Open Journal of Applied Sciences
Vol.4 No.6(2014), Article ID:45854,24 pages DOI:10.4236/ojapps.2014.46030
Role of Complements and Immunoglobulins in Duchenne Muscular Dystrophy
Sanjeev Kumar1*, Reena Mittal2, Sweety1, Depp Chand Jain3
1Department of Physics, Central Medical Physics Research Laboratory, D.A.V. (P.G.) College, Muzaffarnagar, India
2Department of Mathematics, Shri.K.K.JAIN. College, Khatauli, India
3Department of Neurology, Safdarganj Hospital, New Delhi, India
Email: *sanjeev1962kumar@yahoo.co.in, *sanjeev1962kumar@rediffmail.com, reena_math@rediffmail.com, drdcjain@gmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 26 March 2014; revised 29 April 2014; accepted 7 May 2014
ABSTRACT
Muscular dystrophies are myopathies and tend to progressive, with ongoing degeneration and regeneration of muscle fibers. Spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS) and polio myelitis are essentially diseases of the anterior horn cells of the spine. It has been reported in literature that humoral immunity is manifested by the antibodies production. These are special chemical substances that react against foreign body. Antibodies are serum proteins, which are immunoglobulins and possess antibody activity and are classified according to antigens and stimulate their production such as IgA, JgG, IgM, IgD and IgE. All the immunological parameters such as of C3, C4, IgG, IgM and IgA, which are measured in Duchenne muscular dystrophy go down in comparison to healthy subjects. Complement C3 and Complement C4 go down about 44.3% and 78.57% respectively from the normal values. The serum IgG, IgM and IgA levels were also go down about 65%, 84% and 99.56% respectively in comparison to healthy subjects. A trend between all the immunoglobulins has been set up and it is rAG.M wang##Bracket# rMA.G. > rGM.A, while we have a trend in DMD cases is rMA.G. > rAG.M > rGM.A. We are in a position to say that our data have a relevance of high authenticity and reliability to accept that there is a deficit in immunity in DMD cases. The deficit in immunity may be a cause to damage muscle for abnormal functioning.
Keywords:Complements, Immunoglobulins, Duchenne Muscular Dystrophy, Regression Coefficients, Multiple Correlation Coefficients

1. Introduction
Humans are able to freely move their bodies about muscles, which is a precious, complex physiology that should not be taken for granted. There are so many types of muscles in human body like voluntary and involuntary muscles, and smooth and strained muscle fibers that all function in a tight realm with the nervous system and different chemical reactions.
Muscles are composed of protein in a highly organized system from large groups to small fibers. Muscle units are separated from other muscle groups by plasma membranes called sarcolemma and the cytoplasm, which is called sarcoplasm. There are long protein bundles within the sarcoplasm called myofibrils. There are so many ATP producing mitochondria, as well as glycogen and myoglobin. Bundles of paralled myofilaments make up the myofibrils where most of the actions take place. There are contractile proteins in the myofilaments, called myosin, and action. The space between each Z disc, where these filaments interact, is called the sarcomere. When signaled, the actin and myosin interlock and slide over each other to stretch or slide into one another to contraction. They are signaled from the nervous system followed by a series of chemical reactions involving ATP, calcium, sodium and potassium ions.
There are many other proteins involved in the process. Aside from the contractile proteins, there are regulatory proteins called tropomysin and trooponin which act like a switch to determine when to contract and where to relax. On the muscle fiber, the “I band” is the space between the myosin (thick) filaments, where lies only the thin filaments. There is a dark disc in the middle of each “I” band called “Z disc”. This disc is made of titan. This is connected to sarcolemma by the cytoskeleton. As the muscle contracts, the “I band” shrinks and the sarcomere shortens and as the Z disc’s come closer together pulling on the sarcolemma shortening the cell. This is how the muscle contracts?
One of the most clinically important accessory proteins is dystrophin. This is located just under the sarcolemma in the cytoplasm in the area of the “I band”. It is produced by specific genes and links the actin filaments to the protein extracellular matrix in the membrane known as the dystrophin associated protein complex.
Elements of the dystrophin gene and the protein structure have been completely identified. The exact functional role is still a bit unclear. It is thought that its primary function is to provide mechanical reinforcement to the structure of the sarcolemma and thereby protecting the membrane from the stress or tearing during contraction. If dystrophin is defective or absent, the membrane breaks down, which then substances and molecules like proteins and enzyme leak out the fibre into circulation. These enzymes and chemicals that leak out are responsible for certain chemical reactions and necessary to produce for muscle contraction. At the same time the extracellular substances leak into the fiber through the broken down membrane damaging the fiber and disrupting the process of muscle contraction and may cause irreparable damage. The absence or abnormality of dystrophin results in a condition known as muscular dystrophy (MD). Muscular dystrophny is a crippling disease resulting from mutated genes, which showly wastes away muscle tissue. It has been seen that without dystrophin to help to protect the fiber membrane keeping it intact, and assisting to create energy, the muscles begin to degenerate and atrophy, being replaced by fat and fibrous scar tissue creating fascia adhesions throughout the body. It is thought that the major determinate of the membrane damage would be the level of stress associated with contraction rather than the number of muscle actions. Petrof, B., et. al. [1] have given their thoughts to explain why it primarily affects the peripheral limbs? Muscular dystrophies most commonly involve in a genetic mutation in the dystrophin genes preventing the production of dystrophin or limiting the amount in subnormal levels. Generally in muscle tissue it has a normal value for small tares on the sarcolemma to occur as the muscle undergoes excessive strain and there are small molecules that enhance the natural repair process.
However in the absence of dystrophin, the sarcolemma is left unprotected tearing more frequently and more easily therefore muscle degeneration greatly out weights muscle regeneration eventually leading to death and adhesion of the tissue.
Emery, A.E. [2] has given his thought regarding muscular dystrophies, which are characterized by progressive irreversible degeneration process, which results in weakness and wasting of muscle tissue. The muscular dystrophies share clinical symptoms. These differ largely in severity, age of onset, and distribution of affected muscle tissue.
The mechanisms responsible for this divergence in pathology are still not identified in detail. It is useful to understand the basic anatomy of the nerve and muscle disorders. The nerve cells have bodies that contain a nucleus. The nucleus contains a genetic material, projection called dendrites that receive information from other nerves, and axons that transmit information to other nerves, ultimately, to muscles. Whether it be the limb, eyes, bladder, bowel, lungs, or heart, all body movements depend on or regulated by the action of nerves on muscle. The brain is made up of billions of nerve cells that communicate with each other by sending information across interconnecting axons and dendrites, i.e., by synapsing. Some nerve cells in the brain have long axons that leave the brain and descend in the spiral cord to synapse with the cell bodies of other nerves, the anterior horn cells, in the spinal cord. These cell bodies are found at each level of the spine. After receiving the information from the brain, they send these information down their axons (peripheral nerves) to communicate synapse with muscles in the limb, head and diaphragm. The axons of limbs peripheral nerves synapse with muscle cells across meet at a junction called myoneural junction.
By definition, neuropathies are diseases of nerves and myopathies are diseases of muscle. After cystic fibrosis, myopathies are the most commonly inherited diseases. Muscular dystrophies are myopathies and tend to progressive, with ongoing degeneration and regeneration of muscle fibers. Spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS) and polio myelitis are essentially diseases of the anterior horn cells of the spine. Bach, R.J. [3] has given views that people with SMA are born with fewer-than normal anterior horn cells of the spine. Neuromuscular diseases are diseases of the peripheral nerves (neuropathies and anterior horn cell diseases), the myoneural junctions (myasthenia gravis), or the muscles (myopathies) themselves can be understood with the detailing of these diseases. These conditions cause muscle weakness but do not affect sensory functions such as the ability to feel objects, see, hear, or smell, nor do they affect the autonomic nervous system that controls bladder and bowl function.
Every cell in the human body has forty eight chromosomes. Forty six chromosomes are autosomes and two are X or Y sex chromosomes. Forty six autosomes are twenty three pairs of chromosomes; one in each pair is inherited from each parent. Our entire genetic make-up is determined by the deoxyribonucleic acid (DNA) content of the hundreds of thousands of genes in each of the chromosomes. Defective genes result in many of the neuromuscular diseases.
