedback from differences between predicted and actual clinical outcomes allows for an improved parameter set that captures a more complex biology and is a natural aspect of the learn-and-confirm paradigm [75]. This illustrates also the power of such a quantitative systems pharmacology approach; unlike traditional animal models which are hard-wired and resistant to changes, computer models allow one to learn from their erroneous predictions and can improve with each iteration.
The model can then tentatively predict the anticipated outcomes of 5-HT4 receptor activation in conditions of mild or moderate Alzheimer’s disease [39]. The outcome suggests that strong 5-HT4 receptor agonism can benefit patients in the early stages of the disease and this effect is additive to AChE-I, similarly to what is expected for scopolamine-induced cognitive deficit in healthy volunteers.
However, as AD pathology progresses, progressively more excitatory-excitatory pyramidal synapses are eliminated compared to excitatory-inhibitory synapses, leading to gradually more inhibition and silencing of the cortical network. Indeed, specific neuronal cell types, characterized by lipofuscin-laden cortical projection neurons with long, thin, and sparsely myelinated axons were identified as most vulnerable [8], suggesting that the last formed neurons were most susceptible to AD pathology. Neurofilament inclusions have indeed only been found in 10% of myelinated axons [76]. Using anti-pHF/tau antibodies [6] tau alteration have been predominantly found in pyramidal and granule cells of the neocortex and hippocampus.
In addition, there is evidence that GABA receptor levels remain relatively intact or are even upregulated [77- 79]. A detailed study [80] found that small cortical inhibitory interneurons are expressing higher levels of NADPHd/nNO early in the paralimbic-limbic-neocortical sequence of AD progression. HPLC studies of GABA and glutamate [81] showed a somewhat greater decrease of Glu than GABA levels in AD brain, but not Down syndrome brain.
As strong 5-HT4 receptor antagonism in these more severe cases tends to further increase GABA tone by increasing 5-HT and thus increasing 5-HT3 activation levels (opposite from a 5-HT3 antagonism) [82], it is not unexpected to observe a worsening of the cognition. Conversely, with a weak 5-HT4 receptor partial agonist that functions as an antagonist, the reduction in 5-HT3 activation, downstream of reduced DR firing tends to reduce GABA inhibitory tone, thereby partially restoring the pathological changes and increasing the stability of a memory trace. This is completely different from the observations in scopolamine-induced deficit in healthy volunteers and suggests that great care need to be taken to extrapolate any findings from these Phase I studies. It also suggests that the two different patient populations (mild vs. moderate to severe) need different treatment paradigms.
The quantitative systems pharmacology approach has a number of limitations, including the lack of receptor desensitization, uncertainty to the basal level of 5-HT in AD pathology, linear relationships between receptor activation and intracellular events and the absence of disease-modification modeling.
This result assumes the lack of desensitization at the 5-HT4 receptor; experimental evidence however suggests that these receptors can desensitize substantially [83-85]. Indeed the dynamics of the endogenous 5-HT is strictly regulated by the 5-HT transporter, while the partial agonist are cleared on a much slower time scale. Although we don’t expect this to play a role in the acute scopolamine paradigm, it might become more important when considering long term treatment of AD patients.
The basal level of 5-HT4 receptor activation was derived from human imaging studies in schizophrenia patients where no substantial serotonergic deficit is documented. However, given the observation of a prominent neuropathology in Alzheimer’s disease [19,86,87], the 5-HT tone is likely to be lower, especially in more severe cases. Therefore the ambient 5-HT tone in AD patients could be significantly lower so as to modulate the possible negative effect in mild AD or beneficial effect in severe AD of PF-04995274. In addition we don’t assume a progressive decline of 5-HT4 receptor density over time, or a further general decrease in 5-HT tone associated with AD pathology.
The model also provides only symptomatic modeling and its prediction to a functional outcome such as the ADAS-Cog clinical scale. At this point in time, there are no disease modifying processes included which might limit the usefulness of our approach. However, given the latest clinical failures of amyloid-modulating therapies, there is a renewed and increased interest in symptomatic treatments.
