Advances in Alzheimer's Disease
Vol.2 No.2(2013), Article ID:33176,6 pages DOI:10.4236/aad.2013.22008
The role of APP in Alzheimer’s disease
![]()
1Department of Interventional Radiology, Shanghai 10th People’s Hospital, Shanghai Key Laboratory of Signaling and Disease Research, School of Life Science and Technology, Tongji University, Shanghai, China; #Corresponding Author: lei.xue@tongji.edu.cn
2Department of Biomedical Engineering, School of Medicine, Johns Hopkins University, Baltimore, Maryland, USA
3School of Chinese Medicine, Hong Kong Baptist University, Hong Kong
Copyright © 2013 Leland Pung et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 13 March 2013; revised 26 April 2013; accepted 6 May 2013
Keywords: Alzheimer’s disease; APP; Aβ
ABSTRACT
Alzheimer’s disease (AD) is one of the most significant neurodegenerative disorders in terms of both severity and cost. Despite being defined over a century ago, there is currently no cure to this disease that affects an increasing elderly population. The amyloid precursor protein (APP) has been shown to play an important role in AD progression. The amyloid β peptide (Aβ), which accumulates in senile plaques, a central etiological AD factor, is a proteolytic product from APP by the enzymatic action of β- and γ-secretases. In this review, we summarize the current knowledge of the processing and physiological functions of APP, and the involvement of APP and Aβ in AD.
1. INTRODUCTION
Alzheimer’s disease (AD) is currently the most prevalent neurodegenerative disease in the world and is expected to increase exponentially as baby boomers age. Sociologically, AD presents a huge strain on caregivers who face pressures ranging from social and psychological to physical and economic. In the United States alone, AD affected an estimated 5 million people in 2007 and was expected to increase to 13 million by 2050 [1].
Characterized by a progressive cognitive decline leading to social and occupational debilitation, AD is pathologically differentiated from other causes of dementia by a reduction in synaptic contact [2], correlating with impairments of higher cognitive function in addition to the accumulation of neurofibrillary tangles [3,4], aggregates of the microtubule-associated hyper-phosphorylatedprotein tau [5], and extracellular senile plaques (SP) containing the amyloid β peptide (Aβ) as first discovered by Alois Alzheimer [6]. Although senior individuals often develop some plaques and tangles as a consequence of aging, the brains of AD patients have a greater number of them in specific brain regions such as the temporal lobe [3].
The amyloid β peptide found predominantly in the senile plaques was shown to be a 4.2 kDa peptide approximately 40 amino acids in length [7]. It was soon verified that Aβ was cleaved from a larger precursor termed as amyloid precursor protein (APP) [8].
2. STRUCTURE OF APP
APP is a highly conserved transmembrane protein with many analogues in mammalian species as well as two homologues found in invertebrates [9,10]. It belongs to a protein family that also includes two APP-like proteins, APLP1 and APLP2, in mammals [11-14]. By alternative splicing, a single APP gene generates several APP isoforms including APP695, APP750 and APP751 [15,16], most of which contain a protease inhibitor domain and are expressed ubiquitously. However, one isoform, APP- 695, is present mainly in the nervous system and lacks the protease inhibitor domain [17-19].
All members of the APP family are single-pass transmembrane proteins with a large N-terminal extracellular domain and a relative shorter intracellular C-terminus [8]. Cleavage of APP can occur by two separate pathways through α- or β-secretase which both remove nearly the entire extracellular domain, while the membrane-anchored C-terminal fragments may trigger apoptosis [20]. APP is subsequently cleaved by γ-secretase, a large multi-unit complex, within the membrane spanning region [21]. While cleavage of APP by α-secretase precludes the production of Aβ, cleavage by β- and γ-secretases results in the generation of Aβ and its subsequent extracellular deposition (Figure 1).
3. GENETICS OF APP
It has been shown that the pathological characteristics of autosomal dominant AD is similar to that of sporadic AD. In autosomal dominant AD, there are 32 mutations identified in APP, centered primarily around the γ-secretase cleavage site. In addition, there are 179 mutations on the presenilin 1 gene locus and 14 on the presenilin 2 gene locus, which are key components of the γ-secretases [22]. This serves to reinforce the concept that Aβ deposition is crucial in AD development as mutations at these locations have been shown to increase the production of Aβ42, a more toxic and less soluble version of the wild type Aβ40 peptide.
