Neuroscience & Medicine
Vol.2 No.1(2011), Article ID:4275,4 pages DOI:10.4236/nm.2011.21003
An Etiological Model for MyalgicEncephalomyelitis/Chronic Fatigue Syndrome
DePaul University, Center for Community Research, Fullerton Ave., Chicago, USA.
E-mail: ljason@depaul.edu
Received September 25th, 2010; revised December 20th, 2010; accepted January 25th, 2010.
Keywords: Myalgic Encephalomyelitis/Chronic Fatigue Syndrome, Kindling, Limbic-hypothalamic-pituitary Cxis
ABSTRACT
Kindling might represent a heuristic model for understanding the etiology of Myalgic Encephalomyelitis/chronic fatigue syndrome (ME/CFS). Kindling occurs when an organism is exposed repeatedly to an initially sub-threshold stimulus resulting in hypersensitivity and spontaneous seizure-like activity. Among patients with ME/CFS, chronically repeated low-intensity stimulation due to an infectious illness might cause kindling of the limbic-hypothalamic-pituitary axis. Kindling might also occur by high-intensity stimulation (e.g., brain trauma) of the limbic-hypothalamic-pituitary axis. Once this system is charged or kindled, it can sustain a high level of arousal with little or no external stimulus and eventually this could lead to hypocortisolism. Seizure activity may spread to adjacent structures of the limbic-hypothalamic-pituitary axis in the brain, which might be responsible for the varied symptoms that occur among patients with ME/CFS. In addition, kindling may also be responsible for high levels of oxidative stress, which has been found in patients with ME/CFS.
1. Introduction
The kindling theory posits that repeated exposure to an initially sub-threshold stimulus can eventually exceed threshold limits, resulting in persistent hypersensitivity to the stimulus and ultimately, spontaneous seizure-like activity. This hypothesis asserts that induced seizures may increase the likelihood that more seizures will occur, since repeated stimulation lowers the threshold for more seizures to occur spontaneously after repetitive subthreshold stimuli. For example, if rats have their brains electrically or chemically stimulated over a period of weeks at a very low intensity that is known to be sub-threshold for eliciting seizure activity, many of the rats will eventually experience epileptic convulsions [1]. Patients with Myalgic Encephalomyelitis/chronic fatigue syndrome (ME/CFS) often report onset of this illness after exposure to a viral infection. New retroviruses such as the xenotropic murine leukemia virus related virus and polytropic murine leukemia viruses have recently been found among some patients with ME/CFS [2,3]. Bro-derick et al. (2010) applied network analysis to cytokines in patients with ME/CFS and healthy controls, and outcomes were consistent with a latent viral infection (i.e., attenuated Th1 and Th17 immune responses, an established Th2 inflammatory milieu, diminished NK cell responsiveness) [4]. Viruses increase activation of ma-crophages, a primary immune response, which produce a release of interleukin-1beta (IL-1β), a pro-inflammatory cytokine, causing an alteration in the electrical activity of the brain [5]. Chronically repeated low-intensity stimulation due to an infectious illness might cause kindling of the limbic-hypothalamic-pituitary axis among patients with ME/CFS. Kindling might also occur by high intensity stimulation (e.g., brain trauma) of the limbic-hypo-thalamic-pituitary axis.
Kindling involves after-discharge of neuronal cell populations that continue to fire after the initiating stimulation has ceased [6]. The stimulus is followed by a growing EEG 3Hz spiking, which increases and decreases in amplitude many times, something that does not occur in a brain functioning normally. This seizure activity often spreads to adjacent structures in the brain. Once this system is charged, it can sustain a high level of arousal with little or no external stimulus. Excessive aro-usal can lead to an increase in the dendrites of the limbic system, with an increase in excitatory postsynaptic receptors and a decrease in inhibitory presynaptic receptors.
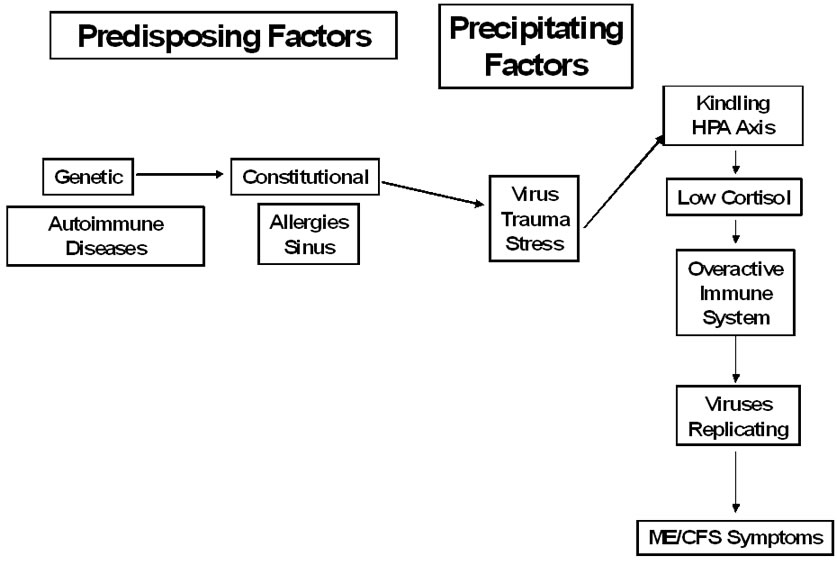
Figure 1. Model for ME/CFS
Arnsten (2009) has found that dendrites in the prefrontal cortex change after only one week of stress, and dendrites in the amygdala expand in response to chronic stress exposure [7]. This can result in excitatory neurotoxicity, or the death of nerve cells due to very high levels of glutamate [8].
Brouwer and Packer (1994) indicated that people with ME/CFS might have “unstable cortical excitability associated with sustained muscle activity resulting in varied magnitudes of descending volleys” [9]. There is evidence of changes in ion transport and ion channel activity among patients with ME/CFS [10]. This kindling might also be what is responsible for high levels of oxidative stress in patients with ME/CFS due to the dysfunction of ion channels in conjunction with ion transport [11,12].
Recently, Jason, Jessen, et al. (2009) developed a scale to measure the duration, severity and frequency of different fatigue-related sensations and symptoms. When factor analyzed, several fatigue factors emerged for individuals with ME/CFS. One factor was considered an over stimulation of the mind or body without the available energy to act out the mental or physiological excited state. This symptom, which could be a manifestation of kindling, was found among patients with ME/CFS but not among healthy individuals [13]. In addition, kindling could also be implicated in several other core symptoms of ME/CFS [14]. Figure 1 presents a model of predisposing and precipitating factors that have been implicated in the onset of ME/CFS as well as how kindling might be part of other factors that will be reviewed below.
2. Neuroendocrine, Autonomic and Immune Consequences of Kindling
Kuratsune and Watanabe (2007) believe that brain dysfunction among patients with ME/CFS is caused by abnormal production of cytokines, proteins expressed by white blood cells that promote the development of T-cells, B-cells, and Hematopoietic cells. They suggest this abnormal production may be due to reactivation of various herpes viral infections and/or chronic mycoplasma infection. Within seconds of exposure to these types of stressors, corticotropin-releasing hormone (CRH), located in the peptidergic neurons in the paraventricular hypothalamic nucleus, is released from nerve terminals to influence hormonal secretion from ACTH in the pituitary and glucocorticoid secretions in the adrenal glands. Infections could cause the sub-threshold kindling effects that eventually increase levels of CRH [15]. CRH induces neuronal excitability, which can lead to epileptic output, or seizure activity [16]. Fevers and trauma can activate CRH receptors in the hippocampus and amygdala to induce seizures in both children and rats. CRH increases the frequency of spontaneous excitatory postsynaptic currents by 252% [16]. Because of this, there is thought to be a reciprocal relationship as limbic seizure kindling results in increased levels of CRH in the hippocampus as well [17]. Once the kindling has occurred, the CRH might play a less critical role in maintaining the kindling, and after time, CRH levels may become depleted.
Thus, kindling could contribute to dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis. Adults with ME/CFS tend to display lower levels of cortisol (hypocortisolism), the main signaling hormone of the HPA axis [18]. Others have found a lack of responsiveness on the part of the HPA axis to challenge [19], a pattern of glucocorticoid resistance [20], and a disruption or dysregulation of the expected diurnal cortisol pattern [21]. These findings have led several investigators to conclude that fatigue in ME/CFS is partially the result of hypocortisolism [22].
Cortisol serves as the main signaling hormone of the HPA axis. This glucocorticoid can suppress or induce the transcription of other substances that serve to signal aspects of the immune system. In turn, the release of select cytokines can further influence the amount of cortisol from the HPA axis [23]. Sorenson et al. (2011) have found increased expression of pro-inflammatory cytokine IL-8 in fatigued patients, which in turn suppresses the expression of cortisol [24]. Cortisol is also influenced by dehydroepiandrosterone (DHEA), an endogenous hormone that has been implicated in the pathogenic process of fatigue [18]. Cleare, Keane and Miell (2004) found individuals with ME/CFS have demonstrated higher levels of DHEA than controls matched for age, gender, weight, body mass index and menstrual history [25].