Though the model adequately predicted the clinical outcomes, a greater degree of granularity would be helpful. As an example, the assumption of a linear relationship between 5-HT4 intrinsic activity and receptor or downstream stimulus was sufficient in this situation given that the intrinsic activity range was 6% - 33%, depending on isoform contribution. A nonlinear model would also be expected to provide the same results, yet may have greater implications on the degree of intrinsic activity required for an effective compound.
The quantitative systems pharmacology model output suggested a beneficial effect of 5-HT4 agonism on working memory. The model also projected no effect or an exacerbation of scopolamine impairment for low intrinsic activity 5-HT4 agonists, which was supported by the subsequent human trial outcome. This example shows how systems pharmacology modeling can aid in the confidence in drug development through early levels of target identification, directed hypothesis testing, and interpretation of data.
5. DISCLOSURE
All authors, unless specified, were employed by Pfizer Inc. at the time of this work. RC, PR, AS and HG were employees of In Silico Biosciences who were contracted by Pfizer for this work.
REFERENCES
- Geerts, H. (2011) Modeling & Simulation as a tool for improving CNS drug research and development. Drug Development Research, 72, 66-73. doi:10.1002/ddr.20403
- van der Graaf, P.H. and Benson, N. (2011) Systems pharmacology: Bridging systems biology and pharmacokinetics-pharmacodynamics (PKPD) in drug discovery and development. Pharmaceutical Research, 28, 1460-1464. doi:10.1007/s11095-011-0467-9
- Alzheimer’s Association (2007) Every 72 seconds someone in America Develops Alzheimer’s: Alzheimer’s disease facts and figures 2007.
- Braak, H. and Braak, E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica ogica, 82, 239-259. doi:10.1007/BF00308809
- Braak, H., Braak, E. and Bohl, J. (1993) Staging of Alzheimer-related cortical destruction. European Neurology, 33, 403-408. doi:10.1159/000116984
- Shin, R.W., Iwaki, T., Kitamoto, T., Sato, Y. and Tateishi, J. (1992) Massive accumulation of modified tau and severe depletion of normal tau characterize the cerebral cortex and white matter of Alzheimer’s disease. Demonstration using the hydrated autoclaving method. American Journal of Pathology, 140, 937-945.
- Thal, D.R., Griffin, S.T., de Vos, R.A. and Ghebremedhin, E. (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathologica, 115, 599-609. doi:10.1007/s00401-008-0366-2
- Braak, H., Tredici, K., Schultz, C. and Braak, E. (2000) Vulnerability of select neuronal types to Alzheimer’s disease. Annals of the New York Academy of Sciences, 924, 53-61. doi:10.1111/j.1749-6632.2000.tb05560.x
- Maher-Edwards, G., G., Dixon, R., Hunter, J., Gold, M., Hopton, G., Jacobs, G., Hunter, J. and Williams, P. (2011) SB-742457 and the donepezil and Alzheimer’s disease: A randomized, placebo-controlled study. International Journal of Geriatric Psychiatry, 26, 536-544. doi:10.1002/gps.2562
- Maher-Edwards, G., Zvartau-Hind, M., Hunter, A.J., Gold, M., Hopton, G., Jacobs, G., Davy, M. and Williams, P. (2010) Double-blind, controlled phase II study of a 5- HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Current Alzheimer Research, 7, 374-385. doi:10.2174/156720510791383831
- Cirrito, J., Disabato, B.M. and Restivo, J. (2011) Serotonin signaling is associated with lower amyloid beta levels and plaques in transgenic mice and humans. Proceedings of the National Academy of Sciences, 108, 14968- 14973. doi:10.1073/pnas.1107411108
- Consolo, S., Arnaboldi, S., Giorgi, S., Russi, G. and Ladinsky, H. (1994) 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. NeuroReport, 5, pp. 1230-1232. doi:10.1097/00001756-199406020-00018
- Eglen, R.M., Wong, E., Dumuis, A. and Bockaert, J. (1995) Central 5-HT4 receptors. Trends in Pharmacological Sciences, 16, pp. 391-398. doi:10.1016/S0165-6147(00)89081-1
- Kahana, M.J. (2006) The cognitive correlates of human brain oscillations. The Journal of Neuroscience, 26, pp. 1669-1672. doi:10.1523/JNEUROSCI.3737-05c.2006
- Megerian, J.T. (2009) Results of a phase 2A study of a novel 5HT4 agonist for the treatment of Alzheimer’s disease. American Society for Experimental Neuro Therapeutics Annual Meeting, Arlington, 6 March 2009.