4.FUNCTION OF APP
4.1. Synapse Development
APP has been known to play an important role in regulating the synapse functions by modulating the synaptic transmission, plasticity, memory formation and maintenance. AD patients exhibited a decrease of synapses with the progression of the disease [23]. In addition, APP showed a strong immunoreactivity in human neuromuscular junctions (NMJs) [24], suggesting a physiological role of APP in synaptic functions. Consistently, the Drosophila homologue APPL modulates the synaptic bouton number and synapse structure in NMJ [25], while APP/APLP2 double knockout mice show dramatic defects in the development of NMJs [26,27]. Interestingly, the conserved C-terminal motif GYENPTY of APP family members is required for synapse development from fly to mammals [25,28].
4.2. Axonal Transport and Phototaxis
Both APP and APPL are involved in axonal transport through the interaction between their C-terminal intracellular domain (AICD) and Kinesin, which is crucial for the vesicle transport responsible for the growth of the synapse [29-33]. In addition, loss of APPL in Drosophila
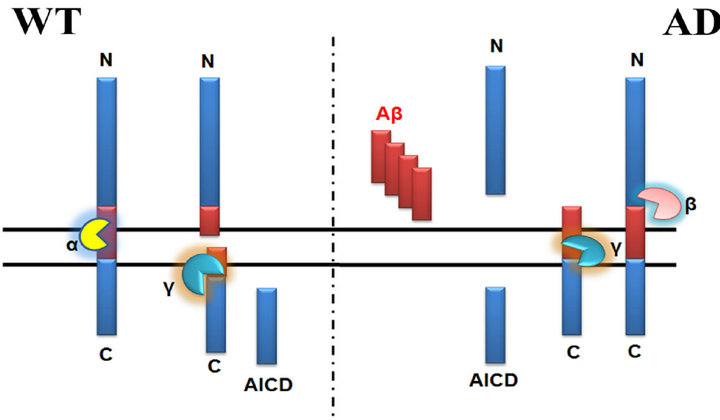
Figure 1. The Structure and Processing of APP.
resulted in a defect in the fast phototaxis, which could be rescued by expressing APPL or human APP [34].
4.3. MSO Development
The APP protein family also regulates development of the peripheral nervous system (PNS). Ectopic expression of APP and its homologues affect mechano-sensory organs (MSOs) development in Drosophila, which resembles Notch gain-of-function phenotype [35]. This function of APP depends on the NPTY motif in the AICD domain, and is mediated by Numb and Dab [35].
4.4. Cerebral Amyloid Angiopathy
AD patients have shown cerebral amyloid angiopathy with deposition of Aβ peptides in the walls of leptomeningeal, cortical arterioles, endothelial cells, smooth muscle cells, adventitial cells and brain pericytes and/or perivascular cells [36]. The deposits, a result of impaired clearance of parenchymal Aβ peptides, will lead to inhibition of angiogenesis, resting hypoperfusion and decreased vascular density [37-40]. Thus, clinical therapies targeting cerebrovascular amyloid angiopathy clearance might provide a potential cure for AD [41].
4.5. Other Functions of APP
APP has been shown to modulate cell growth, motility, neurite outgrowth and cell survival in transfected cell lines [42]. An in vivo study showed that rodents injected APP RNAi developed neuronal migration abnormalities [43]. These functions can be reproduced by the soluble extracellular domain of APP that contains two heparinbinding regions [44]. Recently, the APP extracellular domain was shown to act as a ligand for Death Receptor 6, thus activating caspase 3- triggered cell body apoptotic degeneration and caspase 6- triggered axonal degeneration [45]. The C-terminal intracellular domain of APP (APP intracellular domain, AICD) was also shown to be important by serving as a transcriptional regulator [46]. In addition, the highly conserved YENPTY motif found in AICD is crucially required for the internalization of clathrin-coated pit [47], and thus, modulates the endocytosis and intracellular sorting of APP [48] and Aβ production [49].
5. Aβ CLEARANCE
Despite the enhanced concentration of Aβ production, a subsequent increase in either APP expression or Aβ production has not been reported in patients with sporadic or late onset AD. The concentration of Aβ in the brain thus depends on a maintained balance between its production, influx across the blood brain barrier and clearance from the brain into circulation. While soluble Aβ can be removed from the brain via interstitial fluid bulk flow, this accounts for the removal of only 10% - 15% of the total Aβ within the brain. In addition, the bulk flow of ISF is also responsible for influx of Aβ into the brain from plasma. Recently, receptor mediated Aβ transport has become an important part of the mechanism of Aβ clearance, with several receptors playing key roles [50].