There is a close link between oxidative stress and decreased glucocorticoid sensitivity [26], and elevated markers of oxidative stress are reported in patients with ME/CFS [11]. The mechanism by which this occurs might involve histone deacetylases (HDACs), a group of enzymes that inhibit gene regulation through removal of acetyl groups [27]. HDACs inhibit the process of DNA unwinding initiated by histone acetyltransferase. Histone hypoacetylation is associated with high levels of free radicals, whereas hyperacetylation is observed with weak oxidative stress [28]. A higher level of HDAC activity in this population leading to hypoacteylation, in conjun-ction with a loss of overall antioxidative power, would indicate there is an apparent dysregulation of select gene expression. The expression of genes encoding antioxidative and Phase II detoxification enzymes is regulated at the transcriptional level and is mediated by a specific enhancer, the antioxidant response element (ARE), found in the promoter region of the antioxidant enzyme genes. The transcription factor Nrf2 is implicated as the central protein that interacts with the ARE to activate gene transcription. In that regard, HDACs need to be inhibited to allow for the recruitment of Nrf2 and its co-activators to fully activate gene expression of ARE. As HDACs have been indicated to positively regulate pro-inflammatory gene expression [29], this could account for the increased expression of IL-8 that, in turn, suppresses the expression of cortisol [24]. This dysregulation of gene expression could also account for divergent immune cell class findings in those with fatigue, possibly through influence on coactivator proteins leading to differential cytokine/chemokine expression. Jason, Sorenson et al. (2011) recently found among an elderly sample with ME/CFS increased histone deacetylases activity, lower total ant-ioxidant power, in the context of decreased plasma cort-isol and increased plasma dehydroepiandrosterone conc-omitant with decreased expression of the encoding gene for the glucocorticoid receptor. Therefore, it is possible that increased HDAC activity may in turn contribute to a chronic pro-inflammatory state that may result in the expression of fatigue [30].
Variance in the expression of genes associated with HPA axis function was related with ME/CFS across several studies with adult populations [31,32]. A genetic study by Rajeevan et al. (2006) also implicated cortisol regulatory mechanisms in ME/CFS, finding that single nucleotide polymorphisms in the glucocorticoid receptor (GR) gene were associated with increased risk for ME/CFS [33]. Previous work has found variance in the expression of GR (NR3C1) in individuals with ME/CFS when compared to controls [34]. Those with ME/CFS may have decreased sensitivity to the effects of cortisol due to a down-regulation of GR [20]. In a pediatric ME/CFS sample, Jason et al. (2010) found hypocor-tisolism and the down-regulated expression of NR3C1 (the encoding gene for the glucocorticoid receptor expression GR), NFKB1, and NFKB2 [35]. It is possible that epigenetic alterations in expression of NR3C1 leads to an inflammatory immunologic profile, which further suppresses cortisol levels through a process of feedback through bidirectional pathways. The reduced expression of the gene for the glucocorticoid receptor expression provides evidence for dysfunction of the HPA axis in those with CFS.1
The classic COX inhibitors such as aspirin, which inhibit the formation of prostanoids, may contribute to several dysfunctions observed in patients with ME/CFS, including inflammation, immunity, signaling molecules created by oxidation, central nervous system dysfunction, and even a dysregulation in sex hormones [36]. This, in turn, may work in conjunction with the oxidative stress observed in patients with ME/CFS. Also, because the COX enzyme is associated with glucocorticoid activity, this may provide an explanation for the hypoactivity of cortisol, something that is also exhibited in persons with ME/CFS.
Kindling might cause sympathetic nervous system hyperactivity, which may ultimately result in decreased levels of both serum cortisol and acetylcholine, which is a primary neurotransmitter of the parasympathetic nervous system and is widely distributed throughout the brain and spinal cord [37]. Chaudhuri, Majeed, Dinan, and Behan (1997) believe that ME/CFS entails a depletion of acetylcholine and increased sensitivity of the postsynaptic acetylcholine receptors [38]. Chronic cortisol deficiency can cause an over production of pro-inflammatory cytokine interleukin-6 (Il-6), which has been associated with symptoms of ME/CFS [39]. Lower cortisol [40] as well as an overactive sympathetic nervous system could be responsible for the findings of ejection fraction decreases (fraction of blood pumped out of the ventricles per heartbeat) and lower cardiac output among patients with ME/CFS [41].
Exposure to chronic stressors could eventually lead to hypocortisolism, and glucocorticoids do have an inhibitory effect on serotonin function. Among ME/CFS patients, Cleare et al. (1995) have found hypocortisolism along with increased serotonin neurotransmitter function [42]. Vassallo et al. (2001) found evidence of elevated activity of presynaptic serotonin neurons, which could be an area affected by kindling [43]. The serotonin hy-pothesis links fatigue to increases in serotonin synthesis and elevations of activities of serotonergic neurons. Serotonin, a neurotransmitter derived from the amino acid tryptophan, plays a role in mood, body temperature, physical coordination, appetite and sleep. Serotonin helps keep moods under control by calming anxiety, relieving depression, and helping with sleep. Low serotonin levels are linked with depression and insomnia, and sleep can sometimes be restored when levels of serotonin are brought up to normal [44]. The area of the brain that mediates deep sleep is called the raphe nuclei, and this area contains nerve cells that use serotonin to communicate with each other. Shepherd (1988) found that cats are unable to sleep when this area of the brain is destroyed, and a drug that blocks serotonin synthesis produces insomnia [45]. In contrast to depression, studies have found that patients with ME/CFS have abnormally high level of brain serotonin, and this may contribute to the persistent central fatigue [46]. One pilot study found that medications that block serotonin (5-HT3) receptors were followed by at least a 35% improvement in about one-third of patients [47].
Still, there may be subtypes of ME/CFS patients. One study found decreased brain serotonin levels in patients with ME/CFS [48]. Yamamoto et al. (2004) using PET, found that the density of 5-HTT (the serotonin transporter involved in the reuptake of serotonin from the synaptic cleft immediately after its release) of the rostral subdivision of the anterior cingulate cortex was significantly reduced in patients with ME/CFS. These findings suggest that an alteration in the serotonergic neurons in the anterior cingulate cortex, specifically a depletion of serotonin, might play a role in the pathophysiology of ME/CFS [49]. There are at least seven receptor subtypes (5-HT1A, 5-HT1B, 5-HT1C, 5-HT1D, 5-HT2, 5-HT3, 5-HT4) in the brain, and direct-acting agonists and antagonists can have selective affinity for specific receptor subtypes [50]. Cleare, Messa, Rabiner, and Grasby (2005), for example, found widespread reduction in 5-HT1A receptor binding potential, and this was particularly mar-ked in the hippocampus bilaterally, where a 23% reduction was observed [51]. In a related area, in one of the major genes of the serotonergic neurotransmission sys-tem (serotonin receptor 2A, HTR2A), Falkenberg, Gurbaxani, Unger and Rajeevan (2010) found that HTR2A is up-regulated in patients with ME/CFS [52].
Stressors and viruses can together and separately cause the serotonin and dopamine systems to become activated in the central nervous system [53]. If stressors occur for weeks or months, glucocorticoid, norepinephrine, and epinephrine levels suppress the immune system and the TH1 (pro-inflammatory) immune response, with a shift to a TH2 (anti-inflammatory) immune response [54]. Because of the TH2 shift, the body would not have an effective defense against viral or intracellular bacterial infections. Eventually, the HPA axis might switch from hyperto hypo-functioning [55]. However, when the HPA axis is down-regulated, there would still not be an effective TH1 response to attack the viral infection; now the immune system may cause inflammation, explaining elevated antinuclear antibodies, antibodies that react against components of the cellular nucleus [56].
Of course, there are many other factors that could be affected by kindling and ultimately cause severe fatigue. For example, increases in transforming growth factor- β (TGF-β) have been observed during infection and stress in rats [57]. The increase in TGF-β inhibits the production of DHEA-S, which is related to the dysmetabolism of acetyl-L-carnitine, leading to deterioration of biosynthesis of glutamate in the anterior cingulum. In addition, the effort of going from home to the hospital actually increases TGF-beta levels [58].
Others have proposed alternative explanations for the pathophysiology found in ME/CFS. For example, Bara-niuk et al. (2005) identified proteins that predicted ME/ CFS status with 80% concordance (i.e., alpha-1-macro-globulin, amyloid precursor-like protein 1, keratin 16, orosomucoid 2 and pigment epithelium-derived factor). Baraniuk et al. suggested that patients with ME/CFS had unusual proteins in cerebral spinal fluid, and the aggregation of these abnormal proteins, also known as amyloids, could cause small amounts of bleeding in the brain. Amyloid proteins are folded improperly as they are being made, and because these proteins are not doing the activities they are designed to do, vital processes might be neglected. Baraniuk et al. postulate that ME/CFS symptoms could be caused by these abnormally folded proteins causing small punctures in the blood vessels and then small amounts of blood leak into the brain. Other proteins suggest a protease—antiprotease imbalance, increased free radical production, vasoconstriction of the blood vessels, inflammation, and altered rates of cell suicide. Baraniuk et al. suggest that inflammation, hemorrhagic elements, increased cell death, and free radical production could be by-products of damage of abnormally folded proteins impeding blood flow and ultimately puncturing blood vessels in the brain [59]. Patients with ME/CFS might produce more of these proteins than healthy individuals, and their bodies might not be able to metabolize them, resulting in excessive concentrations of these damaged proteins. In addition, Baraniuk et al. believe that patients with ME/CFS have a longer rather than shorter version of the gene CNDP1 (carnosine dipeptidase). The longer version of this gene impairs the ability of the brain to protect itself from free radicals. CNDP1 degrades carnosine, which is a free radical scavenger and increases corticosterone levels [59]. Therefore, low carnosine levels could contribute to increased oxidative stress. This genetic susceptibility may cause increased viral severity and duration, which in combination with a previous neurotropic viral infection such as Herpes viruses, EBV, XMRV, or HIV, could lead to the results found in kindling studies.