- Robert, S.J. and Lezoualc’h, F. (2008) Distinct functional effects of human 5-HT4 receptor isoforms on beta-amyloid secretion. Neurodegenerative Diseases, 5, pp. 163- 165. doi:10.1159/000113691
- Shen, F., Smith, J., Chang, R., Bourdet, D.L., Tsuruda, P., Obedencio, P. and Beattie, D.T. (2011) 5-HT(4) receptor agonist mediated enhancement of cognitive function in vivo and amyloid precursor protein processing in vitro: A pharmacodynamic and pharmacokinetic assessment. Neuropharmacology, 61, pp. 69-79. doi:10.1016/j.neuropharm.2011.02.026
- Lezoualc’h, F. and Robert, S.J. (2003) The serotonin 5- HT4 receptor and the amyloid precursor protein processing. Experimental Gerontology, 38, 159-166. doi:10.1016/S0531-5565(02)00157-2
- Reynolds, G.P., Mason, S.L., Meldrum, A., De Keczer, S., Parties, H., Eglen, R.M. and Wong, E.H.F. (1995) 5-Hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: Distribution, pharmacology and effects of neurodegenerative diseases. British Journal of Pharmacology, 114, 993-998. doi:10.1111/j.1476-5381.1995.tb13303.x
- Vilaro, M.T., Cortes, R., Gerald, C., Branchek, T., Palacios, J.M. and Mengod, G. (1996) Localization of 5-HT4 receptor mRNA in rat brain by in situ hybridization histochemistry. Molecular Brain Research, 43, 356-360. doi:10.1016/S0169-328X(96)00248-3
- Geerts, H. (2011) Modeling & Simulation as a tool for improving CNS Drug Research and Development. Drug Development Research, 72, 66-73. doi:10.1002/ddr.20403
- Geerts, H., Spios, A. and Roberts, P. (2012) Has the time come for Quantitative Systems Pharmacology on CNS R&D. CPT: Pharmacometrics and Systems Pharmacology, 1, e16. doi:10.1038/psp.2012.17
- Povysheva, N.V., Zaitsev, A.V., Rotaru, D.C., GonzalezBurgos, G., Lewis, D.A. and Krimer, L.S. (2008) Parvalbumin-positive basket interneurons in monkey and rat prefrontal cortex. Journal of Neurophysiology, 100, pp. 2348-2360. doi:10.1152/jn.90396.2008
- Zaitsev, A.V., Povysheva, N., Gonzalez-Burgos, G., Rotaru, A.V., Fish, K.N., Krimer, L.S. and Lewis, D.A. (2009) Interneuron diversity in layers 2-3 of monkey prefrontal cortex. Cerebral Cortex, 19, 1597-1615. doi:10.1093/cercor/bhn198
- Hashimoto, T., et al., (2009) Protracted developmental trajectories of GABAA receptor alpha1 and alpha2 subunit expression in primate prefrontal cortex. Biological Psychiatry, 65, 1015-1023. doi:10.1016/j.biopsych.2009.01.004
- Drachman, D.A. and Leavitt, J. (1974) Human memory and the cholinergic system. A relationship to aging? Archives of Neurology, 30, 113-121. doi:10.1001/archneur.1974.00490320001001
- Rusted, J.M. and Warburton, D.M. (1988) The effects of scopolamine on working memory in healthy young volunteers. Psychopharmacology, 96, 145-152. doi:10.1007/BF00177553
- Ebert, U., Siepmann, M., Oertel, R., Wesnes, K.A. and Kirch, W. (1998) Pharmacokinetics and pharmacodynamics of scopolamine after subcutaneous administration. Journal of Clinical Pharmacology, 38, 720-726. doi:10.1002/j.1552-4604.1998.tb04812.x
- Fredrickson, A., Snyder, P.J., Cromer, J., Thomas, E., Lewis, M. and Maruff, P. (2008) The use of effect sizes to characterize the nature of cognitive change in psy- -chopharmacological studies: An example with scopolamine. Human Psychopharmacology, 23, 425-436. doi:10.1002/hup.942