5.1. Flow Mediation of Aβ across Brain Blood Barrier
Loss of function of low density lipoprotein receptorrelated protein (LRP) has been implicated in decreased Aβ clearance in mice and has been genetically linked in epidemiological studies to AD pathogenesis [51, 52]. LRP expression has also been shown to be repressed by increased Aβ levels, which might contribute to the observation that LRP activity in the brain of AD patients is low [51]. Another molecule, ATP-binding cassette transporter p-glycoprotein (p-gp) has also been shown to be inversely correlated with the increase of Aβ in AD patients, suggesting its role as a mediator of Aβ efflux [53].
Cells with receptor for advanced glycation end products (RAGE) have been shown to assist in the transport of Aβ across the blood to the brain, in apposition to LRP and p-gp [54]. Studies have indicated that RAGE expression is up-regulated by extracellular Aβ in the AD brain, suggesting that these interactions exacerbate AD pathology [55].
5.2. Enzyme Mediated Aβ Clearance
Degradation of Aβ through enzymatic processes also plays an important role in the removal of Aβ from the brain. Neprilysin, an Aβ degrading enzyme in the brain, is extracellularly exposed to sites of Aβ deposits. While expression of neprilysin decreases in the cortex and hippocampus of AD brains [56], transgenic expression of human intracellular neprilysin was able to drastically reduce amyloid deposits in mice [56,58]. Insulin-degrading enzyme (IDE) was also shown to influence the clearance of Aβ in AD patients. In animal studies, impairment of IDE resulted in decreased Aβ degradation, while overexpression of IDE was able to reduce extracellular Aβ levels and completely prevent the formation of amyloid plaque [58,59].
6. Aβ TOXICITY
The role of APP has been primarily focused on the toxicity of Aβ since this peptide was found to be accumulated in the senile plaques [60]. Aβ was shown to be neurotoxic [61], probably through apoptosis triggered by Aβ oxidation [62]. Mice overexpressing mutant human APP quickly developed Aβ plaques and elicited loss of synaptic contact, infections and decreased spatial memory. This is accompanied by release of caspases, including caspase 3, 6, 7, 8, and 9 [63-66]. Caspase induced damage in AD may occur independent of apoptosis, as APP itself is a target of caspase cleavage and produces a potentially toxic intracellular C-terminal fragment [67, 68]. Controversy has arisen over the toxicities of intracellular versus extracellular Aβ, as intracellular Aβ is seen early in AD. Consistently, intracellular injection of Aβ42, but not Aβ40, resulted in neuronal apoptosis [69].
Soluble Aβ has been shown to activate GSK3β and cdk5, two kinases that regulate tau phosphorylation in generating neurofibrillary tangles. In addition, cleavage of tau was shown to be regulated by Aβ through activation of GSK3β, caspase 3 and 9 as well as calpain [70,71]. Consistently, tau appeared to play an important downstream regulatory role in the Aβ toxicity, as transgenic mice overexpressing APP, but lacking endogenous tau, failed to display the loss of spatial memory phenotype despite an abundance of Aβ [72].
Although the neuronal toxicity of accumulated Aβ in the brain has been well documented, its pathological role in AD progression has remained unresolved. Firstly, removal of APP, and thus, Aβ production in mice did not prove beneficial to adults. Secondly, though clearance of amyloid plaques in AD patients was achieved in a clinical study using an active immunization protocol against Aβ42 peptide, this clearance did not prevent progressive neurodegeneration [73]. Finally, peripheral injection of Aβ peptides in an experimental autoimmune encephalomyelitis (EAE) animal model suppressed inflammation in lymphoid tissues, which further led to reduced brain inflammation and delayed occurrence of motor paralysis, suggest a neuroprotective effect of Aβ peptides in autoimmune diseases [74].
Despite the many challenges, significant progress has been made in understanding the progression of AD. While the pathological roles of APP in AD development have not been fully understood, by examining in more detail the molecular process through which APP and Aβ peptides affect neuronal development and death, new drugs could possibly be developed targeting these pathways.
7. ACKNOWLEDGEMENTS
This study was supported by the program for new century excellent talents in university (Grant No. NCET-10-0608) and the specialized research fund for the doctoral program (Grant No. 20120072110023) from the Ministry of Education of China.