3. Targets of Kindling in the Brain
Baraniuk et al. (2005) believes that neural circuits running from the spinal cord to the brainstem are not working properly [59]. In patients with ME/CFS, gates are not filtering out unnecessary information, and the most critical neural circuit, the Papez Circuit, which ties together the anterior cingulate, amygdala and hippocampus, is associated with heightened awareness [60]. The drug Klonopin often helps patients with ME/CFS because it slows down the over activity of the brain by increasing production of GABA, a central nervous system inhibitory neurotransmitter, thereby reducing the ‘set point’ at which it’s neurons are activated.
de Lange et al. (2005) observed significant reductions in grey matter volume in patients with ME/CFS, and this might be due to oxidative stress caused by kindling [61,62]. Biswal, Kunwar, and Natelson (2010) found sig-nificant cerebral blood flow reductions in patients with ME/CFS in nearly every region of the brain assessed [63]. It is critical to attempt to identify where damage has occurred. Magnetic resonance (MR) studies of encephalopathy and encephalomyelitis associated with acute Epstein-Barr Virus (EBV) infection have found T2 prolongation over gray and white matter, brain atrophy, and periventricular leukomalacia [64]. A MR study examining a pediatric population of patients suffering from chronic EBV infection has shown evidence for the presence of lesions in the hippocampal region [65].
Areas of the prefrontal cortex and anterior cingulate influence the amygdala [66], and kindling in these areas and others could cause continuous sympathetic stimulation that would eventually lead to mental and physical exhaustion as well as glandular depletion. Administration of CRH in rats produces seizures in the amygdala and epileptiform discharges in the hippocampus, but the earliest CRH induced epileptiform discharges are produced in the amydgala and propagate to the hippocampus [16]. Another provocative finding is that following injury, the dorsal root ganglion cells are in a state of neuropathic pain, and glia prolong this state of neuronal hypersen-sitization by releasing substances that act on the immune system [67].
Johnson and DeLuca (2005) concluded that functional neuroimaging studies among patients with ME/CFS generally show hypometabolism in the frontal lobes and ganglia [22]. Neary et al. (2008) tested whether patients with ME/CFS have reduced oxygen delivery to the brain during and exercise challenge. They found that in addition to significant exercise intolerance, patients in comparison to controls evidenced reduced prefrontal oxygenation, suggesting altered cerebral oxygenation and blood volume in the brain [68]. Clearly, both kindling and oxidative stress could be implicated in these findings. Abnormal findings in different regions of the brain may be due to kindling that occurs in a secondary manner, separate from the initial kindling. These secondary sites could then affect different parts of the brain.
When imagining fatigue-provoking events, larger observed activity was found using fMRI in the medial parietal cortex and precuneus in patients with ME/CFS compared with healthy controls [69]. In addition, Caseras et al. also found lower cerebral activity in the dorsolateral prefrontal cortex, an area where grey matter reductions have been found among patients with ME/CFS [69,70]. Interestingly, de Lange et al. (2008) found that cognitive behavioral therapy could partly reverse the grey matter volume reduction in the lateral prefrontal cortex [71].
Billiot, Budzynski, and Andrasik (1997) found increased microvolt levels in lower frequencies (5 - 7 Hz) among patients with ME/CFS, and excess theta waves could be related to cognitive problems (delta waves occurs from 0 to 4 Hz, theta from 4 to 8 Hz, alpha from 8 to 13 Hz and beta from 13 to 21 Hz) [72]. Research using low-resolution electromagnetic brain tomography by Sherlin et al. (2006) has found that twins with ME/CFS compared to their healthy co-twins had higher delta waves in the left uncus and parahippocampal gyrus and higher theta waves in the cingulate gyrus and right superior frontal gyrus [73]. It appears that the slowing of the deeper structures of the limbic system is associated with affect. Flor-Henry, Lind, and Koles (2009) were able to successfully differentiate ME/CFS from controls in 83% of cases using the alpha band during a verbal cognitive condition. Significantly greater source-current activity was found in the left frontal-temporal-parietal regions of the cortex among the patients with ME/CFS [74].
Using quantified EEG (qEEG) data, Donati, Fagioli, Komaroff, and Duffy (1994) found spike waves for 44% of the patients with ME/CFS compared to only 1.3% of all others (i.e., patients with depression who were medicated, patients with depression who were not medicated, as well as healthy controls) [75]. Spikes were most common in the temporal regions, which contains the hippocampus. Those in the ME/CFS group also had significantly more sharp waves, more frequent high amplitude alpha, and more frequent bursts of theta waves in the posterior regions. In the patients with ME/CFS, abnormalities were observed that involved high amplitude sharp alpha rhythm (10 Hz) that occurs in the occipital lobes upon closing the eyes. Also, discharges of the type one associates with epilepsy were seen in the temporal lobes. These are typically found after head injury and extreme sleep deprivation [75]. Temporal lobes have a predilection for infection by the herpes virus in acute herpes encephalopathy and encephalitis; therefore, the findings might be related to post-viral mild encephalopathy affecting primarily the temporal lobes, which could cause the self-reported memory and attention problems. In a later study, Duffy et al. (2009) found that factors derived from the EEG data were able to discriminate with nearly 90% accuracy patients with ME/CFS from healthy controls and from those with major depression [76].
Patients with ME/CFS and FM have a propensity toward light sleep (stages 1 and 2) and lack of deep sleep (stage 3), and during sleep there are findings of alpha or awake-like brain waves. Kishi et al. (2010) found that a sleep disruption specific to ME/CFS was a significantly enhanced probability of transition from REM sleep to waking [77]. They also found that those with ME/CFS and FM had greater probabilities of transitioning from waking, REM sleep and Stage 1 to Stage 2, and those from slow-wave sleep to Stage 1 [77]. Using a multiple sleep latency test (MSLT), in which patients are given the opportunity to fall asleep during five 20-minute nap periods, Spitzer and Broadman (2010) found that 80% of FM and ME/CFS patients fell asleep in under 8 minutes, which is an indication of excessive daytime sleepiness [78]. In addition, an immunological marker DQB1*0602 that is present in about half of people with narcolepsy, was present in 43% of patients, which is five times higher than in healthy people. People with narcolepsy sleep poorly at night and have excessive daytime sleepiness [78]. Spitzer and Broadman believe that patients with ME/CFS and FM have a lesion in the ventral-lateral preoptic nucleus of hypothalamus, which is critical to initiating and maintaining sleep. Sodium oxybate (Xyrem) has been used for narcolepsy, as it greatly enhances deep level sleep, and Spitzer and Broadman report that 59% of ME/CFS and FM patients who tried this medication reported substantial relief of pain, and 75% had significant reduction of fatigue [78]. Moldofsky, Inhaber, Guinta, and Alvarez-Horine (2010) also found improved EEG sleep physiology and sleep-related FM symptoms after treatment with Xyrem [79]. Also of interest is that stress can trigger mast cells, which are heavily populated in the thalamus (that is located next to the sleep/wake center in the hypothalamus), to release the stimulant histamine. Excessive mast cells and their release of stimulants could be one of the reasons that sleep is interrupted in patients with ME/CFS and FM. Mast cells act as an immunologic defense against external pathogens, and they are five to 14 fold increases of these cells in the top layer of the skin in 100% of FM patients [80]. Kindling might impact mast cells and the release of histamine in the thalamus, resulting in disrupted sleep patterns among patients with ME/CFS.
4. Risk Factors
Because viruses are so prevalent, it is important to understand why some individuals might be resistant, and others at higher risk for developing kindling and chronic activation. Hickie et al. (2006) followed up with people who had cases of mononucleosis (glandular fever), Q fever, and Ross River virus, respectively, who later met the criteria for ME/CFS [81]. They found that the percentage who went on to have ME/CFS was the same for the three infectious diseases (11% at 6 months), suggesting that the reason these people develop ME/CFS is not associated with the particular pathogen, but rather with their host response [81].
In animal models, kindling and related seizure activity is associated with production of pro-inflammatory proteins within the brain [82]. For example, the admi-nistration of pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α) increases seizure activity in animals [83]. TNF-α is a cytokine involved in systemic inflammation, and it stimulates the release of CRH, therefore stimulating the HPA axis. Among patients with ME/CFS, the production of pro-inflammatory cytokines (IL-1β and IL-6) is correlated with acute sickness behavior (i.e., fever, malaise, pain, fatigue, and poor concentration). As mentioned earlier, prolonged exposure to these cytokines might induce a state of chronic activation and kindling [84]. Individuals who have higher levels of these pro-inflammatory cytokines might be at greater risk of developing ME/CFS. In support of this proposition, Voll-mer-Conna et al. (2008) found that severe illness following an infection was more likely to occur among individuals with high levels of IFN-γ (a pro-inflammatory cytokine) and low levels of IL-10 (an anti-inflammatory cytokine) [85]. In addition, elevated levels of pro-inflammatory cytokines can lead to increases in levels of nitric oxide, and this nitric oxide can, in turn, react with superoxide to form the powerful oxidant peroxynitrite, resulting in oxidative stress. Some investigators, however, have not been able to find increased levels of nitric oxide in patients with ME/CFS [86].