Recessive diseases require that the abnormal gene by inherited from both parents. When an abnormal gene needs to be present on the autosome inherited from one parent only, the disorder is called autosomal dominant. Thus, neuromuscular diseases inherited from defective genes on autosomes are either autosomal recessive or autosomal dominant disorders. One of about every 40 - 60 adults carries the defective gene for SMA on one of his or her two number 5 chromosomes. For a child to inherit SMA, an autosomal recessive disorder, both mother or father must carry the defective gene and transmit it to the child. There is one out of four chance that a child of parent carriers of SMS will inherit the abnormal gene from both parents and develop SMA. There is a fifty percent chance that the child will be a carrier but not develop SMA. There will be twenty five percent chance that the child will neither be a carrier nor develop SMA. Children with SMA must inherit defective genes from both parents. For an autosomal dominant condition like facioscapulohumeral muscular dystrophy, on the other hand, only one parent’s affected gene needs to be transmitted. Each patient must have one affected parent, and there is a fifty percent chance of each child inheriting the disease. Neuromuscular disease can also be inherited from defects on the X (Sex) chromosome. Children with sex-linked recessive disorders, such as Duchenne, Becker, and Emery-Dreifus muscular dystrophies, inherit a single defective gene on the X chromosome from the mother and a normal Y chromosome from the father. Because Y chromosome does not have the genetic material to offset the presence of the defect on the X-chromosome, the child inherits the disease. This is called sex-linked recessive disorder.
A female child with a defective X-chromosome from the mother and a normal X chromosome from the father does not develop the disease. She is a disease carrier who can transmit it to her own male children. When the mother is a carrier, her sons will have a fifty percent chance of being affected, while her daughters will have the same chance of being heterozygous carriers. Mother’s male relatives may be affected. A survey has been made and it has been reported in the literature that 70% of the carriers of the abnormal gene for Duchenne muscular dystrophy (DMD) actually have some degree of muscle weakness.
There are nine kinds of muscular dystrophies. These are classified by the depending on distribution of affected muscle groups, severity and prognosis, genetic defects and the means of inheritance. Duchenne muscular dystrophy is the most common and most severe. It affects not just all the voluntary muscles but also the heart and respiratory muscles as well shortening one’s life span drastically. This type of dystrophy is due to genetic mutations on the twenty third chromosome, the sex determining chromosome, being an X linked recessive or sex-linked recessive trait. Therefore the males are affected from the gene passed down by their mothers. Mother was only a carrier.
Other types of dystrophies are those inherited through X-linked recessive trait are Becker and Emery-Dreifuss muscular dystrophy. Becker muscular dystrophy is a different mutuated gene. It is located on the same gene locus as Duchenne. This type of dystrophy affects the heart tissue in general. It is less severe. It has a longer life expectancy. Autosomal dominate trait may inherit in some different types of dystrophies. These are facioscapulohumeral, distal and occulopharyngeal muscular dystrophies as well as myotonic dystrophy. Facioscapulohumeral muscular dystrophy is caused by a missing piece of DNA on chromosome 4. This type of dystrophy affects the face, shoulders and upper arms but later affects certain muscles of the legs, the abdominal and pelvic girdle leading to extreme lordosis which may require a wheel chair. This type of dystrophy rarely affects the heart and respiratory muscles. It cannot short the span of life. The most of this type begins at the age of 20 years. It spreads very slowly due to rapid bursts of muscle deterioration.
Duchenne muscular dystrophy is the most common neuromuscular disease. A case of DMD was first reported in 1836 in Italy. It was recognized and described as an inherited disorder of boys by the English physician Meryan, E. [4] in 1852. The condition was in appropriately named after “Duchenne”. Duchenne, G.B.A. [5] was a senior neurologist practicing in Paris. Duchenne was disparaged Meryan’s work and first wrote about the condition in 1868. Duchenne at first confused it with cerebral palsy, poliomyelitis, and other conditions.
Although muscle strength may be slightly decreased from birth, infants with DMD are thought to have no physical problems. It has been reported in the literature that the children with DMD begin to walk at the age of one and half years. Thirty percent children can walk independently before fifteen months of age. Some children can begin to walk only when they are three years old. There is no correlation found between the late walking and rapid progression or severity.
Muscle weakness is generally first recognized between ages of three and five years. It progress symmetrically and in a predictable pattern. Children tend to walk on their toes. Some children walk up on their toes from outset and are never able to walk with their feet flat. They also tend to waddle and have to climb up their legs, thighs, and hips when getting up from the floor or from the chair. The ability to run, which is generally achieved by two years of age, is not attained.
If the children of DMD are not properly treated they develop an imbalance in muscle strength at every joint. Hip flexors remain stronger than hip extensors. Knee flexors remain stronger than knee extensors. The ankle plantar flexors remain stronger than ankle dorsiflexors. The imbalance in strength at any joint leads to contracture of the joints muscle and soft tissues.
The stronger muscles are shorten. The muscles and their surrounding connective tissues become tight. The joint’s range of motion is decreased. The stronger muscles on one side of the joint stretch the weaker muscles on the other side of the joint. This not only causes contracture of the stronger muscles but also makes the weaker muscles get weaker even faster, because they are stretched and no longer at their ideal length for contracting. Severe muscle weakness can prevent ambulation. It has been seen that the children with DMD and other neuromuscular diseases lose the ability to walk prematurely because of joint contractures.
Moser, H. [6] has studied Duchenne muscular dystrophy: Pathogenic aspects and genetic prevention. The author has put forward his views before the medical science as the patients are an wheel chair bound at the age of eight to ten years. They die before the age of twenty years generally. The mutation rate is in the order of 7×10–5. This rate is higher than for any other X-linked genetic disease. There is no structurally or functionally abnormal protein known that might represent the primary gene product. It has no any pathogenic mechanism leading to the observed biochemical and histochemical alterations been elucidated. The functional defect of the muscular plasma membrane is still most attractive. It would be able to explain both the excess of muscular constituents found in serum of patients and carriers such as creating kinase (CK), as well as the excessive calcium uptake by dystrophic muscle fibres, which, prior to necrosis, could lead to hyper contractions rupture of myofilaments in adjacent sarcomas and by excessive calcium uptake to mitochondrial damage causing crucial energy loss.
Eemery, A.E. [7] and Abood, E.A. et al. [8] have studied and given their thought as the incidence of DMD is approximately 1 in every 3000.
The first symptoms of DMD are characterized by frequent falling, difficulty of getting up from a standing or lying position and a waddling gait. The calf musculature is significantly enlarged by hypertrophy, fat, infiltration, and accumulation of connective tissue. It has been seen that twenty percent patients of DMD has a mental impairment. Affected skeletal musculature is mainly proximal, and results in wheel chair dependence during the early teenage. Patients die with cardiae failure as an adolescent. Simands, A.K. et al. [9] have studied DMD and found that infections, leading to pneumonia as a result of lack of ventilation are the cause of death. The care of respiration may increase the survivals. Somer, H., et al. [10] have studied DMD and found that a major characteristic of DMD and other muscular dystrophies is displayed of muscle necrosis, which results in high serum levels of the isoenzyme creatine kinase (CK). The leaks of CK from affected degeneration myofibers to the blood stream in addition to other enzymes such as perovative Kinase.
Mokri, B., et al. [11] have found that plasma membrane defects are early and basic pathologic alterations and are represented by lesions of various sizes. He also found that there are some ultra structural abnormalities of the membrane. Engel, J., et al. [12] have studied regions near small lesions contain dilated endocytotic vesicles, whereas large lesions harbor dilated SR Vesicles, irregularly positioned sarcotubular components depending mitochondria, and small clusters of glycogen.
Gillis, T.N. [13] has studied and found that extracellular calcium can enter the myobiber and cause as imbalance of the calcium homeostasis, which results in myofiber degeneration. The imbalance of calcium homeostasis might be that degeneration preludes the extracellular calcium influx.
The detection of apoptotic nuclei demonstrating DNA fragmentation in muscle biopsies from DMD patients lead to another hypothesis. The absence of the DGC leads to sarcolemmal rupture and subsequent leaking of intracellular proteins, which are unknown to the immune system.
Spencer, M.J., et al. [17] have studied DMD and found that the leaking of intracellular proteins attracts and activates cytotoxic lymphocytes and helper T-cells, which initiate the apoptotic program in ruptures myofibers.
Bradley, W.G., et al. [18] have studied dystrophin deficiency and found that this deficiency leads to irreversible myofiber cell death or necrosis. The first necrotic myofiber can be demonstrated in the neonatal period. The myobiber at this stage is single. The necrotic myobibers appear in groups of 2 - 15 myofibers with the increase of age of the patient.
Bowman, W. [19] has studied muscular dystrophy and found that myofiber necrosis is segmental, which means that not the entire myofiber is affected by necrosis. The basal lomina surrounding the necrotic myofiber is not affected. It remains empty shell. The affected myofibers contain membrane attack complexes, which are the result of complement activation. The insertion of this in the sarcolemma creates holes and causes cell-lysis.