REFERENCES
- Hebert, L.E., Scherr, P.A., Bienias, J.L., Bennett, D.A. and Evans, D.A. (2003) Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Archives of Neurology, 60, 1119-1122. doi:10.1001/archneur.60.8.1119
- DeKosky, S.T. and Scheff, S.W. (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Annals of Neurology, 27, 457-464. doi.:10.1002/ana.410270502
- Bouras, C., Hof, P.R., Giannakopoulos, P., Michel, J.P. and Morrison, J.H. (1994) Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex, 4, 138-150. doi:10.1093/cercor/4.2.138
- Trojanowski, J.Q. and Lee, V.M. (1994) Paired helical filament tau in Alzheimer’s disease. The kinase connection. The American Journal of Pathology, 144, 449-453.
- Goedert, M., Spillantini, M.G. and Crowther, R.A. (1991) Tau proteins and neurofibrillary degeneration. Brain Pathology, 1, 279-286. doi:10.1111/j.1750-3639.1991.tb00671.x
- Alzheimer, A., Stelzmann, R.A., Schnitzlein, H.N. and Murtagh, F.R. (1995) An English translation of Alzheimer’s 1907 paper, Uber eine eigenartige Erkankung der Hirnrinde. Clinical Anatomy, 8, 429-431. doi:10.1002/ca.980080612
- Glenner, G.G. and Wong, C.W. (1984) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications, 120, 885-890. doi:10.1016/S0006-291X(84)80190-4
- Kang, J., Lemaire, H.G., Unterbeck, A., Salbaum, J.M., Masters, C.L., Grzeschik, K.H., Multhaup, G., Beyreuther, K. and Muller-Hill, B. (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature, 325, 733-736. doi:10.1038/325733a0
- Daigle, I. and Li, C. (1993) Apl-1, a Caenorhabditis elegans gene encoding a protein related to the human β-amyloid protein precursor. Proceedings of the National Academy of Sciences of the United States of America, 90, 12045-12049. doi:10.1073/pnas.90.24.12045
- Rosen, D.R., Martin-Morris, L., Luo, L.Q. and White, K. (1989) A Drosophila gene encoding a protein resembling the human β-amyloid protein precursor. Proceedings of the National Academy of Sciences of the United States of America, 86, 2478-2482. doi:10.1073/pnas.86.7.2478
- Wasco, W., Bupp, K., Magendantz, M., Gusella, J.F., Tanzi, R.E. and Solomon, F. (1992) Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid β protein precursor. Proceedings of the National Academy of Sciences of the United States of America, 89, 10758-10762. doi:10.1073/pnas.89.22.10758
- Wasco, W., Gurubhagavatula, S., Paradis, M.D., Romano, D.M., Sisodia, S.S., Hyman, B.T., Neve, R.L. and Tanzi, R.E. (1993) Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid β protein precursor. Nature Genetics, 5, 95-100. doi:10.1038/ng0993-95
- Sprecher, C.A., Grant, F.J., Grimm, G., O'Hara, P.J., Norris, F., Norris, K. and Foster, D.C. (1993) Molecular cloning of the cDNA for a human amyloid precursor protein homolog: Evidence for a multigene family. Biochemistry, 32, 4481-4486. doi:10.1021/bi00068a002
- Bayer, T.A., Cappai, R., Masters, C.L., Beyreuther, K. and Multhaup, G. (1999) It all sticks together—The APPrelated family of proteins and Alzheimer’s disease. Molecular Psychiatry, 4, 524-528. doi:10.1038/sj.mp.4000552
- Tanaka, S., Shiojiri, S., Takahashi, Y., Kitaguchi, N., Ito, H., Kameyama, M., Kimura, J., Nakamura, S. and Ueda, K. (1989) Tissue-specific expression of three types of β-protein precursor mRNA: Enhancement of protease inhibitor-harboring types in Alzheimer’s disease brain. Biochemical and Biophysical Research Communications, 165, 1406-1414. doi:10.1016/0006-291X(89)92760-5
- Sola, C., Mengod, G., Probst, A. and Palacios, J.M. (1993) Differential regional and cellular distribution of β- amyloid precursor protein messenger RNAs containing and lacking the Kunitz protease inhibitor domain in the brain of human, rat and mouse. Neuroscience, 53, 267-295. doi:10.1016/0306-4522(93)90304-X
- Ponte, P., Gonzalez-DeWhitt, P., Schilling, J., Miller, J., Hsu, D., Greenberg, B., Davis, K., Wallace, W., Lieberburg, I. and Fuller, F. (1988) A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature, 331, 525-527. doi:10.1038/331525a0