Two receptors residing on the cell surface membranes of neurons are GABA (gamma aminobutyric acid), which inhibits neuronal firing, and NMDA (N-methyl-D-aspartate), which excites neuronal firing. The GABA and NMDA receptors are normally balanced, but after an injury or viral attack, NMDA fires more rapidly than GABA, increasing neuronal firing. Minor and Hunter (2002) have proposed that prolonged exposure to inescapable stressors will eventually deplete GABA, thus reducing an important form of inhibition on excitatory glutamate transmission [87]. Doi, Ueda, Nagatomo, and Willmore (2009) found that rats with diminished GABA functioning were more likely to develop kindling [88]. Winkelman et al. (2008) have found that brain GABA levels were nearly 30% lower in patients with primary insomnia [89]. Recent findings indicate that glial cells or astrocytes (glial cells in the spine or brain) produce adenosine, an inhibitory neurotransmitter that promotes sleep and suppresses arousal, which appears to be implicated in controlling wake to sleep transitions [90]. High adenosine levels can even suppress epileptic seizures, and in rats the adenosine A(2A) receptor (A(2A)R) agonist induces sleep by inhibiting the histaminergic system, an integral part of the immune system, through increasing GABA release [91]. It is also possible that kindling could result in low levels of adenosine over time, causing the development of sleep difficulties, although adenosine is only one of many substances that promotes sleep [92].
In addition, Glass et al. (2004) found that healthy individuals with certain biological patterns (i.e., lower cortisol, more heart rate variability, and NK attenuated response to stress) developed somatic symptoms when asked to stop exercising for a week [93]. These might be some of the other predisposing neuroendocrine and immunologic irregularities of individuals who are at increased risk for developing ME/CFS.
Previous research has found a genetic link to some seizures [94,95]. For example, Haug et al. found three heterozygous mutations in ten genes causing distinct forms of idiopathic epilepsy. The three mutations caused the following: loss of function of CIC-2 channels, decreased transmembrane chloride gradient needed for inhibition of GABA, and changes in voltage gating which may cause membrane depolarization and neuronal hyperexcitability, the latter of which is exhibited in patients with ME/CFS [94]. These genetic mutations may be applicable to the kindling found in patients with ME/CFS patients. In addition, studies have been performed on both monozygotic and dizygotic twins to see if there was a genetic link to seizure activity. Berkovic et al. found that 94% of monozygotic pairs and 71% of dizygotic pairs of twins had major epilepsy syndrome in both twins. The researchers concluded that genetics play a crucial role in epilepsy [95]. There is a need for research on possible genetic connections between seizures and/or neural hyperactivity among patients with ME/CFS.
5. Discussion
Neurotropic viral infections could be responsible for the appearance of lesions in the brain and the presence of focal epileptiform seizure activity. It is possible that kindling could play a major role in the promotion of these seizures. Reactivation of various herpes viral infections and/or chronic mycoplasma infection cause abnormal production of cytokines, which can lead to increases in levels of CRH. CRH elevates the frequency of spontaneous excitatory postsynaptic currents by 252% [16]. Consequently, CRH receptors in the amygdala may induce seizures [16], and stressors may even cause dendrites in the amygdala to expand [7]. The earliest CRH induced epileptiform discharges in rats are produced in the amydgala and propagate to the hippocampus [16]. In some cases, cortical lesions caused by herpes viridae infections fade before MR documentation can take place. Lesions may then reappear under specific conditions of environmental stimuli, a process that fits well with the relapsing and remitting nature of ME/CFS. Within the brain, areas of the prefrontal cortex and anterior cingulate influence the amygdala [66], and kindling in these areas could cause continuous sympathetic nervous stimulation that would eventually lead to glandular depletion.
Evidence reviewed above supports a pattern of HPA axis dysfunction in individuals with ME/CFS. Cortisol regulates several physiologic states, including the prod-uction of immune proteins. In turn, cortisol is influenced by immune proteins called cytokines. Decreased levels of salivary cortisol are associated with insomnia, while the production of select pro-inflammatory cytokines (TNF-α and IL-6) is associated with excessive daytime sleepiness [96]. Hypocortisolism is a finding in individuals with ME/CFS, along with research demonstrating increased levels of DHEA [97,98]. There is some evidence of genetic variance in terms of the glucocorticoid receptor (GR) to which cortisol binds.
It can be hypothesized that COX dysregulation combined with oxidative stress, the dysregulation of the HPA axis, and interactions with serotonin may cause the TH1 to TH2 immune shift. Dysregulation of the HPA axis [22] is related to lower levels of cortisol [18]. In addition, those with ME/CFS may have decreased sensitivity to the effects of cortisol due to a down-regulation of GR [20]. Decreased glucocorticoid sensitivity is associated with oxidative stress [26], and increased markers of oxidative stress are reported in patients with ME/CFS [11]. As HDACs are involved in deacetylation of non-histone proteins, including various transcription factors, higher HDAC activity mediated by oxidative stress could adversely affect the binding of GR to DNA, which requires an acetylated form of GR.
There are constant interactions between the sympathetic and central nervous systems, the immune system, and the sensory systems. Kindling could cause the enhancement of peripheral sensory signals, which activate sympathetic nervous system reflexes. Long-term sensory receptor activation can lead to sensitization of spinal cord and brain systems that transmit fatigue signals, causing long-term fatigue enhancement within the central nervous system [99]. In addition, vascular smooth muscle adrenergic receptors desensitize due to the constant release of catecholamines [100]. Therefore, this dysregulation could lead to bouts of increased metabolites that would further activate sensory receptors. Light et al. (2009) maintain that exercise could send a continuous signal of muscle sensory fatigue to the central sympathetic nervous system causing dysregulation of sympathetic nervous system reflexes, and ultimately producing the recognition of enhanced fatigue [101]. Light et al. found patients with ME/CFS demonstrated increases after exercise that reliably exceeded responses of control subjects in mRNA for genes receptors that can detect muscle produced metabolites, genes that are essential for sympathetic nervous system processes, and immune function genes. About 90% of the ME/CFS patients could be distinguished from control subjects using just 4 of the genes measured (i.e., P2X4, adrenergic b-1, adrenergic b-2, IL-10). The researchers concluded that ME/ CFS patients might have enhanced sensory signal for fatigue that is increased after exercise [101]. These findings all indicate persistent changes in cell membrane function, which are compatible with kindling theory that maintains that excessive arousal can lead to an increase in the dendrites of the limbic system, with an increase in excitatory postsynaptic receptors and a decrease in inhibitory presynaptic receptors.
Recently, Jason, Benton, Torres-Harding, and Muldowney (2009) found that the two groups of patients with ME/CFS had different outcomes on measures of physical functioning and fatigue severity after participating in a non-pharmacological intervention [102]. In general, those patients who exerted more energy than they had available did not improve, whereas those patients who were able to stay within their energy boundaries made significant improvements over time. These findings suggest that when an individual with ME/CFS avoids over-exertion, maintaining an optimal level of activity over time, it might be associated with some improvements in physical functioning and fatigue. This study suggests that being overextended and going beyond energy reserves can be an impediment to improving functionality and fatigue levels [102]. Kindling is an explanation for what might occur when patients with ME/CFS overexert themselves and deplete energy reserves. The kindling hypothesis suggests that once this system is charged, either by high-intensity stimulation or by chronically repeated low-intensity stimulation, activities that involve going beyond energy reserves might enhance an already high level of arousal. In a sense, patients with ME/CFS might have this type of cortical excitability that might be due to kindling, and then when they go beyond their energy reserves, the kindling produces high arousal that has implications for the hypothalamus, the autonomic nervous system, as well as the immune system. Other approaches might include neurofeedback facilitating thalamocortical inhibitory mechanisms, which has been applied to patients with Fibromyalgia and has resulted in reductions in pain and fatigue [103].
ME/CFS could have a genetic predisposition based on Baraniuk and colleagues (2005) finding concerning the CNDP1 gene, which is abnormally long in patients with ME/CFS [60]. A predisposition to develop seizures could be associated with CNDP1. There are a number of documented genetic links to seizures [95,104], the longer gene could be connected with a predisposition to hyperneural activity. It is at least possible that ME/CFS is a genetically transmitted disease that may be triggered by specific environmental cues if these potential coding regions become expressed due to an identical or similar promoter region. This might be similar to what has been found in Crohn’s disease, where about one in three people have a mutated gene Atg16L1, and yet Crohn’s occurs in only about 500,000 Americans. Cadwell et al.. (2010) found that three factors were necessary in mice to create a condition similar to the human bowel disorder Crohn’s disease: a mutated gene, exposure to a damaging chemical and infection with a specific virus [105].