The interests of brevity and accuracy are both properly served by a simple statement that we do not know the etiology of DMD. There has been so much discussion in the last few years challenging the traditional concept of muscular dystrophy as a primary illness of the muscle. Murphy, D.L., et al. [20] have given their views regarding the search for abnormalities in catecholamine metabolism and it was accelerated by the findings that there was a reduced initial rate of accumulation of serotonin in platelets from the DMD patients.
Paulson, O.F., et al. [21] have studied blood flow in muscle of DMD patients and could not find any abnormality. Jerusalem, F., et al. [22] have studied electron microscopic and morphometric studies of the capillaries in muscle from patients with the disease have been unrevealing.
Mc Comas, A.J., et al. [23] have suggested that the muscle fibers in Duchenne muscular dystrophy were not being lost in a random fashion but disappearing in groups associated with the loss of motor units. They also proposed that DMD might have its origin in the malign influence of a sick motor neuron upon the muscle fibers.
Panayiotopoulos, C.P., et al. [24] and Ballantyne, J.P., et al. [25] have studied DMD and could not find any abnormality in the muscle fibers. The disease may be associated with an abnormal neural influence.
Matheson, D.W., et al. [26] have studied the changes in the behaviour of different cell membranes and critically described the DMD and concluded that an abnormal surface deformation of erythrocytes produced by washing the red blood cells of DMD patients in saline.
Miale, T.D., et al. [27] and Miller, S.E., et al. [28] have studied DMD patients and could not attain the results of Matheson, D.W., et al. [26] and proposed that it may be a non-specific outcome of Matheson, D.W., et al. [26] work.
The behaviour of some of the enzymes, which are associated with sarcolemmal or erythrocyte membranes are anomalous.
Brown, H.D., et al. [29] have studied the activity of Na, K, ATPase of erythrocyte membranes in DMD found that this is stimulated by ouabain, rather than being inhibited as in the normal situation.
Peter, J.B., et al. [30] have shown that a factor present in the serum of DMD patients may contribute to the alteration given by Brown, H.D., et al. [29] . Roses, A.D., et al. [31] have studied endogenoces phospnorylation of a group of membrane proteins. This protein activity is called as protein Kinase activity and it is found abnormal high in erythrocyte membranes.
Mawatari, S., et al. [32] have studied adenyl cyclase which is an enzyme and it is found to be abnormal. It is associated with sarcolemmal membranes. The enzyme is stimulated by epinephrine and sodium fluoride but in DMD patients this stimulation is less than normal .Most of the efforts in carrier detection have been directed to the detection of subclinical abnormalities. Muscle biopsy can be done. Pearce, G.W., et al. [33] and Roy, S., et al. [34] have studied muscle biopsies in DMD patients and one may see foci of necrotic fibers, moth eaten and whorled changes in intermyfibrillar network pattern. The internal nuclei are in abnormal number. These changes were also found in normal healthy persons. These patients are not carriers and the gastro enemius muscle is biopsied. Their changes are found in patients who are at the risk. This is the strong presumptive evidence of the carrier state.
Ionasescu, V., et al. [35] have found an abnormality in ribosomal protein synthesis of DMD patients and found that isolated ribosomes were synthesized and are in increased amounts of collagen. Ionasescu, V., et al. [36] have also found similar situations in the carriers of DMD patients. Ribosomes were found to synthesize abnormally higher amounts of noncollagenous protein as well as collagen. Roses, A.D., et al. [37] have studied and concluded that the increased ability of the erythrocyte membrane to phosphorylate protein is found in carriers of the illness. The mean values for groups of definite, probable and possible carriers were found to be similar and different from the mean value for matched healthy controls. The mutation rate must be very low. If there were non-carrier mothers in the possible and probable groups, their normal values of protein Kinase should have reduced the mean value of these two groups compared to the definite carriers.
Hobbins, J.C., et al. [38] have studied an interesting concept of the detection of DMD patients. They have performed amneucontesis at the fifteen weeks of pregnancy and a determination of the sex of the fetus. If the fetus is female, the child will not suffer the clinical symptoms of DMD. If the fetus of the examination uterus is male, then mother may elect to have the pregnancy terminated. It is the question of emotional aspects of the mother and can be left to decide what should be done? Analysis of amniotic fluid CPK is not a useful test to detect whether a male fetus has the illness or not. It has become feasible to obtain fetal blood at the eighteen weeks during fetuscopy prenatal diagnosis of the illness.
Bodensteiner, J.B., et al. [39] have studied calcium in muscle of DMD patients and found that there is a tendency of calcium level to be on the higher side. Bertorini, T.E., et al. [40] have studied calcium and magnesium content in muscle of DMD patients and found that the levels of calcium were higher and levels of magnesium were lower. Some of the researchers have applied Freeze fracture technique (FFT) to study the muscle membrane and splits the surface membrane, peeling the outer surface away from the inner surface along a line, which passes through the interior of the membrane. Both the faces can be looked at under the electron microscope and these faces are found to be covered with particles of various types, some of which are arranged with a rectangular distribution. These are called square or orthogonal arrays.
Schotland, D.L., et al. [41] [42] applied FFT to the DMD patients and found that there is a significant diminution in the number of orthogonal arrays. Peluchetti, D., et al. [43] have studied this technique and suggested that there may be a decrease in the intermembranous particles on both surfaces.
The cell surface membrane is not homogenous. There are so many proteins and other compounds embedded in cell membrane, which serving different aspects of membrane activity. The glycoproteins and glycolipids of cell membranes perform important functions including a role in the receptor sites. There are some probes to identify these constituents by virtue of the selective binding of the probe to a specific compound. One such probe is Concanavalin A. This probe is a member of group of plant proteins called lections which have a specific affinity for sugar residues. Concanavalin A binds specifically with mannose, glucose, and fructose residues. Heimann-Patterson, T.D., et al. [44] have studied the pattern of Concanavalin A binding in DMD patients and found that it was patchy and irregular. It was regular and has a good pattern in normal muscle. They have also found an abnormality in the cell membrane.
Brooke, M.H. [45] has written in his book on muscular dystrophy as if DMD is a primary abnormality in the muscle, then we would suppose to the change might be reproduced by culturing muscles cells from patients with the illness. Some of the authors [46] [47] have studied DMD patients and find some abnormalities in the growth pattern which are able to change in the isoenzyme pattern of CK in the myotubes. It has also been noticed that this growth pattern is suspiciously normal in the cultured muscle. FFt does not reveal any abnormality in the inter membranous particles.
Rottman, S.M., et al. [48] and Mawatri, S., et al. [49] have found that the cultured cells seem to behave normally electro-physiologically. Cerri, C.A., et al. [50] have found that there is abnormality in adenyl cyelase. It was not the same as that found in the dystrophic muscle itself.
It is believed that the possibility of hypothetical membrane abnormality might affect other tissues then muscle alone has been raised. DMD is a genetic disease and different cell surface membranes share many similar properties. Rowland, L.P. [51] has studied the relatively in exhaustible supply of membrane in the red blood cell and this has led to a legion of studies.
Some of the authors [52] -[56] have studied the red cell membranes have been both inculpated and exculpated of all of the following such as changes in size, shape, deformability and other physical properties, abnormalities in different ATPases, changes in Calcium transport, abnormalities of electrolyte content and permeability, abnormal phospnorylation of protein, abnormalities of adenyl cyelase.
Arthur, H., et al. [57] have found that there is an abnormality in low density lipoproteins in the plasma in DMD children leading to a suggestion that there transport capacity was impaired. Fibroblasts cultured from the skin of patients with the illness have also been harvested in an attempt to detect abnormalities. A decreased adhesiveness between fibroblasts has been described using a technique in which the cells are encouraged to collide with each other and the stickiness is measured in such collisions. The adhesiveness has been found to be reduced in the fibroblasts from Duchenne patients. There is an overlap between the normal healthy controls and the DMD patients. Cultured skin fibroblasts also contain CK, which might make this a useful tool for studying the enzyme in disease states.
Incurable and untreatable are not synonymous and much can be done to make the life of a patient more comfortable. Sometimes the parents of dystrophic children faced with the problem of illness in their child, manifest one of two reactions. They either try to ignore the child or they build the things for his aid so that he becomes encased in mechanical contraptions.
The proper management of DMD depends on a team approach. The team may be made in ideal situation and neurologist, orthopaedic surgeon, physical and occupational therapist, social worker and the orthotist can take part.
It has been reported in literature that humoral immunity is manifested by the antibodies production. These are special chemical substances that react against foreign body. Antibodies are named as immunoglobulins, which are serum proteins. The immunoglobulins possess antibody activity.We can classify according to the antigens and stimulate their production such as IgA, JgG, IgM, IgD and IgE.