- Liem-Moolenaar, M., et al. (2010) The effects of the glycine reuptake inhibitor R213129 on the central nervous system and on scopolamine-induced impairments in psychomotor and cognitive function in healthy subjects. Journal of Psychopharmacology, 24, 1671-1679. doi:10.1177/0269881109106942
- Liem-Moolenaar, M., et al., (2011) Pharmacokineticpharmacodynamic relationships of central nervous system effects of scopolamine in healthy subjects. British Journal of Clinical Pharmacology, 71, 886-898. doi:10.1111/j.1365-2125.2011.03936.x
- Molchan, S.E., et al. (1992) Increased cognitive sensitivity to scopolamine with age and a perspective on the scopolamine model. Brain Research Reviews, 17, pp. 215-226. doi:10.1016/0165-0173(92)90017-G
- Patat, A., Klein, M.J., Surjus, A., Hucher, M. and Granier, J. (1991) RU 41,656 does not reverse the scopolamineinduced cognitive deficit in healthy volunteers. European Journal of Clinical Pharmacology, 41, 225-231. doi:10.1007/BF00315434
- Snyder, P.J., Bednar, M., Cromer, J.R. and Maruff, P. (2005) Reversal of scopolamine-induced deficits with a single dose of donepezil, an acetylcholinesterase inhibitor. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 1, 126-135. doi:10.1016/j.jalz.2005.09.004
- Thomas, E., et al. (2008) Specific impairments in visuospatial working and short-term memory following lowdose scopolamine challenge in healthy older adults. Neuropsychologia, 46, 2476-2484. doi:10.1016/j.neuropsychologia.2008.04.010
- Vitiello, B., et al. (1997) Cognitive and behavioral effects of cholinergic, dopaminergic, and seotonergic blockade in humans. Neuropsychopharmacology, 16, 15-24. doi:10.1016/S0893-133X(96)00134-0
- Roof, R., Sawant, A., Zaleska, M. and Iredale, P. (2011) Assessment of PF-04995274 in scopolamine disrupted Morris water maze compared to and in combination with donepezil. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 7, s772. doi:10.1016/j.jalz.2011.05.2219
- Geerts, H. (2012) alpha7 Nicotinic receptor modulators for cognitive deficits in schizophrenia and Alzheimer’s disease. Expert Opinion on Investigational Drugs, 21, 59- 65. doi:10.1517/13543784.2012.633510
- Roberts, P.D., Spiros, A. and Geerts, H. (2012) Simulations of symptomatic treatments for Alzheimer’s disease: Computational analysis of pathology and mechanisms of drug action. Alzheimer’s Research & Therapy, 4, 50. doi:10.1186/alzrt153
- Spiros, A., Carr, R. and Geerts, H. (2010) Not all partial dopamine D2 receptor agonists are the same in treating schizophrenia. Exploring the effects of bifeprunox and aripiprazole using a computer model of a primate striatal dopaminergic synapse. Neuropsychiatric Disease and Treatment, 6, 589-603. doi:10.2147/NDT.S12460
- Spiros, A. and Geerts, H. (2012) A quantitative way to estimate clinical off-target effects for human membrane brain targets in CNS research and development. Journal of Experimental Pharmacology, 4, 53-61. doi:10.2147/JEP.S30808
- Montague, P.R., et al. (2004) Dynamic gain control of dopamine delivery in freely moving animals. The Journal of Neuroscience, 24, 1754-1759. doi:10.1523/JNEUROSCI.4279-03.2004
- Ogren, S.O., et al. (2008) The role of 5-HT1A receptors in learning and memory. Behavioural Brain Research, 195, 54-77. doi:10.1016/j.bbr.2008.02.023
- John, C.E., Budygin, E.A., Mateo, Y. and Jones, S.R. (2006) Neurochemical characterization of the release and uptake of dopamine in ventral tegmental area and serotonin in substantia nigra of the mouse. Journal of Neurochemistry, 96, 267-282.