Chow et al. (2010) recently found a gene called Arch in a type of bacteria, which responds to yellow light and a gene called Mac in a fungus that responds to blue light [106]. They found that yellow light silences Arch, while blue light silences Mac. The investigators used a harmless virus to deliver these genes into specific brain cells. When hit with a certain color of light, the gene instructs the cells to make pumps that pump protons out of the cell and this changes the electrical charge of the cell, preventing it from sending signals. In other words, light activates the proteins, which lowers the voltage in the neurons and inhibits their activity and prevents them from firing. The light only affects those neurons sensitized to specific colors of light [106]. Up to now, experiments have been conducted with only animals, but in the future, it is possible that people with ME/CFS might have their brains reprogrammed so that different colors of light correct the corrupted neural components that maintain ME/ CFS.
6. Conclusions
In summary, kindling might represent an approach for better understanding the etiology of ME/CFS. Patients might be exposed to repeated low-intensity stimulation due to an infectious illness or by high-intensity stimulation which kindles aspects of the limbic-hypothalamic-pituitary axis. A high level of arousal with little or no external stimulus can ultimately induce seizure activity, which could spread to adjacent structures in the brain, which may be responsible for the varied symptoms that occur among patients with ME/CFS.
As there are many types of cancer and heart disease, ultimately we might find that there are also different types of ME/CFS, each with their own set of manifestations and treatment needs, some types might be initiated and affected by kindling and other subtypes might have other causes. Landmark-Høyvik et al. (2010) in a recent review article on the genetics and epigenetics of fatigue concluded that there is a need for studies with larger sample sizes with more a clearly defined phenotype [107]. With studies involving larger samples, it would be possible to identify many subtypes of ME/CFS. In addi-tion, as recommended by Landmark-Høyvik et al., we need studies based on systems biology that explains the illness, in combination with more details about the environmental contributors to the illness as well as validation of findings with functional studies.
REFERENCES
- G. V. Goddard, “Development of Epileptic Seizures Through Brain Stimulation at Low Intensity,” Nature, Vol. 214, No. 5092, 1967, pp. 1020-1021. doi:10.1038/2141020a0
- S. Lo, N. Pripuzova, B. Li, A. L. Komaroff, G. Hung, W. R. Richard and H. J. Alter, “Detection of MLV-Related Virus Gene Sequences in Blood of Patients with Chronic Fatigue Syndrome and Healthy Blood Donors,” Pro-ceedings of the National Academy of Sciences, Vol. 107, No. 36, 2010, pp. 15874-15879. doi:10.1073/pnas.1006901107
- V. C. Lombardi, F. W. Ruscetti, J. D. Gupta, M. A. Pfost, K. S. Hagen, D. L Peterson, S. K. Ruscetti, R. K. Bagni, C. Petrow-Sadowski, B. Gold, M. Dean, R. H. Silverman and J. A. Mikovits, “Detection of an Infectious Retrovirus, XMRV, in Blood Cells of Patients with Chronic Fatigue Syndrome,” Science.
- G. Broderick, J. Fuite, A.Kreitz, S. D. Vernon, N. Klimas and M. A. Fletcher, “A Formal Analysis of Cytokine Networks in Chronic Fatigue Syndrome,” Brain, Behavior, and Immunity, 2010. [Epub ahead of print].
- S. F. Maier, L. R. Watkins and M. Fleshner, “Psychoneuroimmunology: The Interface between Behavior, Brain, and Immunity,” American Psychologist, Vol. 49, No. 12, 1994, pp. 1004-1017. doi:10.1037/0003-066X.49.12.1004
- W. Loescher and U. Ebert, “The Role of the Piriform Cortex in Kindling,” Progress in Neurobiology, Vol. 50, No. 5, 1996, pp. 427-482. doi:10.1016/S0301-0082(96)00036-6
- A. F. T. Arnsten, “Stress Signaling Pathways that Impair Prefrontal Cortex Structure and Function,” Nature Revi-ews Neuroscience, Vol. 10, No. 6, 2009, pp. 410-422. doi:10.1038/nrn2648
- D. A. Girdano, G. S. Everly Jr., and D. E. Dusek, “Controlling Stress and Tension,” Englewood Cliffs, N.J.: Prentice Hall, 1990.
- B. Brouwer and T. Packer, “Corticospinal Excitability in Patients Diagnosed with Chronic Fatigue Syndrome,” Muscle and Nerve, Vol. 17, No. 101994, pp. 1210-1212.
- J. W. Gow, C. Cannon, W. M. H. Behan, P. Herzyk, S. Keir, G. Riboldi-Tunnicliffe, et al., “Whole-Genome (33,000 genes) Affymetrix DNA Microarray Analysis of Gene Expression in Chronic Fatigue Syndrome,” Paper presented at the International Conference on Fatigue Science, Karuizawa, Japan, 2005.
- G. Kennedy, V. A. Spence, M. McLaren, A. Hill, C. Underwood and J. Belch, “Oxidative Stress Levels are Raised in Chronic Fatigue Syndrome and are Associated with Clinical Symptoms,” Free Radical Biology & Medicine, Vol. 39, No. 5, 2005, pp. 584-589.doi:10.1016/j.freeradbiomed.2005.04.020
- M. Robinson, S. R. Gray, M. S.Watson, G. Kennedy, A. Hill, J. J. Belch, et al., “Plasma IL-6, its Soluble Receptors and F-Isoprostanes at Rest and during Exercise in Chronic Fatigue Syndrome,” Scandinavian Journal of Medicine & Science in Sports, Vol. 13, No. 3, 2009, pp. 1-9.
- L. A. Jason, T. Jessen, N. Porter, A. Boulton, M. G. Njoku, and F. Friedberg, “Examining Types of Fatigue Among Individuals with ME/CFS,” Disability Studies Quarterly, Vol. 29, No. 3, 2009. http://www.dsq-sds org/ article/view/938/1113.
- B. M. Carruthers, A. K. Jain, K. L. DeMeirleir, D. L. Peterson, N. G. Klimas, A. M. Lerner, et al., “Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Clinical Working Case Definition, Diagnostic and Treatments Protocols,” Journal of Chronic Fatigue Syndrome, Vol. 11, No. 1, 2003, pp. 7-115. doi:10.1300/J092v11n01_02
- H. Kuratsune and Y. Watanabe, “Chronic Fatigue Synd-rome,” In: Y. Watanabe, B. Evengard, B. H. Natelson, L. A. Jason & H. Kuratsune, Eds., Fatigue Science for Human Health, Springer, Tokyo, 2007, pp. 67-88.
- T. Z. Baram and C. G. Hatalski, “Neuropeptide-Mediated Excitability: A Key Triggering Mechanism for Seizure Generation in the Developing Brain,” Trends in Neuro-science, Vol. 21, No. 11, 1998, pp. 471-476. doi:10.1016/S0166-2236(98)01275-2
- S. C. Heinrichs and G. F. Koob, “Corticotropin-Releasing Factor in Brain: A Role in Activation, Arousal, and Affect Regulation,” Perspectives in Pharmacology, Vol. 311, No. 2, 2004, pp. 427-440.
- A. J. Cleare, “The Neuroendocrinology of Chronic Fatigue Syndrome,” Endocrine Reviews, Vol. 24, No. 2, 2003, pp. 236-252.
- T. G. Dinan, T. Majeed, E. Lavelle, L. V Scott, C. Berti and P. Behan, “Blunted Serotonin Mediated Activation of the Hypothalamic-Pituitary-Adrenal Axis in Chronic Fati-gue Syndrome,” Psychoneu-roendocrinology, Vol. 22, No. 4, 1997, pp. 261-267.doi:10.1016/S0306-4530(97)00002-4
- A. Kavelaars, W. Kuis, L. Knook, G. Sinnema, and C. J. Heijnen, “Disturbed Neuroendocrine-Immune Interacti-ons in Chronic Fatigue Syndrome,” Journal of Clinical Endocrinology and Metabolism, Vol. 85, No. 2, 2000, pp. 692-696. doi:10.1210/jc.85.2.692
- S. R. Torres-Harding, M. Sorenson, L. Jason, N. Reynolds, M. Brown, K. Maher and M. A. Fletcher, “The Associations between Basal Salivary Cortisol and Illness Symptomatology in Chronic Fatigue Syndrome,” Journal of Applied Biobehavioral Research, Vol. 13, No. 3, 2008, pp. 157-180. doi:10.1111/j.1751-9861.2008.00033.x
- S. K. Johnson and J. DeLuca, “Chronic Fatigue Syndrome and the Brain,” In: J. DeLuca, Ed., Fatigue as a Window to the Brain, MIT Press: Cambridge, Vol. 1, No. 4, MA, 2005, pp. 137-156.
- A. V. Turnbull and C. L. Rivier, “Regulation of the Hypothalamic-Pituitary-Adrenal Axis by Cytokines: Actions and Mechanisms of Action,” Physiological Reviews, Vol. 79, No. 1, 1999, pp. 1-71.
- M. Sorenson, N. Porter, L. A. Jason, A. Lerch and H. Matthews, “IL-8 Increased in Patients with CFS,” Manuscript Submitted for Publication, 2011.
- A. J. Cleare, V. O. O’Keane and J. P. Miell, “Levels of DHEA and DHEAS and Responses to CRH Stimulation and Hydrocortisone Treatment in Chronic Fatigue Syndrome,” Psychoendocrinology, Vol. 29, No. 6, 2004, pp. 724-732.doi:10.1016/S0306-4530(03)00104-5
- I. M. Adcock, B. Cosio, L. Tsaprouni, P. J. Barnes and K. Ito, “Redox Regulation of Histone Deacetylases and Glucocorticoid-Mediated Inhibition of the Inflammatory Response,” Antioxidants & Redox Signaling, Vol. 7, No. 1-2, 2005, pp. 144-152. doi:10.1089/ars.2005.7.144
- Z. Yuan, N. Rezai-Zadeh, X. Zhang and E. Seto, “Histone Deacetylase Activity Assay,” In: S. Chellappan, Ed., Chromatin Protocols, Springer, 2009, pp. 279-293.