An extra substance is required for antibody to have a cytotoxic effect called complement. The presence of antigens in the body stimulates certain types of defense cells to produce antibodies. Humoral immune response depends on a group of small lymphocytes called B-lymphocytes (B-cells). B-cells originate in bone marrow and travel directly into lymphoid tissues. Cells mediated immunity is directly expressed by certain type of defense against the antigens. These cells are also lymphocytes, but with a slightly different maturation having the influence of the thymus gland. These are called T-lymphocytes in contrast to B-lymphocytes, which produce humoral immunity. T-cells also originate in bone marrow but unlike B-cells, they do not travel directly into lymphoid tissues, but first center the thymus where they undergo a conditioning process, hence the name thymus dependent lymphocytes. After leaving the thymus thus migrate to special regions known as thymus-dependent areas, namely the paracortical area of lymph.
CNS is inaccessible to the immune system in human beings. The concept of immune privilege originated from four historical findings. Ehrlich [58] reported some of the main findings regarding parenterally administration of aniline dyes, which stained almost all body tissues except CNS. Due to this magnificent observation concept of blood brain barrier (BBB) physiologically separating central nervous tissue from the systemic circulation and systemic immune response.
Murphy, J.B., et al. [59] have studied mouse sarcoma tissue, which was found not to be rejected implantation into rat brain and given their views that the CNS was exempt from the immunological processes responsible for graft rejection. Sabin, F.R. [60] has also studied the absence in brain of direct lymphatic drainage and by implication of circulating lymphocytes-depriving the CNS. Ediden, M. [61] showed that normal nervous tissue, which was not to express major histocompatibility complex (MHC) antigens and exempts the brain from participation in cell-medicated immune reactions.
Some of the authors [62] -[65] have studied and reported that immune system and CNS both cannot be separated but rather as elements of a vast commutation system in which they may exchange information both via an atomic connections and though the hormones and mediators released by hypothalamopituitary axis. Any uncontrolled or excessive immune reaction may be expected to interfere with the brain’s physiology. Any neurologic dysfunction carries the risk of disturbing the delicate equilibrium of the immune network. This is conversely also true.
Sternberg, E.M., et al. [64] have given a statement, which is helpful to provide the rationale for the therapeutic concept of immunomodulation of certain immune disorders. Reichlin, S., et al. [63] and Sternberg, E.M., et al. [64] have given their views regarding abnormalities of the immune system, which are indeed increasing number of neuropathies as well as in some psychiatric syndromes. It has been established that pharmocologic manipulations of immune pathologies such as rheumatoid arthritis, systemic lupus erythmutius etc, by means of neurohormones have been routinely performed. This is well established that the process of inflammations increases vascular permeability and allows antibody, complement and other proteins to pass out the circulation and enter the extravascular space also induce inflammatory cells, including lymphocytes, to cross the vascular endothelium and accumulate in the tissues. The total net effect is to deploy all the resources of the immune system at the site of injury. Cells antibody and complement leave the blood and go into action where the demand is high. It may be in the affected tissue outside the vessel wall. Tourtellotte, W. [66] has given a statement related to the effect is to abrogate in CNS, if only temporarily, its isolation from the immune processes of the body. The barrier, which excludes plasma proteins from the brain breaks down, allowing antibody to enter the extra vascular space. The amount of protein in CSF increases and with it the level of immunoglobulin. Immuno competent cells enter the CNS. CNS now becomes capable to generating an immune response. Antibody, which synthesized locally, adds to that leaking from vessels and increases the level of immunoglobulin. This is a complete reversal of the normal stage and the antibody levels very low. Lymphoid cells may be excluded. Inflammation has three main consequences. It allows lymphocytes and antibody forming cells to enter the CNS and immunoglobulin to be synthesized in the brain and spinal cord.
There are so many inflammatory diseases found in the CNS due to lymphocytes accumulation. They enter from the blood stream, which passed out of the capillary wall between the endothelial cells through altered tight junctions by emperopolesis. Emperopolesis is a process, which describes the behaviour of lymphocytes in tissue culture. They appear to move about inside the cytoplasm of the sebbile cells.
Oldstone, et al. [67] have reported that albumin is a small molecule and convenient as a marker, but larger protein such as fibrinogen leaks from versel as well. It may be deposited as fibrin in and around the cerebral vessel. Immunoglobulin and complement C3 also pass out of the vessel and diffuse into the brain parenchyma. This is well written that a vascular leak directly may be seen in human diseases, which changes the composition of CSF and inflammation may increase the total protein level. This level is found normally (200 - 400) mg/l and raise in much neurological diseases. CSF protein level is not a measure of the efficiency of the blood brain barrier (BBB). Many proteins may have an involvement in the process of fluctuation of their levels independently and some of them may be produced within the nervous system. Many of proteins may be released from damaged brain tissue. It was believed that only foreign proteins are considered true antigens. Ehrlich, P. [58] had introduced horror autotoxicus, which may generate antibodies against itself. Autoimmunity suggests that an appropriate immune response is directed against a normal tissue component and leads directly to disease in the absence of persisting infection. Microbes are responsible for autoimmunity. If it persists during the disease and found to be in autoimmune disease.
Miller, S.D., et al. [68] have given a statement the distinction between autoimmune response triggered by an infection and inflammation directed against a persistent microbe. T-cell reactivity against the major encephalitogenic peptides of proteolipid proteins and myelin basis protein.
1.1. Mechanism of Auto Immunity
Auto immunity may be initiated in the following ways.
1) A self-antigen may be modified and appear as foreign.
2) Ignorant clones may be educated. Microbes may cross-react with self antigens to which the immune system is ignorant. Epitope is at low concentration perhaps. A cross-reacting microbes present in heavy numbers than the original value of antigen and is able to activate and prime T-cells.
3) Removal of suppression of auto reactive processes. Microbes might cross-react with idiotope and so disrupt the anti-idiotypic network in favour of immunity. The normal regulatory mechanisms of the immune response should restore self tolerance after the initiation of autoimmunity. The maintenance of autoimmune disease must require either multiple rounds of autoimmunity to different self-antigens or a single autoimmune response that is perpetrated by defective regulation. A different form of altered immune regulation is an abnormal cytokine response.
Majority of autoimmune processes are driven by T-cellular processes. Important exceptions are the anti acetyl choline receptor antibody of myasthenia gravis and antibodies against epithelial adhesion molecules in the buleous skin disease. In the CNS, the pathogenicity of auto antibodies, such as those associated with the paraneoplastic syndrome or stiff man syndrome is not clearly well documented.
1.2. Antigen Specific Tolerance to Unknown Auto Antigens
It has been seen in many autoimmune diseases of the nervous system, driving auto antigen is not known. A treatment is required that makes no assumptions about the provoking antigen and induces antigen-specific tolerance. A short pulse of antigen nonspecific therapy set up a sequence of events leading to the perpetuation of antigen, specific tolerance. This strategy has been used to justify the use of humanized monoclonal antibody. Angello, V. [69] has given his views on complement deficiency of specific complement components, which is responsible for a couple of diseases. It is very important to remember that this is similar to the disorders, which occur with selective deficiencies of the immune system.
Fearon, D.T. [70] has given information regarding complement system and he suggested that this complement system is the most complex of the several protein activation system in blood, but this complexity can be reduced by considering the system to be comprised of their functional division: two pathways for activation the classical and alternative pathways and a common effect or sequence to which the activating pathways are directed and from which are derived many of the biological activities of complement. Molecular weight of complement C3 is 185,000 while that of C4 is 180,000. Serum concentration of complement C3 and C4 are 1500 μg/ml and 400 µg/ml respectively.
Some of the important phenomena have been studied by Valanakis, J.E. [71] occurring during activation of the complement sequence and these are related to 1) Acute inflammation 2) Cell killing namely, which has the following procedure a) increased vascular permeability;
b) chemo taxis of leukocytes;
c) enhanced phagocytosis;
d) membrane damage.
The limited proteolysis reactions, which characterize the complement system, are essentially not reversible. These reactions lead to cleaved proteins, which are recognized by the body as altered or foreign. These proteins are rapidly cleared from circulation. Despite compensatory increase in synthesis the result is usually a decrease in plasma levels. Thus, an ongoing immunologic event, which is activating the complement system in vivo, is likely to generate a decrease in plasma levels of these proteins. Conversely, the abatement of the complement activating stimulus may be paralleled by a reform of the complement levels toward normal has been reported by Stein, J.H. [72] .