- Pendyam, S., Mohan, A., Kalivas, P.W. and Nair, S.S. (2009) Computational model of extracellular glutamate in the nucleus accumbens incorporates neuroadaptations by chronic cocaine. Neuroscience, 158, 1266-1276. doi:10.1016/j.neuroscience.2008.11.014
- Howes, O.D., Allan, V., McGuire, P., Stokes, P. and Kapur, S. (2009) Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: Insights from PET and SPECT imaging. Current Pharmaceutical Design, 15, 2550-2259. doi:10.2174/138161209788957528
- Durstewitz, D., Seamans, J.K. and Sejnowski, T.J. (2000) Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. Journal of Neurophysiology, 83, 1733-1750.
- DeFelipe, J. (2002) Cortical interneurons: From Cajal to 2001. Progress in Brain Research, 136, 215-238. doi:10.1016/S0079-6123(02)36019-9
- Sidiropoulou, K., et al. (2009) Dopamine modulates an mGluR5-mediated depolarization underlying prefrontal persistent activity. Nature Neuroscience, 12, 190-199.
- Isaacson, J.S. and Scanziani, M. (2011) How inhibition shapes cortical activity. Neuron, 72, 231-243. doi:10.1016/j.neuron.2011.09.027
- Bijak, M., Zahorodna, A. and Tokarski, K. (2001) Opposite effects of antidepressants and corticosterone on the sensitivity of hippocampal CA1 neurons to 5-HT1A and 5-HT4 receptor activation. Naunyn-Schmiedeberg’s Archives of Pharmacology, 363, 491-498. doi:10.1007/s002100000389
- Bockaert, J., Claeysen, S., Compan, V. and Dumuis, A. (2004) 5-HT4 receptors. Current Drug Target-CNS & Neurological Disorders, 3, 39-51. doi:10.2174/1568007043482615
- Ansanay, H., Dumuis, A., Sebben, M., Bockaert, J. and Fagni, L. (1995) cAMP-dependent, long-lasting inhibittion of a K+ current in mammalian neurons. Proceedings of the National Academy of Sciences of the United States of America, 92, 6635-6639. doi:10.1073/pnas.92.14.6635
- Torres, G.E., Arfken, C.L. and Andrade, R. (1996) 5-Hydroxytryptamine4 receptors reduce afterhyperpolarization in hippocampus by inhibiting calcium-induced calcium release. Molecular Pharmacology, 50, 1316-1322.