- M. Berthiaume, N. Boufaied, A. Moisan and L. Gaudreau, “High Levels of Oxidative Stress Globally Inhibit Gene Transcription and Histone Acetylation,” DNA Cell Biology, Vol. 25, No. 2, 2006, pp. 124-134.
- K. A. Bode, K. Schroder, D. A. Hume, T. Ravasi, K. Heeg, M. J. Sweet and A. H. Dalpke, “Histone Deacetylase Inhibitors Decrease Toll-Like Receptor-Mediated Activation of Proinflammatory Gene Expression by Impairing Transcription Factor Recruitment,” Immunology, Vol. 122, No. 4, 2007, pp. 596-606. doi:10.1111/j.1365-2567.2007.02678.x
- L. A. Jason, M. Sorenson, N. Porter, M. Brown, A. Lerch, J. Mikovits, L. J. Roberts, K. Sebally, D. Alkazemi and S. Kubow, “Increased HDAC is Associated with Hypocor-tisolism in Older Adults,” Manuscript Submitted for Publication, 2011.
- J. R. Kerr, R. Petty, B. Burke, J. Gough, D. Fear, L. I. Sinclair, D. L. Mattey, S. C. Richards, J. Montgomery, D. A. Baldwin, P. Kellam, T. J. Harrison, G. E. Griffin, J. Main, D. Enlander, D. J. Nutt and S. T. Holgate, “Gene Expression Subtypes in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis,” Journal of Infec-tious Diseases, Vol. 197, No. 8, 2008, pp. 1171-1184. doi:10.1086/533453
- T. Saiki, T. Kawai, K. Morita, M. Ohta, T. Saito, K. Rokutan and N. Ban, “Identification of Marker Genes for Differential Diagnosis of Chronic Fatigue Syndrome,” Molecular Medicine, 2008. PMID: 18596870
- M. S. Rajeevan, A. K. Smith, I. Dimulescu, E. R. Unger, S. D. Vernon, C. Heim and W. C. Reeves, “Glucocorticoid Receptor Polymorphisms and Haplotypes Associated with Chronic Fatigue Syndrome,” Genes, Brain,& Behavior, Vol. 6, 2006, pp. 167-176.doi:10.1111/j.1601-183X.2006.00244.x
- A. K. Smith, P. D. White, E. Aslakson, U. Vollmer-Conna and M. S. Rajeevan, “Polymorphisms in Genes Regulating the HPA Axis Associated with Empirically Delineated Classes of Unexplained Chronic Fatigue,” Pharma-cogenomics, Vol. 7, 2006, pp. 387-394. doi:10.2217/14622416.7.3.387
- L. A. Jason, M. Sorenson, N. Porter, M. Brown, A. Lerch, C. Van der Eb and J. Mikovits, “Possible Genetic Dysre-gulation in Pediatric CFS,” Psychology, Vol. 1, 2010, pp. 247-251. doi:10.4236/psych.2010.14033
- A. J. Dunn, “Cytokine Activation of the HPA Axis,” Annals New York Academy of Sciences, Vol. 917, 2006, pp. 608-617.doi:10.1111/j.1749-6632.2000.tb05426.x
- S. Kizildere, T. Gluck, B. Zietz, J. Scholmerich and R. H. Straub, “During a Corticotropin-Releasing Hormone Test in Healthy Subjects, Administration of a Beta-Adrenergic Antagonist Induced Secretion of Cortisol and Dehydroepiandrosterone Sulfate and Inhibited Secretion of ACTH,” European Journal of Endocrinology, Vol. 148, 2003, pp. 45-53. doi:10.1530/eje.0.1480045
- A. Chaudhuri, T. Majeed, T. Dinan and P. O. Behan, “Chronic Fatigue Syndrome: A Disorder of Central Cholinergic Transmission,” Journal of Chronic Fatigue Syndrome, Vol. 3, 1997, pp. 3-16. doi:10.1300/J092v03n01_02
- M. C. Arnold, D. A. Papanicolaou, J. A. O’Grady, A. Lots-ikas, J. K. Dale, S. Straus, et al., “Using an Interleukin-6 Challenge to Evaluate Neuropsychological Performance in Chronic Fatigue Syndrome,” Psychological Medicine, Vol. 32, 2002, pp. 1075-1089. doi:10.1017/S0033291702006086
- M. Pall, “Explaining ‘Unexplained Illnesses: Disease Paradigm for Chronic Fatigue Syndrome, Multiple Che-mical Sensitivity, Fibromyalgia, Posttraumatic Stress Disorder, Gulf War Syndrome and Others,” Bighamton, N.Y.: Haworth Press, 2007.
- A. Peckerman, R. Chemitiganti, C. Zhao, K. Dahl, B. H. Natelson, L. Zuckler, et al., “Left Ventricular Function in Chronic Fatigue Syndrome (CFS): Data from Nuclear Ventriculography Studies of Responses to Exercise and Portural Stress,” FASEB, Vol. 17 (F Suppl: Part 2), 2003, p. A853.
- A. J. Cleare, J. Bearn, T. Allain, A. McGregor, S. Wessely, R. M. Murray, et al., “Contrasting Neuroendocrine Responses in Depression and Chronic Fatigue Syndrome,” Journal of Affective Disorders, Vol. 34, 1995, pp. 283-289. doi:10.1016/0165-0327(95)00026-J
- C. M. Vassallo, E. Feldman, T. Peto, L. Castell, A. L. Sharpley and P. J. Cowen, “Decreased Tryptophan Availability but Normal Post-Synaptic 5-HT Receptor Sensitivity in Chronic Fatigue Syndrome,” Psychological Medicine, Vol. 31, No. 4, 2001, pp. 585-591.
- S. H. Vashadze, “Insomnia, Serotonin and Depression,” Georgian Medical News, Vol. 150, 2001, pp. 22-24.
- G. M. Shepherd, “Neurobiology,” Oxford University Press, 1988, pp. 517-528.
- A. M. Bakheit, P.O Behan, T. G. Dinan and V. O’Keane, “Possible Upregulation of Hypothalantic 5-Hydroxytry-Ptamine Receptors in Patients with Postviral Fatigue Syndrome,” British Medical Journal, Vol. 304, 1992, pp. 1010-1012.
- M. Spath, D. Welzel and L. Farber, “Treatment of Chr-onic Fatigue Syndrome with 5-HT3 Receptor Antagonists Preliminary Results,” Scandinavian Journal of Rheumatology Supplement, Vol. 113, 2000, pp. 72-77.
- A. B. Badawy, C. J. Morgan, M. B. Llewelyn, R. J. Selwyn, S. R. J. Albuquerque and A. Farmer, “Heterogeneity of Serum Tryptophan Concentration and Availability to the Brain in Patients with the Chronic Fatigue Syndrome,” Journal of Psychopharmacology, Vol. 19, 2005, pp. 385-391. doi:10.1177/0269881105053293
- S. Yamamoto, Y. Ouchi, H. Onoe, E. Yoshikawa, H. Tsu-kada, H. Takahashi, et al., “Reduction of Serotonin Tran-sporters of Patients with Chronic Fatigue Syndrome,” Brain Imaging, Vol. 15, No. 17, 2004, pp. 2571-2574.
- R. W. Fuller, “Role of Serotonin in Therapy of Depression and Related Disorders,” Journal of Clinical Psychiatry, Vol. 52, 1991, pp. 52-57.
- A. J. Cleare, C. Messa, E. A. Rabiner and P. M. Grasby, “Brain 5-HT1A Receptor Binding in Chronic Fatigue Syndrome Measured Using Positron Emission Tomography and [11C]WAY-100635,” Biological Psychiatry, Vol. 57, No. 3, 2005, pp. 239-246.
- V. R. Falkenberg, B. M. Gurbaxani, E. R. Unger and M. S. Rajeevan, “Functional Genomics of Serotonin Receptor 2a (HTR2A): Interaction of Polymorphism, Methylation, Expression and Disease Association,” Neuromolecural Medicine, 2010.
- M. Tanaka and Y. Watanabe, “Mechanism of Fatigue Studied in a Newly Developed Animal Model of Combined (Mental and Physical) Fatigue,” In: Y. Watanabe, B. Evengard, B.H. Natelson, L. A. Jason & H. Kuratsune, Eds., Fatigue Science for Human Health, Springer, Tokyo, 2007, pp. 203-212,.
- I. J. Elenkov, R. L. Wilder, G. P. Chrousos and E. S. Vizi, “The Sympathetic Nerve—An Integrative Interface Between Two Supersystems: The Brain and the Immune System,” Pharmacological Reviews, Vol. 52, No. 2, 2000, pp. 595-638.