- Tanzi, R.E., McClatchey, A.I., Lamperti, E.D., Villa-Komaroff, L., Gusella, J.F. and Neve, R.L. (1988) Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature, 331, 528-530. doi:10.1038/331528a0
- Kitaguchi, N., Takahashi, Y., Tokushima, Y., Shiojiri, S. and Ito, H. (1988) Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature, 331, 530-532. doi:10.1038/331530a0
- Zheng, H. and Koo, E.H. (2006) The amyloid precursor protein: Beyond amyloid. Molecular Neurodegeneration, 1, 5. doi:10.1186/1750-1326-1-5
- Selkoe, D.J. (1991) The molecular pathology of Alzheimer’s disease. Neuron, 6, 487-498. doi:10.1016/0896-6273(91)90052-2
- Shepherd, C., McCann, H. and Halliday, G.M. (2009) Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathologica, 118, 37-52. doi:10.1007/s00401-009-0521-4
- Terry, R.D., Masliah, E., Salmon, D.P., Butters, N., DeTeresa, R., Hill, R., Hansen, L.A. and Katzman, R. (1991) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology, 30, 572-580. doi:10.1002/ana.410300410
- Askanas, V., Engel, W.K. and Alvarez, R.B. (1992) Strong immunoreactivity of β-amyloid precursor protein, including the β-amyloid protein sequence, at human neuromuscular junctions. Neuroscience Letters, 143, 96-100. doi:10.1016/0304-3940(92)90241-X
- Torroja, L., Packard, M., Gorczyca, M., White, K. and Budnik, V. (1999) The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. The Journal of Neuroscience, 19, 7793-7803.
- Wang, B., Yang, L., Wang, Z. and Zheng, H. (2007) Amyolid precursor protein mediates presynaptic localization and activity of the high-affinity choline transporter. Proceedings of the National Academy of Sciences of the United States of America, 104, 14140-14145. doi:10.1073/pnas.0704070104
- Wang, P., Yang, G., Mosier, D.R., Chang, P., Zaidi, T., Gong, Y.D., Zhao, N.M., Dominguez, B., Lee, K.F., Gan, W.B., et al. (2005) Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. The Journal of Neuroscience, 25, 1219-1225.doi:10.1523/JNEUROSCI.4660-04.2005
- Wang, Z., Wang, B., Yang, L., Guo, Q., Aithmitti, N., Songyang, Z. and Zheng, H. (2009) Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. The Journal of Neuroscience, 29, 10788-10801. doi:10.1523/JNEUROSCI.2132-09.2009
- Gunawardena, S. and Goldstein, L.S. (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron, 32, 389-401. doi:10.1016/S0896-6273(01)00496-2
- Kamal, A., Almenar-Queralt, A., LeBlanc, J.F., Roberts, E.A. and Goldstein, L.S. (2001) Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature, 414, 643- 648. doi:10.1038/414643a
- Kamal, A., Stokin, G.B., Yang, Z., Xia, C.H. and Goldstein, L.S. (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron, 28, 449-459. doi:10.1016/S0896-6273(00)00124-0
- Sisodia, S.S. (2002) Biomedicine. A cargo receptor mystery APParently solved? Science, 295, 805-807. doi:10.1126/science.1069661
- Torroja, L., Chu, H., Kotovsky, I. and White, K. (1999) Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Current Biology, 9, 489-492. doi:10.1016/S0960-9822(99)80215-2
- Luo, L., Tully, T. and White, K. (1992) Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron, 9, 595-605. doi:10.1016/0896-6273(92)90024-8
- Merdes, G., Soba, P., Loewer, A., Bilic, M.V., Beyreuther, K. and Paro, R. (2004) Interference of human and Drosophila APP and APP-like proteins with PNS development in Drosophila. The EMBO Journal, 23, 4082-4095. doi:10.1038/sj.emboj.7600413
- Natte, R., De Boer, W.I., Maat-Schieman, M.L., Baelde, H.J., Vinters, H.V., Roos, R.A. and van Duinen, S.G. (1999) Amyloid β precursor protein-mRNA is expressed throughout cerebral vessel walls. Brain Research, 828, 179-183. doi:10.1016/S0006-8993(99)01361-X
- Girouard, H. and Iadecola, C. (2006) Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. Journal of Applied Physiology, 100, 328-335.doi:10.1152/japplphysiol.00966.2005
- Miao, J., Xu, F., Davis, J., Otte-Holler, I., Verbeek, M.M. and Van Nostrand, W.E. (2005) Cerebral microvascular amyloid β protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β precursor protein. The American Journal of Pathology, 167, 505-515. doi:10.1016/S0002-9440(10)62993-8