The main mediator mechanism of humeral immunity is the complement system. It is an important and essential mechanism for the destruction both of foreign organisms and immuncomplexes in the presence or absence of specific antibodies. It is a double edged sword and may also destroy host tissue. Fishman, R.A. [73] reported that CNS is immunologically unusual tissue do to the presence of BBB. It may exclude many components of the immune system like antibodies and complement system. These studies imply a suppressive environment for immune reactivity in the CNS. Monocytes or macro phases are the important and major extra hepatic sources of complement components. Microglia or astrocytes have been suspected to be the cells which produce complement protein in brain such as C3.
Milica, T.C., et al. [74] have been studied the role of complement system. This system does not support in chronic and neurodegenerative disorders. There are two separate complement pathways active by distinctly different classes of activators, specific identification of complement factors in CSF promises to be diagnostic not only of complement activation but also of the mode of activation.
Isenberg, D.A. [75] has studied muscle immunoglobulin deposition in skeletal muscle in primary muscle disease. He added here that immunoglobulins and complements deposition in skeletal muscle was an abnormal finding. On the basis of these depositions the direct immunofluoresence detection may be used as a diagnostic tool. Spuler, S., et al. [76] studied unexpected sarcolemmal complement membrane attack complex deposits on non necrotic muscle fibers in muscular dystrophies. It has been reported that antibody—dependent complement —mediated muscle fiber injury as a hypothetical immune effector response in inflammatory muscle disease. Moreover, a sarcolemental alteration in muscular dystrophies might trigger anti body—independent activation of the alternative component pathway. Their findings do not support a role for antibody—dependent component— mediated muscle fiber injury in the major inflammatory muscle diseases. Fengel, A.G., et al. [77] have studied component activation in muscle fiber necrosis and demonstrated the membrane attack complex of component in necrotic fibers. They have pointed out that none of the non-necrotic fibers reacted for immunoglobulin or component. On the basis of detection of membrane attack complex neo antigens in necrotic fibers in a wide variety of muscle disease shows that the lytic component pathway is consistently activated and participates in muscles fiber necrosis in vivo. They have also given that the complement reaction products are generated that can stimulate cellular infiltration and phagocytosis of the necrotic fiber. Their findings also suggest that cell necrosis in general may involve participation of complement.
Irena Niebroj, D., et al. [78] have studied immunoblot analysis of sarcoplasmic calcium binding proteins in Duchenne muscular dystrophy and found that the decrease in the proteins may contribute to the elevation of free intracellular 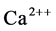 level in the sarcoplasm of dystrophic muscle. They add here that due to the regulation process of the proteins abnormalities will occur. Minetti, C., et al. [79] have given immunologic study of vinculin in Duchenne muscular dystrophy. They have reported that DMD patients show patchy and low-intensity immunostain at the sarcolemma of most fibers. Vinculin content with low intensity shows that dystrophin is absent in muscle.
level in the sarcoplasm of dystrophic muscle. They add here that due to the regulation process of the proteins abnormalities will occur. Minetti, C., et al. [79] have given immunologic study of vinculin in Duchenne muscular dystrophy. They have reported that DMD patients show patchy and low-intensity immunostain at the sarcolemma of most fibers. Vinculin content with low intensity shows that dystrophin is absent in muscle.
Wochner, R.D., et al. [80] have studied accelerated break down of immunogloblulinG (IgG) in myotonic dystrophy. The plasma proteins that possess immunologic activity have been subject of intensive study nowadays. These proteins as a group are referred to as immunoglobulins and can be divided into at least three major classes. These are IgG (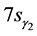 -globulins), IgA (
-globulins), IgA (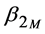 -globulins), and IgM (
-globulins), and IgM (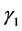 -macroglobulins or
-macroglobulins or 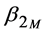 -globulins).
-globulins).
Each of these fractions possess antibody activity and certain structural features in common, but can be distinguished from the others by physicochemical and immunochemical technique. They have found that the concentrations of IgG were significantly reduced with a mean of (7.2 ± 2.6) mg per mole compared to (12.1 ± 2.6) mg per mole in controls. Normal serum concentrations and rates of catabolism were also observed for albumin, IgM and IgA. Myotonic dystrophic patients have an unique immunoglobulin abnormality—an isolated hypercatabolism of IgG.
Larsen, B., et al. [81] have studied immunoglobulin concentration and Gm allotypes in myotonic dystrophy and find that there was not any proof of sub class of IgG, which is being hypercatabolised in patients.
Sanders, B.G., et al. [82] have studied serum IgG levels in the storrs strain of hereditary muscular dystrophic chickens. They have reported that the reduction in IgG levels in the storrs strain of muscular dystrophic chickens are due to strain difference. The authors also found that the mode of inheritance of serum IgG levels in the stross strain of muscular dystrophic chickens was polygenic.
2. Materials and Method
2.1. Selection and Exclusion of Patients
We have selected Duchenne muscular dystrophy diseased patients and normal healthy controls whose aged group has a range of 10 to 18 years. The patients were on standard medicines. We did not select above this age group. Blood samples of these patients were collected from the Department of Neurology, Safdarjang Hospital, New Delhi 110 016 after the approval of ethical committee of the hospital. 10 milliliters freshly drawn blood from each patient was collected in clean and dry test tube without any anti-coagulant. The test tube was kept for 45 minutes at room temperature (22˚C ± 2˚C) for the formation of clot. Sera of different patients and normal healthy controls were separated by centrifugation at 1500 r.p.m. up to 15 minutes and were collected in screw capped test tubes.
The immunological parameters (IgA, IgG, IgM, C3 & C4) were quantitied by using singles radial immunodiffusion method of Mancini, G., et al. [83] using commercially available antibody-agar plates. The plates were standardized with purified immunoglobulins. The purpose is to quantitate serum levels of immunoglobulins (IgG, IgA, IgM) and complements (C3 and C4). These measurements aid in the clinical diagnosis, assessment of disease activity, response to treatment, and follow-up in patients with various clinical conditions. Measurements of immunoglobulin A (IgA) and immunoglobulin M (IgM) aids in the diagnosis of abnormal protein metabolism and the body’s lack of ability to resist infectious agents. Measurements of IgG aids in the diagnosis of autoimmune diseases, sarcoidosis, chronic liver disease, chronic and recurrent infections, lymphoid malignancies, multiple myeloma and severe and variable immunodeficiency. The complements system is the main mediator mechanism of humoral immunity. To study the levels of complements C3 and C4 is also our obient regarding the complete knowledge of immune system in Duchenne muscular dystrophy
2.2. Statistical Analysis
We have used a regression analysis regarding the validity of our data. A multiple correlation coefficients analysis has also been applied to the study. Regression analysis with different equations has also applied to calculate multiple correlation coefficients.
(a) Regression analysis is used to find equations that fit data. Once we have the equation, we can use the statistical model to make predictions. One type of regression analysis is linear analysis. When a correlation coefficient shows that data is likely to be able to predict future outcomes and a scatter graph of the data appears to form a straight line, statisticians may use linear regression to find a predictive function. The equation for a line is
y = mx + b.
We can take data to calculate linear regression, and we can find the regression equation as

Simple linear regression is a way to describe a relationship between two variables through an equation of a straight line, called line of best fit, that most closely models to this relationship. Linear regression formula can be used to derive the equation for the line of best fit:
y = a + bx
where
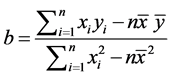
and
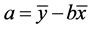
(b) The Simple multiple correlation coefficient (R) is a measure of the strength of the association between independent (explanatory) variables and the one dependent (prediction) variable. Interpretation of R can be understood with the strength of the association. The strength of the association is measured by the sample multiple correlation coefficient; R. The correlation coefficient R may any value from 0 to +1.
• The closer R is to one, the stronger the linear association is.
• If R equals to zero, then there is no linear association between the dependent variable and the independent variables. Unlike the simple correlation coefficient, r, which tells both the strength and direction of the association, R tells only the strength of the association. R cannot be a negative value. This can be seen from the formula below, since the square root of this value indicates it has positive root. Calculation of R can be done for two independent variables, X1 and X2 as
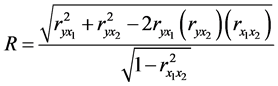
3. Results
The measured values of immunological parameters in the present work along with other earlier studies by different researchers are given in Table 1.
The mean and standard deviations in DMD and controls are given in Table 2.
Statistical analysis like regression analysis and multiple correlations were also tabulated in Table 3, Table 4.

Table 1. Experimental findings along with earlier work carried out by researchers.
Continued
Continued
Continued
Continued
Continued
Table 2. Mean levels and standard deviation of C3, C4, IgG, IgM, IgA in DMD patient and normal healthy control.
Table 3. Regression and correlation coefficient studies on C3, C4, IgG, IgM and IgA in normal blood samples.