- B. V. Van Houdenhove, F. Van Den Eede and P. Luyten, “Does Hypothalamic-Pituitary-Adrenal Axis Hypofunction in Chronic Fatigue Syndrome Reflect a Crash in the Stress System?” Medical Hypothesis, Vol. 72, No. 6, 2009, pp. 701-705. doi:10.1016/j.mehy.2008.11.044
- R. A. Van Konynenburg, “Comments Posted on Esther Sternberg’s Presentation at the NIH CFS Workshop Health Consequences of a Dysregulated Stress Response,” Posted on Co-Cure@LISTSERV. NODAK. EDU on July 1, 2003.
- K. Inoue and T. Fushiki, “Exercise Fatigue,” In: Y. Watanabe, B. Evengard, B. H. Natelson, L. A. Jason, & H. Kuratsune, Eds., Fatigue Science for Human Health, Springer, Tokyo, 2007, pp. 187-201,.
- P. D. White, K. E. Nye, A. J. Pinching, T. M. Yap, N. Power, V. Vleck, D. J. Bentley, J. M. Thomas, M. Buckland and J. M. Parkin, “Immunological Changes After Both Exercise and Activity in Chronic Fatigue Syndrome: A Pilot Study,” Journal of Chronic Fatigue Syndrome, Vol. 12, No. 2, 2004, pp. 51-66. doi:10.1300/J092v12n02_06
- J. N. Baraniuk, B. Casado, H. Maibach, D. J. Clauw, L. K. Pannell and S. S. Hess, “A Chronic Fatigue Syndrome-Related Proteome in Human Cerebrospinal Fluid,” BioMedCentral Neurology, Vol. 1, 2005, PMCID: PMC 1326206
- C. Johnson, “Proteins on the Brain: Spinal Tapping for ME/CFS,” Phoenix Rising Website, 2010, Retrieved on April 9, 2010. http://www.aboutmecfs.org/News/BrainPr-oteomeMar10.aspx.
- F. P. de Lange, J. S. Kalkman, G. Bleijenberg, P. Hagoort, J. W. M. van der Meer and I. Toni, “Gray Matter Volume Reduction in the Chronic Fatigue Syndrome,” Neuro-Image, Vol. 26, No. 3, 2005, pp. 777-781. doi:10.1016/j.neuroimage.2005.02.037
- L. A. Jason, N. Porter, J. Herrington, M. Sorenson and S. Kubow, “Kindling and Oxidative Stress as Contributors to Myalgic Encephalomyelitis/Chronic Fatigue Syndrome,” Journal of Behavioral and Neuroscience Research, Vol. 7, 2009, pp. 1-17.
- B. Biswal, P. Kunwar and B. H. Natelson, “Cerebral Blood Flow is Reduced in Chronic Fatigue Syndrome as Assessed by Arterial Spin Labeling,” Journal of the Neurologic Sciences, 2010. [Epub ahead of print]
- W. J. Shian and C. S. Chi, “Epstein-Barr virus Encephalitis and Encephalomyelitis: MR Findings,” Pediatric Ra-diology, Vol. 26, No. 9, 1996, pp. 690-693.
- M. Hausler, V. T. Ramaekers, M. Doenges, K. Schweizer, K. Ritter and L. Schaade, “Neurological Complications of Acute and Persistent Epstein-Barr Virus Infection in Paediatric Patients,” Journal of Medical Virology, Vol. 68, No. 2, 2002, pp. 253-263. doi:10.1002/jmv.10201
- A. Gupta, “Unconscious Amygdalar Fear Conditioning in a Subset of Chronic Fatigue Syndrome Patients,” Medical Hypotheses, Vol. 59, No. 6, 2002, pp. 727-735.
- B. S. McEwen and M. Kalia, “The Role of Corticosteroids and Stress in Chronic Pain Conditions,” Metabolism, Vol. 59, 2010, pp. S9-15.
- J. P. Neary, A. D. W. Roberts, N. Leavins, M. F. Harrison, J. C. Croll and J. R. Sexsmith, “Prefrontal Cortex Oxygenation During Incremental Exercise in Chronic Fatigue Syndrome,” Clinical Physiology and Functional Imaging, 2008.
- X. Caseras, D. Mataix-Cols, K. A. Rimes, V. Giampietro, M. Brammer, F. Zelaya, et al., “The Neural Correlates of Fatigue: An Exploratory Imaginal Fatigue Provocation Study in Chronic Fatigue Syndrome,” Psychological Medicine, Vol. 38, No. 7, 2008, pp. 941-951.
- T. Okada, M. Tanaka, H. Kuratsune, Y. Watanabe and N. Sadato, “Mechanisms Underlying Fatigue: A Voxel-Based Morphometric Study of Chronic Fatigue Syndrome,” BioMedCentral Neurology, Vol. 4, No. 1, 2004, pp. 14-20.doi:10.1186/1471-2377-4-14
- F. P. de Lange, H. Knoop, G. Bleijenberg and J. W. van der Meer, “The Experience of Fatigue in the Brain (Letter to the Editor),” Psychological Medicine, Vol. 38, 2008, pp. 523-524.
- K. M. Billiot, T. H. Budzynski and F. Andrasik, “EEG Patterns and Chronic Fatigue Syndrome,” Journal of Neurotherapy, Vol. 2, 1997, pp. 20-30. http://www.snr-jnt.org/JournalNT/JNT(22)4.html.8004844doi:10.1300/J184v02n02_04
- L. Sherlin, T. Budzynski, H. Kogan-Budzynski, M. Con-gedo, M. E. Fischer and D. Buchwald, “Low-Resolution Brain Tomography (LORETA) of Monozygotic Twins Disconcordant for Chronic Fatigue Syndrome,” Neuro-Image, Vol. 34, No. 4, 2006, pp. 1438-1442.
- P. Flor-Henry, J. C. Lind and Z. J. Koles, “EEG Source Analysis of Chronic Fatigue Syndrome,” Psychiatric Research, 2009. PMID: 20006474.
- F. Donati, L. Fagioli, A. L. Komaroff and F. H. Duffy, “Quantified EEG Findings in Patients with Chronic Fatigue Syndrome,” Paper Presented at the American Association for Chronic Fatigue Syndrome, Ft. Lauderdale, Florida, 1994.
- F. H. Duffy, G. B. McAnulty, M. McCreary, M. S. Albert, G. Cucharal, A. Shatzberg, et al., “Electroencephalogra-phic Data Distinguish Patients with CFS from Healthy and Depressed Controls,” Paper Presented at the 9th International Association of CFS/ME, Reno, NV, March, 2009.
- A. Kishi, B. H. Natelson, F. Togo, Z. R. Struzik, D. M. Rapoport and Y. Yamamoto, “Sleep Stage Transitions in Chronic Fatigue Syndrome Patients with or without Fibromyalgia,” Proceedings of the International Conference of the IEEE Engineering in Medicine and Biology Society, Vol. 1, 2010, pp. 5391-5394.
- A. R. Spitzer and M. Broadman, “Treatment of the Narcoleptiform Sleep Disorder in Chronic Fatigue Syndrome and Fibromyalgia with Sodium Oxybate,” Pain Practice, Vol. 10, No. 1, 2010, pp. 54-59.
- H. Moldofsky, N. H. Inhaber, D. R. Guinta and S. B. Alvarez-Horine, “Effects of Sodium Oxybate (Xyrem) on Sleep Physiology and Sleep/Wake-Related Symptoms in Patients with Fibromyalgia Syndrome: A Double-Blind, Randomized, Placebo-Controlled Study,” Journal of Rheumatology, 2010. [Epub ahead of print]
- I. Blanco, N. Béritze, M. Argüelles, V. Cárcaba, F. Fern-ández, S.Janciauskiene, K. Oikonomopoulou, F. J. de Serres, E. Fernández-Bustillo and M. D. Hollenberg, “Abnormal Overexpression of Mastocytes in Skin Biop-sies of Fibromyalgia Patients,” Clinical Rheumatology, 2010. [Epub ahead of print]doi:10.1007/s10067-010-1474-7
- I. Hickie, T. Davenport, D. Wakefield, U. Vollmer-Conna, B. Cameron, S. D. Vernon, et al., “Post-Infective and Chronic Fatigue Syndromes Precipitated by Viral and Non-Viral Pathogens: Prospective Cohort Study,” British Medical Journal, Vol. 333, No. 7568, 2006, pp. 575-578. doi:10.1136/bmj.38933.585764.AE
- C. R. Plata-Salaman, S. E. Ilyin, P. Nicolas, N. P. Turrin, D. Gayle, M. C. Flynn, et al., “Kindling Modulates the IL-1b System, TNF-a, TGF-b1 and Neuropeptide mRNAs in Specific Brain Regions,” Molecular Brain Research, Vol. 75, No. 2, 2000, pp. 248-258.
- A. A. Shandra, L. S. Godlevsky, R. S. Vastyanov, A. A. Oleinik, V. L. Konovalenko, E.N. Rapoport, et al., “The Role of TNF-Alpha in Amygdala Kindled Rats. Neuroscience Research, Vol. 42, No. 2, 2002, pp. 147-153. doi:10.1016/S0168-0102(01)00309-1
- U. Vollmer-Conna, C. Fazou, B. Cameron, H. Li, C. Brennan, L. Luck, et al., “Production of Pro-Inflammatory Cytokines Correlates with Symptoms of Acute Sickness Behaviour in Humans,” Psychological Medicine, Vol. 34, 2004, pp. 1-9.doi:10.1017/S0033291704001953
- U. Vollmer-Conna, B. F. Piraino, B. Cameron, T. Davenport, I. Hickie, D. Wakefield, et al., “Cytokine Polymorphisms Have a Synergistic Effect on Severity of the Acute Sickness Response to Infection,” Clinical Infectious Diseases, Vol. 47, 2008, pp. 1418-1425. doi:10.1086/592967
- M. Meeus, I. Van Eupen, J. Hondequin, L. de Hauwere, D. Kos and J. Nijs, “Nitric Oxide Concentrations are Normal and Unrelated to Activity Level in Chronic Fatigue Syndrome: A Case-Control Study,” In Vivo, Vol. 24, No. 6, 2010, pp. 865-869.