- Cai, X., Flores-Hernandez, J., Feng. J. and Yan, Z. (2002) Activity-dependent bidirectional regulation of GABAA receptor channels by the 5-HT4 receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. The Journal of Physiology, 540, 743-759. doi:10.1113/jphysiol.2001.013391
- Lucas, G., et al. (2005) Frontocortical 5-HT4 receptors exert positive feedback on serotonergic activity: Viral transfections, subacute and chronic treatments with 5-HT4 agonists. Biological Psychiatry, 57, 918-925. doi:10.1016/j.biopsych.2004.12.023
- Lucas, G. and Debonnel, G. (2002) 5-HT4 receptors exert a frequency-related facilitatory control on dorsal raphe nucleus 5-HT neuronal activity. European Journal of Neuroscience, 16, 817-822. doi:10.1046/j.1460-9568.2002.02150.x
- Gulledge, A.T. and Stuart, G.J. (2005) Cholinergic inhibittion of neocortical pyramidal neurons. The Journal of Neuroscience, 25, 10308-10320. doi:10.1523/JNEUROSCI.2697-05.2005
- Timofeeva, O.A. and Levin, E.D. (2011) Glutamate and nicotinic receptor interactions in working memory: Importance for the cognitive impairment of schizophrenia. Neuroscience, 195, 21-36. doi:10.1016/j.neuroscience.2011.08.038
- Zappettini, S., Grilli, M., Salamone, A., Fedele, E. and Marchi, M. (2010) Pre-synaptic nicotinic receptors evoke endogenous glutamate and aspartate release from hippocampal synaptosomes by way of distinct coupling mechanisms. British Journal of Pharmacology, 161, 1161- 1171. doi:10.1111/j.1476-5381.2010.00958.x
- Parikh, V., Ji, J., Decker, M.W. and Sarter, M. (2010) Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. The Journal of Neuroscience, 30, 3518-3530. doi:10.1523/JNEUROSCI.5712-09.2010
- Alkondon, M., Pereira, E., Eisenberg, H. and Albuquerque, E. (2000) Nicotinic receptor activation in human cerebral cortical interneurons: A mechanism for inhibition and disinhibition of neuronal networks. The Journal of Neuroscience, 20, 66-75.
- Brodney, M., et al. (2012) Identification of multiple 5- HT4 partial agonist clinical candidates for the treatment of Alzheimer’s disease. Journal of Medicinal Chemistry, 55, 9240-9254. doi:10.1021/jm300953p
- Collie, A., et al. (2007) Cognitive testing in early-phase clinical trials: Development of a rapid computerized test battery and application in a simulated Phase I study. Contemporary Clinical Trials, 28, 391-400. doi:10.1016/j.cct.2006.10.010
- Woodruff-Pak, D.S. and Gould, T.J. (2002) Neuronal nicotinic acetylcholine receptors: Involvement in Alzheimer’s disease and schizophrenia. Behavioral and Cognitive Neuroscience Reviews, 1, 5-20. doi:10.1177/1534582302001001002
- Geerts, H., Finkel, L., Carr, R. and Spiros, A. (2002) Nicotinic receptor modulation: Advantages for successful Alzheimer’s disease therapy. Journal of Neural Transmission Supplementum, 62, 203-216.
- Kadir, A., et al. (2008) PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiology of Aging, 29, 1204-1217. doi:10.1016/j.neurobiolaging.2007.02.020
- Tariot, P.N., et al. (2000) A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology, 54, 2269-2276. doi:10.1212/WNL.54.12.2269
- Raskind, M.A., Peskind, E., Truyen, L., Kershaw, P. and Damaraju, C. (2004) The cognitive benefits of galantamine are sustained for at least 36 months: A long-term extension trial. Archives of Neurology, 61, 252-256. doi:10.1001/archneur.61.2.252
- Slutsky, I., et al. (2003) Use of knockout mice reveals involvement of M2-muscarinic receptors in control of the kinetics of acetylcholine release. Journal of Neurophysiology, 89, 1954-1967. doi:10.1152/jn.00668.2002
- Bolden, C., Cusack, B. and Richelson, E. (1992) Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells. Journal of Pharmacology and Experimental Therapeutics, 260, 576-580.
- Carson, R.E., et al. (1998) Muscarinic cholinergic recaptor measurements with [18F]FP-TZTP: Control and competition studies. Journal of Cerebral Blood Flow & Metabolism, 18, 1130-1142. doi:10.1097/00004647-199810000-00010
- Matsumoto, M., et al. (2001) Evidence for involvement of central 5-HT4 receptors in cholinergic function associated with cognitive processes: Behavioral, electrophysiological, and neurochemical studies. Journal of Pharmacology and Experimental Therapeutics, 296, 676-682.