Table 4. Regression and correlation coefficient studies on C3, C4, IgG, IgM and IgA in DMD blood samples.
4. Discussion
Central nervous system (CNS) is relatively isolated from systemic immune response in the absence of disease. There is no mechanism found for antibody production within the normal condition of CNS. This system has been described as an immunologically privileged site due to paucity of normal immune surveillance. If a virus penetrates the blood brain barrier (BBB) that excludes most infectious agents, the same barrier may stop viral clearance. Immune responses in the CNS during infection are recruited from the systemic circulation in a relatively selective and specific fashion. Cells and antibodies found in the CNS during infections differ from these that follow non specific rupture in the BBB. During the stage of viral infection, we have an early increase in permeability of vessels, which allow transudation of serum proteins, cell entry is immunologically specific. The cells, which are entering have a specific kinetics and do not simply mirror the proportions of cell phenotypes in the blood. These cells in turn are caused to replicate cells, which may persist for long periods of time within CNS. On the basis of the above mentioned facts a review immune response in the CNS is quit necessary and required. Some of the researchers have proposed the hypothesis of immunological mechanism for the involvement of pathogenesis in muscular dystrophy. Many of the patients of DMD have immune deficient state in the present work.
It is believed that the changes in the levels of immunological parameters in the present study and after the review of the literature are due to manifestation of the factors, which are responsible for this disease. Immunity is related with the food, which we eat. It has already been established that content of the food have some trace elements. The trace elements play a role in human immunity. If the level of these elements goes beyond the limit of normalcy even death may occur. On the other hand if the levels are lower side of the normal range something unnatural can happen. A relation between immunoglobulin complement and trace element is considered in the future preview of the study.
Our findings show that the estimated values of all the immunological parameters such as of C3, C4, IgG, IgM and IgA go down in comparison to healthy subjects. The value of complement C3 go down about 44.3% from the normal values. The value of complement C4 go down to 78.57% from the normal values. The serum IgG, IgM and IgA levels were also go down about 65%, 84% and 99.56% respectively in comparison to healthy subjects. These estimations were observed in all the valuable parameters of immunology. We were very much surprised to note that the decrease in IgG is very high.
Regression and multiple correlation analysis give a good presentation of our work. It has been found that the partial correlation coefficient rAG.M, which has value 0.5699 in normal patients and 0.4528 in DMD patients. We have seen that both the correlations have different values. The value of this correlation is higher in normal persons. It means that the correlation specifically tells that the values in DMD are going in the decreasing directions. The correlation rMA.G in controls is 0.4230, While in DMD patients is 0.7416. This correlation is very high in comparison to healthy persons. A typical correlation, which is negative correlation rGM.A in normal cases is found −0.256 and −0.2252 in DMD cases. Both these correlations are negatively related with each other. We may state here that during negative correlation the value of rGM.A in normal cases is higher than DMD cases. It means that a great impact in DMD cases has been properly seen. A trend between all the immunoglobulins has been set up and it is rAG.M > rMA.G. > rGM.A
while we have a trend in DMD cases is rMA.G. > rAG.M > rGM.A
Multiple correlation between all the immunoglobulins is also a major part of analysis during the study on statistical bases. It has been found that the multiple correlation RG.MA in normal has value 0.2462, while in DMD cases it goes down up to 0.2364. The value of RA.GM is found in normal healthy persons as 0.3476 and in DMD cases it has a value 1.0283, and it is very absurd and not correlated with each other because correlation cannot take the value more than one. It can go either negative side or positive side of 1 but never above the value of 1. The third multiple correlation RM.AG has value 0.1939 for normal cases and 0.5544 in DMD cases. This value of correlation is very high in comparison to healthy person. On the basis of present findings we have seen that a correlation in normal cases is in the form of a new trend, which is given below RA.GM > RG.MA > RM.AG while in DMD it has RA.GM > RM.AG > RG.MA
We are in a position to say that our data has relevance of high authenticity and reliability to accept there is a deficit in immunity in DMD cases. The deficit in immunity may be a cause to damage muscle for abnormal functioning. Immunity is related to trace elemental intake in the human system. There are so many causative factors to down the immune system of the body. We are not going to discuss here because our main focus is on the immune complex system of DMD people by giving suitable supplementation so that immunity may not affect the span of life of DMD people.
Regression coefficients and regression lines with equations are basic foundations of measurements of correlations and show a specific character of variations.
5. Conclusion
The present work is a step to go further investigations to establish the balance of immunity in DMD disease. Our work is based on the factual position in the DMD cases. Multiple correlations and regression coefficients show a proper relation between all these parameters of immunology. Statistically verified data reveal the factual position of the stage of disease. We have to maintain the immune imbalance in DMD. The trends found in the study are very sensitive and we must take into account to go further establishment in the direction of pharmacology to investigate a new medicine to give a long life to the patients of this disorder. The effect of trace elements may be included in the investigations related to new medicines.
6. Acknowledgements
The authors are thankful to Dr. P. K. Saxena, Principal, D.A.V. College, Muzaffarnagar for providing the facility of doing work. We are also thankful to Medical Superintendent, Safdarjang Hospital, New Delhi, for arranging the blood samples of the diseased and healthy controls. Our thanks go to Dr. Manju Chauhan, Head, Department of Biosciences, D.A.V. College, Muzaffarnagar for providing the facilities of estimation of the immunological parameters from sera of different blood samples of the human disorders and controls.
References
- Petrof, B., Shrager, J., Stedman, H., Kelley, A. and Sweeney, H. (1993) Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle Contraction. Proceedings of the National Academy of Sciences of the United States of America, 90, 3710-3714. http://www.pnas.org/content/90/8/3710.full.pdf
- Emery, A.E. (2002) The Muscular Dystrophies. The Lancet, 359, 687-695. http://dx.doi.org/10.1016/S0140-6736(02)07815-7
- Bach, R.J. (1999) Introduction In Guide to the Evaluation and Management of Neuro Muscular Disease. Hanley & Belfus Inc., Philadelphia, 1-4.
- Meryon, E. (1852) On Grannular and Fatty Degeneration of the Voluntary Muscles. Medico-Chirurgical Transactions, 35, 72-84.
- Duchenne, G.B.A. (1868) Researches Sur la paralysic musculaire pseudo hypertrophic or paralysie myo-sclero sique. Archives of General Psychiatry, 11, 5-25.
- Moser, H. (1984) Duchenne Muscular Dystrophy: Pathogenetic Aspects and Genetic Prevention. Human Genetics, 66, 17-40. http://dx.doi.org/10.1007/BF00275183
- Emery, A.E. (1991) Population Frequencies of Inherited Neuromuscular Disease—A World Survey. Neuromuscular Disorders, 1, 19-29. http://dx.doi.org/10.1016/0960-8966(91)90039-U
- Abood, E.A. and Jones, M.M. (1991) Macrophages in Developing Mammalian Skeletal Muscle: Evidence for Muscle Fiber Death as a Normal Developmental Event. Acta Anatomica, 140, 201-212. http://dx.doi.org/10.1159/000147059
- Simonds, A.K., Muntani, F., Heather, S. and Fielding, S. (1998) Impact of Nasal Ventilation on Survival in Hypercapnic Duchenne Muscular Dystrophy. Therax, 53, 949-952. http://dx.doi.org/10.1136/thx.53.11.949
- Somer, H., Donner, M., Murros, J. and Konttinen, A. (1973) A Serum Isoenzyme Study in Muscular Dystrophy. Particular Reference to Creatine Kinase, Aspartate Aminotransferase, and Lactic Acid Dehydrogenase Isozymes. Archives of Neurology, 29, 343-345.
- Mokri, B. and Engel, A.G. (1975) Duchenne Dystrophy: Electron Microscopic Findings Pointing to a Basic or Early Abnormality in the Plasma Membrane of Muscle Fiber. Neurology, 25, 1111-1120. http://dx.doi.org/10.1212/WNL.25.12.1111
- Engel, J., Furthmayr, H., Odermatt, E., von der Mark, H., Aumailley, M., Fleischmajer, R. and Timpl, R. (1985) Structure and Macromolecular Organization of Type VI Collagen. Annals of the New York Academy of Sciences, 460, 25- 37.
- Gillis, J.M. (1999) Understanding Dystrophinopathies: An Inventory of the Structural and Functional Consequences of the Absence of Dystrophin in Muscles of the Mdx Mouse. Journal of Muscle Research & Cell Motility, 20, 605-625. http://dx.doi.org/10.1023/A:1005545325254
- Sandri, M., Minetti, C., Pedemonte, M. and Carraro, U. (1998) Apoptotic Myonuclei in Human Duchenne Muscular Dystrophy. Laboratory Investigation, 78, 1005-1016.