- T. R. Minor and A. M Hunter, “Stressor Controllability and Learned Helplessness Research in the United States: Sensitization and Fatigue Processes,” Integrative Physiological & Behavioral Science, Vol. 37, No. 1, 2002, pp. 44-58. doi:10.1007/BF02688805
- T. Doi, Y. Ueda, K. Nagatomo and L. J. Willmore, “Role of Glutamate and GABA Transporters in Development of Pentylenetetrazol-Kindling,” Neurochemistry Research, Vol. 34, 2009, pp. 1324-1331.doi:10.1007/s11064-009-9912-0
- J. W. Winkelman, O. M. Buxton, J. E. Jensen, K. L. Benson, S. P. O’Connor, W. Wang and P. F. Renshaw, “Reduced Brain GABA in Primary Insomnia: Preliminary Data from 4T Proton Magnetic Resonance Spectroscopy (1H-MRS),” Sleep, Vol. 31, No. 11, 2008, pp. 1499-1506.
- M. M. Halassa, C. Florian, T. Fellin, J. R. Munoz, S. Y. Lee, T. Abel, P. Haydon and M. G. Frank, “Astrocytic Modulation of Sleep Homeostasis and Cognitive Consequences of Sleep Loss,” Neuron, Vol. 61, No. 2, 2009, pp. 213-219. doi:10.1016/j.neuron.2008.11.024
- Z. Y. Hong, Z. L. Huang, W. M. Qu, N. Eguchi, Y. Urade and O. Hayaishi, “An Adenosine A Receptor Agonist Induces Sleep by Increasing GABA Release in the Tuberomammillary Nucleus to Inhibit Histaminergic Systems in Rats,” Journal of Neurochemistry, Vol. 92, 2005, pp. 1542-1549.doi:10.1111/j.1471-4159.2004.02991.x
- J. Krueger, “The Brain that Never Fully Sleeps,” Fibromyalgia Network, Vol. 85, 2009, pp. 12-15.
- J. M. Glass, A. K. Lyden, F. Petzke, P. Stein, G. Whalen, K. Ambrose, et al., “The Effect of Brief Exercise Cessation on Pain, Fatigue and Mood Symptom Development in Healthy, Fit Individuals,” Journal of Psychosomatic Research, Vol. 57, 2004, pp. 391-398.
- K. W. Haug, M. Warnstedt, Alekov, T. Sander, A. Ramirez, B. Poser, S. Maljevic, S. Hebeisen, C. Kubisch, J. Rebstock, S. Horvath, K. Hallmann, J. S. Dullinger, B. Rau, F. Haverkamp, S. Beyenburg, H. Schulz, D. Janz, B. Geise, G. Muller-Newen, P. Propping, C. E. Elger, C. Fahlke, H. Lerche and A. Heils, “Mutations in Clcn2 Encoding a Voltage-Gated Chloride Channel are Associated with Idiopathic Generalized Epilepsies,” Nature Genetics, Vol. 33, 2003.
- [95] S. F. Berkovic, R. A. Howell, D. A. Hay and J. L. Hopper, “Epilepsies in Twins: Genetics of the Major Epilepsy Syndromes,” Annals of Neurology, Vol. 43, 1998, pp. 435-445. doi:10.1002/ana.410430405
- A. N. Vgontzas, V. M. Zoumakis D. A. Papanicolaou, E. O. Bixler, P. Prolo, H. M. Lin, A. Vela-Bueno, A. Kales and G. P. Chrousos, “Chronic Insomnia is Associated with a Shift of Interleukin-6 and Tumor Necrosis Factor Secretion from Nighttime to Daytime,” Metabolism, Vol. 51, 2002, pp. 887-892.doi:10.1053/meta.2002.33357
- M. Demitrack and L. Crofford, “Evidence for and Pathophysiologic Implications of Hypothalamic-Pituitary—Adrenal Axis Dysregulation in Fibromyalgia and Chronic Fatigue Syndrome,” Annals of the New York Academy of Sciences, Vol. 840, 1998, pp. 684-697. doi:10.1111/j.1749-6632.1998.tb09607.x
- L. V. Scott, S. Medbak and T. G. Dinan, “Blunted Adre-nocorticotropin and Cortisol Responses to Cortic-otropin-Releasing Hormone Stimulation in Chronic Fati-gue Syn-drome,” Acta Psychiatrica Scandinavica, Vol. 97, 1998, pp. 450-457. doi: 10.1111/j.1600-0447.1998. tb10030.x
- D. B. Cook, P. J. O’Connor, G. Lange and J. Steffener, “Functional Neuroimaging Correlates of Mental Fatigue Induced by Cognition among Chronic Fatigue Syndrome Patients and Controls,” NeuroImage, Vol. 36, 2007, pp. 108-122.doi:10.1016/j.neuroimage.2007.02.033
- M. P. Kaufman and S. G. Hayes, “The Exercise Pressor Reflex,” Clinical Autonomic Research, Vol. 12, 2002, pp. 429-439. doi:10.1007/s10286-002-0059-1
- A. R. Light, A. T. White, R. W. Hughen and K. C. Light, “Moderate Exercise Increases Expression for Sensory, Adrenergic and Immune Genes in Chronic Fatigue Syndrome Patients but not in Normal Subjects,” The Journal of Pain, Vol. 10, No. 10, 2009, pp. 1099-1112.
- L. A. Jason, M. Benton, S. Torres-Harding and K. Muldowney, “The Impact of Energy Modulation on Physical Functioning and Fatigue Severity Among Patients with ME/CFS,” Patient Education and Counseling, Vol. 77, No. 8, 2009, pp. 237-241. PMCID: PMC2767446
- K. Dursun, D. Ermutlu and K. Karamursel, “Neurofeedback Intervention in Fibromyalgia Syndrome: A Randomized, Controlled, Rater Blind Clinical Trial,” Applied Psychophysiology Biofeedback, 2010.
- J. C. Mulley, I. E. Scheffer, S. Petrou and S. F. Berkovic, “Channelopathies as a Genetic Cause of Epilepsy,” Current Opinion in Neurobiology, Vol. 16, No. 2, 2003, pp. 171-176.doi:10.1097/00019052-200304000-00009
- K. Cadwell, K. K. Patel, N. S. Maloney, T. C. Liu, A. C. Y. Ng, C. E. Storer, R. D. Head, R. Xavier, T. S. Stappenbeck and H. W. Virgin, “Virus Plus Susceptibility Gene Interaction Determines Crohn’s Disease Gene Atg16L1 Phenotypes in Intestine,” Cell, 2010.
- B. Y. Chow, X. Han, A. S. Dobry, X. Qian, A. S. Chuong, M. Li, M. A. Henninger, G. M. Belfort, Y. Lin, P. E. Monahan and E. S. Boyden, “High-Performance Genetically-Targetable Optical Neural Silencing by Light-Driven Proton Pumps,” Nature, Vol. 463, No. 7277, 2010, pp. 98-102. doi:10.1038/nature08652
- H. Landmark-Høyvik, K. V. Reinertsen, J. H. Loge, V. N. Kristensen, V. Dumeaux, S. Dosså, A. L. Børresen-Dale and H. Edvardsen, “The Genetics and Epigenetics of Fatigue,” Physical Medicine and Rehabilitation, Vol. 2, No. 5, 2010, pp. 456-465.
- S. Gupta, E. Aslakson, B. M. Gurbaxani and S. D. Vernon, “The Inclusion of the Glucocorticoid in a Hypothalamic Pituitary Adrenal Axis Model Reveals Bistability,” Theo-retical Biology and Medical Modeling, Vol. 4, 2007. PMID: 17300722.
- J. Visser, E. Lentjes, I. Haspels, W. Graffelman, B. Blauw, R. DeKloet and L. Nagelkerken, “Increased Sen-sitivity to Glucocorticoids in Peripheral Blood Mono-nuclear Cells of Chronic Fatigue Syndrome Patients, without Evidence for Altered Density or Affinity of Glucocorticoid Receptors,” Journal of Investigative Medicine, Vol. 49, No. 2, 2001, pp. 195-204.
NOTES
Funding was provided by NIAID.
1Other studies, however, have found either equivalent or increased expression of GR in hypocortisolic conditions such as ME/CFS [108,109]. The Gupta et al. article, however, is a mathematical theoretical construct that compared the developed model to a standard cortisol curve. It did not utilize a disease population [108]. The Visser et al. article incubated cells for a far longer duration than Jason et al., and it appears that they had stimulated the cell population through the use of a phorbol ester. Jason et al. used unstimulated cells at 48 hours, versus the 96 hours of incubation used by Visser. Visser et al. also incubated cells in the presence of two differing glucocorticoids in order to determine response. They apparently did not use an untreated control [109].