- Shin, H.K., Jones, P.B., Garcia-Alloza, M., Borrelli, L., Greenberg, S.M., Bacskai, B.J., Frosch, M.P., Hyman, B.T., Moskowitz, M.A. and Ayata, C. (2007) Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain, 130, 2310- 2319. doi:10.1093/brain/awm156
- Takano, T., Han, X., Deane, R., Zlokovic, B. and Nedergaard, M. (2007) Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Annals of the New York Academy of Sciences, 1097, 40-50. doi:10.1196/annals.1379.004
- Dorr, A., Sahota, B., Chinta, L.V., Brown, M.E., Lai, A.Y., Ma, K., Hawkes, C.A., McLaurin, J. and Stefanovic, B. (2012) Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain, 135, 3039-3050. doi:10.1093/brain/aws243
- Karaulana, E., Gramatikoff, K. and Milev, P. (1992) Amyloid precursor protein might be a receptor for basic fibroblast growth factor. The International Journal of Neuroscience, 66, 93-95. doi:10.3109/00207459208999793
- Young-Pearse, T.L., Bai, J., Chang, R., Zheng, J.B., LoTurco, J.J. and Selkoe, D.J. (2007) A critical function for β-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. The Journal of Neuroscience, 27, 14459-14469. doi:10.1523/JNEUROSCI.4701-07.2007
- Mok, S.S., Sberna, G., Heffernan, D., Cappai, R., Galatis, D., Clarris, H.J., Sawyer, W.H., Beyreuther, K., Masters, C.L. and Small, D.H. (1997) Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer’s disease. FEBS Letters, 415, 303-307. doi:10.1016/S0014-5793(97)01146-0
- Nikolaev, A., McLaughlin, T., O'Leary, D.D. and TessierLavigne, M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature, 457, 981-989. doi:10.1038/nature07767
- Cao, X. and Sudhof, T.C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase, Tip60. Science, 293, 115-120. doi:10.1126/science.1058783
- Chen, W.J., Goldstein, J.L. and Brown, M.S. (1990) NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. The Journal of Biological Chemistry, 265, 3116-3123.
- Perez, R.G., Soriano, S., Hayes, J.D., Ostaszewski, B., Xia, W., Selkoe, D.J., Chen, X., Stokin, G.B. and Koo, E.H. (1999) Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. The Journal of Biological Chemistry, 274, 18851-18856. doi:10.1074/jbc.274.27.18851
- Ring, S., Weyer, S.W., Kilian, S.B., Waldron, E., Pietrzik, C.U., Filippov, M.A., Herms, J., Buchholz, C., Eckman, C.B., Korte, M., et al. (2007) The secreted β-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiologycal abnormalities of APP-deficient mice. The Journal of Neuroscience, 27, 7817-7826. doi:10.1523/JNEUROSCI.1026-07.2007
- Crossgrove, J.S. et al. (2005) The choroid plexus removes β-amyloid from braincerebrospinal fluid. Experimental Biology and Medicine (Maywood), 230, 771-776.
- Shibata, M. et al. (2000) Clearance of Alzheimer’s amyloid-β(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. Journal of Clinical Investigation, 106, 1489-1499. doi:10.1172/JCI10498
- Lambert, J.C. et al. (1998) Association at LRP gene locus with sporadic late-onset Alzheimer’s disease. Lancet, 351, 1787-1788. doi:10.1016/S0140-6736(05)78749-3
- Lam, F.C. et al. (2001) β-Amyloid efflux mediated by pglycoprotein. Journal of Neurochemistry, 76, 1121-1128. doi:10.1046/j.1471-4159.2001.00113.x
- Yan, S.D. et al.(2000) Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Lancet, 6, 643-651. doi:10.1038/76216
- Deane, R. et al. (2003) RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nature Medicine, 9, 907-913. doi:10.1038/nm890
- Caccamo, A. et al. (2005) Ageand region-dependent alterations in Aβ-degrading enzymes: Implications for Aβ-induced disorders. Neurobiology of Aging, 26, 645- 654. doi:10.1016/j.neurobiolaging.2004.06.013
- Kanemitsu, H. et al. (2003) Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neuroscience Letters, 350, 113-116. doi:10.1016/S0304-3940(03)00898-X
- Marr, R.A. et al. (2003) Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. The Journal of Neuroscience, 23, 1992-1996.