- Tidball, J.G., Albrecht, D.E., Lokensgard, B.E. and Spencer, M.J. (1995) Apptasis Precedes Necrosis of DystrophinDeficient Muscle. Journal of Cell Science, 108, 2197-2204.
- Matsuda, R., Nishikawa, A. and Tanaka, H. (1995) Visualization of Dystrophic Muscle Fibers in Mdx Mouse by Vital Staining with Evans Blue: Evidence of Apoptosis in Dystrophin-Deficient Mouscle. The Journal of Biochemistry, 118, 959-963. http://dx.doi.org/10.1093/jb/118.5.959
- Spencer, M.J., Walsh, C.M., Dorshkind, K.A., Rodriguez, E.M. and Tidball, J.G. (1997) Myonuclear Apoptosis in Dystrophic Mdx Muscles Occurs by Perforin-Mediated Cytotoxicity. Journal of Clinical Investigation, 99, 2745-2751. http://dx.doi.org/10.1172/JCI119464
- Bradley, W.G., Hudgson, P., Larson, P.F., Papapetropoulas, T.A. and Jenkison, M. (1972) Structural Changes in the Early Stages of Duchenne Muscular Dystrophy. Journal of Neurology, Neurosurgery & Psychiatry, 35, 451-455. http://dx.doi.org/10.1136/jnnp.35.4.451
- Bowman, W. (1840) On the Minute Structure and Movements of Voluntary Muscle. The Philosophical Transactions of the Royal Society, 130, 457-501.
- Murphy, D.L., Mendell, J.R. and Engel, W.K. (1973) Serotonin and Platelet Function in Duchenne’s Muscular Dystrophy. Archives of Neurology, 28, 239-242. http://dx.doi.org/10.1001/archneur.1973.00490220047006
- Paulsan, O.F., Engel, A.G. and Gomez, M.R. (1974) Muscle Blood Flow in Duchenne Type Muscular Dystrophy, Limb-Gridle Dystrophy, Polymyositis and in Normal Controls. Journal of Neurology, Neurosurgery & Psychiatry, 37, 685- 690. http://dx.doi.org/10.1136/jnnp.37.6.685
- Jerusalem, F., Engel, A.G. and Gomez, M.R. (1974) Duchenne Dystrophy. I. Morphometric Study of the Muscle Microvascular. Brain, 97, 115-122. http://dx.doi.org/10.1093/brain/97.1.115
- McComas, A.J., Sica, R.E.P. and Currie, S. (1971) An Electrophysiological Study of Duchenne Dystrophy. Journal of Neurology, Neurosurgery & Psychiatry, 34, 461-468. http://dx.doi.org/10.1136/jnnp.34.4.461
- Panayiotopoulos, C.P., Scarpalezos, S. and Papapetropoulos, T. (1974) Electrophysiological Estimation of Motor Units in Duchenne Muscular Dystrophy. Journal of the Neurological Sciences, 23, 89-98. http://dx.doi.org/10.1016/0022-510X(74)90145-2
- Ballantyne, J.P. and Hansen, S. (1974) New Method for Estimation of the Number of Motor Units in a Muscle. 2. Duchenne, Limb-Girdle and Facioscapulohumerol, and Myotonic Muscular Dystrophies. Journal of Neurology, Neurosurgery & Psychiatry, 37, 1195-1201. http://dx.doi.org/10.1136/jnnp.37.11.1195
- Matheson, D.M. and Howland, J.L. (1974) Erythrocyte Deformation in Human Muscular Dystrophy. Science, 184, 165-166. http://dx.doi.org/10.1126/science.184.4133.165
- Miale, T.D., Frias, J.L. and Lawson, D.L. (1975) Erythrocytes in Human Muscular Dystrophy. Science, 187, 453-454. http://dx.doi.org/10.1126/science.1111115
- Miller, S.E., Roses, A.D. and Appel, S.H. (1975) Letter: Erythrocytes in Human Muscular Dystrophy. Science, 188, 1131. http://dx.doi.org/10.1126/science.1215993
- Brown, H.D., Chattopadyay, S.K. and Patel, A.B. (1967) Erythrocyte Abnormality in Human Myopathy. Science, 157, 1577-1578. http://dx.doi.org/10.1126/science.157.3796.1577
- Peter, J.B., Worsfold, M. and Pearson, C.M. (1969) Erythrocyte Ghost Adenosine Triphosphate (AT Pase) in Duchenne Dystrophy. Journal of Laboratory and Clinical Medicine, 74, 103-108.
- Roses, A.D., Herbstreith, M.H. and Appel, S.H. (1975) Membrane Protein Kinease Alteration in Duchenne Muscular Dystrophy. Nature, 254, 350-351. http://dx.doi.org/10.1038/254350a0
- Mawatari, S., Takagi, A. and Rowland, L.P. (1974) Adenyl Cyclase in Normal and Pathologic Human Muscle. Archives of Neurology, 30, 96-102.
- Pearce, G.W., Pearce, J.M.S. and Walton, J.N. (1966) The Duchenne Type Muscular Dystrophy: Histopathological Studies of the Carrier State. Brain, 89, 109-120. http://dx.doi.org/10.1001/archneur.1974.00490310098016
- Roy, S. and Dubowitz, V. (1970) Carrier Detection in Duchenne Muscular Dystrophy: A Comparative Study of Electron Microscopy, Light Microscopy and Serum Enzymes. Journal of the Neurological Sciences, 11, 65-79. http://dx.doi.org/10.1016/0022-510X(70)90041-9
- Ionasescu, V., Zellweger, H. and Conway, T.W. (1971) Ribosomal Protein Synthesis in Ducheme Muscular Dystrophy. Archives of Biochemistry and Biophysics, 144, 51-58. http://dx.doi.org/10.1016/0003-9861(71)90453-X
- Ionasescu, V., Zellweger, H., Shirk, P. and Conway, T.W. (1973) Identification of Carriers of Duchenne Muscular Dystrophy by Muscle Protein Synthesis. Neurology, 23, 497-501. http://dx.doi.org/10.1212/WNL.23.5.497
- Roses, A.D., Roses, M.J., Miller, S.E., Hull, K.L. and Appel, S.H. (1976) Carrier Detection in Duchenne Muscular Dystrophy. New England Journal of Medicine, 294, 193-198. http://dx.doi.org/10.1056/NEJM197601222940404
- Hobbins, J.C. and Mahoney, M.J. (1974) In Utero Diagnosis of Hemoglobinopathies—Technique for Obtaining Fetal Blood. New England Journal of Medicine, 290, 1065-1067. http://dx.doi.org/10.1056/NEJM197405092901908
- Bodensteiner, J.B. and Engel, A.G. (1978) Intracellular Calcium Accumulation in Duchenne Dystrophy and Other Myopathies. A Study of 56, 7,000 Muscle Fibers in 114 Biopsies. Neurology, 28, 439-446. http://dx.doi.org/10.1212/WNL.28.5.439
- Bertorini, T.E., Bhattacharya, S.K., Palmieri, G.M.A., Chesney, C.M., Pifer, D. and Baker, B. (1982) Muscle Calcium and Magnesium Content in Duchenne Muscular Dystrophy. Neurology, 32, 1088-1092. http://dx.doi.org/10.1212/WNL.32.10.1088
- Schotland, D.L., Bonilla, E. and Wakayama, Y. (1980) Application of the Freeze Fracture Technique to the Study of Human Neuromuscular Diseases. Muscle & Nerve, 3, 21-27. http://dx.doi.org/10.1002/mus.880030104
- Schotland, D.L., Bonilla, E. and Van Meter, M. (1977) Duchenne Dystrophy: Alteration in Plasma Membrane Structure. Science, 196, 1005-1007. http://dx.doi.org/10.1126/science.860127
- Peluchretti, D., Mora, M., Protti, A. and Cornelio, F. (1985) Freeze-Fracture Analysis of the Muscle Fiber Plasma Membrane in Duchenne Dystrophy. Neurology, 35, 928-930. http://dx.doi.org/10.1212/WNL.35.6.928
- Heimann-Patterson, T.D., Bonilla, E. and Schotland, D.L. (1982) Cancanavalin A Binding of the Cell Surfac of Duchenne Muscle in Vitro. Annals of Neurology, 12, 305-307. http://dx.doi.org/10.1002/ana.410120317
- Brooke, M.H. (1986) Muscular Dystrophies. In: McSherry, N.C. and Victoria, M.V., Eds., A Clinician’s View of Neuromuscular Disease, Williams & Wilkins, Baltimore, London, Los Angles, Sytdney, 117-154.