- de Vin, F., De Maeyer, J.H. and Lefebvre, R.A. (2011) In-vitro acetylcholine release is not a straightforward model to study hippocampal 5-HT4 receptors. Neuroreport, 22, 892-896. doi:10.1097/WNR.0b013e32834c7fd4
- Sheiner, L.B. (1997) Learning versus confirming in clinical drug development. Clinical Pharmacology & Therapeutics, 61, 275-291.
- Perry, M.J., Lawson, S.N. and Robertson, J. (1991) Neurofilament immunoreactivity in populations of rat primary afferent neurons: A quantitative study of phosphorylated and non-phosphorylated subunits. Journal of Neurocytology, 20, 746-758. doi:10.1007/BF01187848
- Iwakiri, M., et al. (2009) An immunohistochemical study of GABAA receptor gamma subunits in Alzheimer’s disease hippocampus: Relationship to neurofibrillary tangle progression. Neuropathology, 29, 263-269. doi:10.1111/j.1440-1789.2008.00978.x
- Rissman, R.A., et al. (2003) Biochemical analysis of GABAA receptor subunits alpha 1, alpha 5, beta 1, beta 2 in the hippocampus of patients with Alzheimer’s disease neuropathology. Neuroscience, 120, 695-704. doi:10.1016/S0306-4522(03)00030-7
- Mizukami, K., Grayson, D., Ikonomovic, M., Sheffield, R. and Armstrong, D. (1998) GABAA receptor beta 2 and beta 3 subunits mRNA in the hippocampal formation of aged human brain with Alzheimer-related neuropathology. Molecular Brain Research, 56, 268-272. doi:10.1016/S0169-328X(97)00347-1
- Koliatsos, V.E., et al. (2006) Early involvement of small inhibitory cortical interneurons in Alzheimer’s disease. Acta Neuropathologica ogica, 112, 147-162. doi:10.1007/s00401-006-0068-6
- Seidl, R., Cairns, N., Singewald, N., Kaehler, S. and Lubec, G. (2001) Differences between GABA levels in Alzheimer’s disease and Down syndrome with Alzheimerlike neuropathology. Naunyn-Schmiedeberg’s Archives of Pharmacology, 363, 139-145. doi:10.1007/s002100000346
- Puig, M.V., Santana, N., Celada, P., Mengod, G. and Artigas, F. (2004) In vivo excitation of GABA interneurons in the medial prefrontal cortex through 5-HT3 receptors. Cerebral Cortex, 14, 1365-1375. doi:10.1093/cercor/bhh097
- Grider, J.R. (2006) Desensitization of the peristaltic reflex induced by mucosal stimulation with the selective 5-HT4 agonist tegaserod. American Journal of Physiology-Gastrointestinal and Liver Physiology, 290, G319- G327. doi:10.1152/ajpgi.00326.2005
- Mialet, J., Fischmeister, R. and Lezoualc’h, F. (2003) Characterization of human 5-HT4(d) receptor desensitization in CHO cells. British Journal of Pharmacology, 138, 445-452. doi:10.1038/sj.bjp.0705061
- Lefebvre, H., et al. (1998) Effect of prolonged administration of the serotonin4 (5-HT4) receptor agonist cisapride on aldosterone secretion in healthy volunteers. Endocrine Research, 24, 749-752. doi:10.3109/07435809809032681
- Ouchi, Y., et al. (2009) Altered brain serotonin transporter and associated glucose metabolism in Alzheimer disease. The Journal of Nuclear Medicine, 50, 1260-1266. doi:10.2967/jnumed.109.063008
- Hendricksen, M., Thomas, A., Ferrier, I.N., Ince, P. and O’Brien, J. (2004) Neuropathological study of the dorsal raphe nuclei in late-life depression and Alzheimer’s disease with and without depression. American Journal of Psychiatry, 161, 1096-1102. doi:10.1176/appi.ajp.161.6.1096