- Leissring, M.A. et al. (2003) Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron, 40, 1087-1093. doi:10.1016/S0896-6273(03)00787-6
- [61] Goldgaber, D., Lerman, M.I., McBride, O.W., Saffiotti, U. and Gajdusek, D.C. (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science, 235, 877-880. doi:10.1126/science.3810169
- [62] Yankner, B.A., Dawes, L.R., Fisher, S., Villa-Komaroff, L., Oster-Granite, M.L. and Neve, R.L. (1989) Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science, 245, 417-420. doi:10.1126/science.2474201
- [63] Deshpande, A., Mina, E., Glabe, C. and Busciglio, J. (2006) Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. The Journal of Neuroscience, 26, 6011-6018. doi:10.1523/JNEUROSCI.1189-06.2006
- [64] Irizarry, M.C., Soriano, F., McNamara, M., Page, K.J., Schenk, D., Games, D. and Hyman, B.T. (1997) Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. The Journal of Neuroscience, 17, 7053-7059.
- [65] Chen, G., Chen, K.S., Knox, J., Inglis, J., Bernard, A., Martin, S.J., Justice, A., McConlogue, L., Games, D., Freedman, S.B., et al. (2000) A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer’s disease. Nature, 408, 975-979. doi:10.1038/35050103
- [66] Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., Sisodia, S. and Malinow, R. (2003) APP processing and synaptic function. Neuron, 37, 925- 937. doi:10.1016/S0896-6273(03)00124-7
- [67] Spires, T.L. and Hyman, B.T. (2005) Transgenic models of Alzheimer’s disease: Learning from animals. NeuroRx, 2, 423-437. doi:10.1602/neurorx.2.3.423
- [68] Hyman, B.T. (2011) Caspase activation without apoptosis: Insight into Aβ initiation of neurodegeneration. Nature neuroscience, 14, 5-6. doi:10.1038/nn0111-5
- [69] Gervais, F.G., Xu, D., Robertson, G.S., Vaillancourt, J.P., Zhu, Y., Huang, J., LeBlanc, A., Smith, D., Rigby, M., Shearman, M.S., et al. (1999) Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell, 97, 395-406. doi:10.1016/S0092-8674(00)80748-5
- [70] LaFerla, F.M., Green, K.N. and Oddo, S. (2007) Intracellular amyloid-β in Alzheimer’s disease. Nature Reviews Neuroscience, 8, 499-509. doi:10.1038/nrn2168
- [71] Chung, S., Lee, J., Joe, E.H. and Uhm, D.Y. (2001) β-amyloid peptide induces the expression of voltage dependent outward rectifying K+ channels in rat microglia. Neuroscience Letters, 300, 67-70. doi:10.1016/S0304-3940(01)01516-6
- [72] Cho, J.H. and Johnson, G.V. (2004) Glycogen synthase kinase 3β induces caspase-cleaved tau aggregation in situ. The Journal of Biological Chemistry, 279, 54716-54723. doi:10.1074/jbc.M403364200
- [73] Roberson, E.D., Scearce-Levie, K., Palop, J.J., Yan, F., Cheng, I.H., Wu, T., Gerstein, H., Yu, G.Q. and Mucke, L. (2007) Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science, 316, 750-754. doi:10.1126/science.1141736
- [74] Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., Jones, R.W., Bullock, R., Love, S., Neal, J.W., et al. (2008) Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet, 372, 216-223. doi:10.1016/S0140-6736(08)61075-2
- [75] Grant, J.L., Ghosn, E.E., Axtell, R.C., Herges, K., Kuipers, H.F., Woodling, N.S., Andreasson, K., Herzenberg, L.A. and Steinman, L. (2012) Reversal of paralysis and reduced inflammation from peripheral administration of β-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Science Translational Medicine, 4, 145ra105.
NOTES
*These authors contribute equally to this work.