- Ionasescu, R., Kaeding, L., Feld, R., Write, D., Cancilla, P., Kaeding, L. and Stern, L.Z. (1991) Alterations in Creatine Kinase in Fresh Muscle and Cell Cultures in Duchenne Dystrophy. Annals of Neurology, 9, 394-399. http://dx.doi.org/10.1002/ana.410090413
- Thompson, E.J., Yasin, R., Van Beers, G., Nurse, K. and Al-Ani, S. (1977) Myogenic Defect in Human Muscular Dystrophy. Nature, 268, 241-243. http://dx.doi.org/10.1038/268241a0
- Rothman, S.M. and Bischoff, R. (1983) Electrophysiology of Duchenne Dystrophy Myotubes in Tissue Culture. Annals of Neurology, 13, 176-179. http://dx.doi.org/10.1002/ana.410130212
- Mawatari, S., Miranda, A. and Rowland, L.P. (1976) Adenyl Cyelase Abnormality in Duchenne Muscular Dystrophy: Muscle Cells in Culture. Neurology, 26, 1021-1026. http://dx.doi.org/10.1212/WNL.26.11.1021
- Cerri, C.A., Willner, J.H. and Miranda, A.F. (1982) Adenylate Cyclase in Duchenne Fibroblasts. Journal of the Neurological Sciences, 53, 181-185. http://dx.doi.org/10.1016/0022-510X(82)90004-1
- Rowland, L.P. (1980) Biochemistry of Muscle Membranes in Duchenne Dystrophy. Muscle & Nerve, 3, 3-20. http://dx.doi.org/10.1002/mus.880030103
- Falk, R.S., Campion, D., Guthrie, B., Sparkes, R.S. and Fox, C.F. (1979) Phospharylation of the Red-Cell Membrane Proteins in Duchenne Muscular Dystrophy. New England Journal of Medicine, 300, 258-259. http://dx.doi.org/10.1056/NEJM197902013000514
- Fischer, S., Tortolero, M., Piau, J.P., Delaunay, J. and Schapira, G. (1978) Protein Kinase and Adanylate Kinase of Erythrocyte Membrane from Patients with Duchenne Dystrophy. Clinica Chimica Acta, 88, 437-440. http://dx.doi.org/10.1016/0009-8981(78)90278-4
- Roses, A.D., Mabry, M.E., Herbstreith, M.H., Shile, E.V. and Balakrishnan, O.V. (1982) Increased [32P]-Phospharylation of Spectrin Peptides in Duchenne Muscular Dystrophy. In: Schotland, D.L., Ed., Disorders of the Motor Unit, John Wiley & Sons, New York, 413-420.
- Mabry, M.E. and Roses, A.D. (1981) Increased 32P-Phoshorylase of Triptic Peptides of Erythrocytes Spectrin in Duchenne Muscular Dystrophy. Muscle & Nerve, 4, 489-493. http://dx.doi.org/10.1002/mus.880040605
- Roses, A.D., Shile, P.E., Herbstreith, M.H. and Balakrishnan, C.V. (1981) Identification of Abnormality [32P]-Phoshorylated Cyanogen Bromide Cleavage Product of Erythrocyte Membrane Spectrin in Duchenne Muscular Dystrophy. Neurology, 31, 1026-1030. http://dx.doi.org/10.1212/WNL.31.8.1026
- Arthur, H., de Niese, M., Jeffrey, P.L. and Austin, L. (1983) Plasma Lipoprotein in Duchenne Muscular Dystrophy. Biochemistry International, 6, 307-313.
- Ehrlich, P. (1885) Das sauerstufbudurfnis des organismus, in Eine Farbenanalytische Studies. Hirschwald, Berlin, pp. 167.
- Murphy, J.B. and Sturm, E. (1923) Conditions Determining the Transplantability of Tissues in the Brain. The Journal of Experimental Medicine, 39, 183-197. http://dx.doi.org/10.1084/jem.38.2.183
- Sabin, F.R. (1916) The Origin and Development of the Lymphatic System. Johns Hopkins Hospital Reports, Johns Hopkins Press, Baltimore, 347-440.
- Ediden, M. (1972) Transplantation Antigens. Academic Press, New York, 125-140.
- Blalock, J.E. (1984) The Immune System as a Sensory Organ. The Journal of Immunology, 132, 1067-1070.
- Reichlin, S. (1993) Neuroendocrine-Immune Interactions. New England Journal of Medicine, 329, 1246-1253. http://dx.doi.org/10.1056/NEJM199310213291708
- Sternberg, E.M., Chrousos, G.P., Wilder, R.L. and Gold, P.W. (1992) The Stress Response and the Regulation of Inflammatory Disease. Annals of Internal Medicine, 117, 854-866. http://dx.doi.org/10.7326/0003-4819-117-10-854
- Weigent, D.A. and Blalock, J.E. (1987) Interactions between the Neuroendocrine and Immune Systems: Common Hormones Receptors. Immunological Reviews, 100, 79-108. http://dx.doi.org/10.1111/j.1600-065X.1987.tb00528.x
- Tourtellotte, W. (1970) On Cerebrospinal Fluid Immunoglobulin-G (IgG) quotIents in Multiple Sclerosis and Other Disease. A Review and a New Formula to Estimate the Amount of IgG Synthesized Per Day by the Central Nervous System. Journal of the Neurological Sciences, 10, 279-304. http://dx.doi.org/10.1016/0022-510X(70)90156-5
- Oldstone, M.B. and Dixon, F.J. (1968) Immunohistochemical Study of Allergic Encephalomyelitis. American Journal of Pathology, 52, 251-263.
- Miller, S.D., Vanderlugt, C.L., Begolka, W.S., Pao, W., Yauch, R.L., Neville, K.L., Katz-Levy, Y., Carrizosa, A. and Kim, B.S. (1997) Presistent Infection with Theiler’s Virus Leads to CNS Autoimmunity via Epitope Spreading. Nature Medicine, 3, 1133-1136. http://dx.doi.org/10.1038/nm1097-1133
- Agnello, V. (1978) Complement Deficiency States. Medicne, 57, 1-23.
- Fearon, D.T. (1987) Structure and Functions of Compliment. In: Ataasi, M.Z., Vanoss, C.J. and Absolon, D.R., Eds., Molecular Immunology, Marcel Dekker Inc., New York, pp. 511.
- Volanakis, J.E. (1975) The Human Complement System. Journal of Oral Pathology & Medicine, 4, 195-221. http://dx.doi.org/10.1111/j.1600-0714.1975.tb01742.x
- Stein, J.H. (1987) Complement Measurement. In: Puddy, S., Ed., Internal Medicine, 2nd Edition, Little Brown and Company Boston, Toronto, pp. 1184.
- Fishman, R.A. (1980) Cerebrospinal Fluid in Disorders of the Nervous System. Armarou, A., Hrtncry, M. and Shapiro, K., Eds., Sounders, Philadelphia, London, Toronto, 95-107.
- Milica, T.Č., Brinar, V., Pauro, M., Vogrinc, Ž. and Štambuk, N. (1998) Cerebrospinal Fluid Complement Activation in Neurological Diseases. Journal of the Neurological Sciences, 154, 173-181. http://dx.doi.org/10.1016/S0022-510X(97)00225-6
- Isenberg, D.A. (1982) Immunoglobulin Deposition in Skeletal Muscle in Primary Muscle Disease. QJM: An International Journal of Medicine, 52, 297-310.
- Spulre, S. and Engel, A.G (1988) Unexpected Sarcolemental Complement on Membrane Attack Complex Deposits on Nonnecrotic Muscle Fibers in Muscular Dystrophies. Neurology, 50, 41-46. http://dx.doi.org/10.1212/WNL.50.1.41
- Engel, A.G. and Biesecker, G. (1982) Complement Activation in Muscle Fiber Necrosis Demonstration of the Membrane Attack Complex of Complement in Necrotic Fibers. Annals of Neurology, 12, 289-296.
- Niebrój-Dobosz, I., Łukasiuk, M. and Niebrój-Dobosz, I. (1995) Immunoblot Analysis of Sarcoplasmic Calcium Binding Proteins in Duchenne Muscular Dystrophy. Journal of Neurology, 242, 82-86. http://dx.doi.org/10.1007/BF00887821
- Minetti, C., Tanjii, K. and Bonilla, E. (1992) Immunologic Study of Vinculin in Duchenne Muscular Dystrophy. Neurology, 42, 1751-1754. http://dx.doi.org/10.1212/WNL.42.9.1751
- Wochner, R.D., Drews, G., Strober, W. and Warren, S. (1966) Accelerated Breakdown of Immunoglobulin G (IgG) in Myotonic Dystrophy: A Hereditary Error of Immunoglobulin Catabolism. Journal of Clinical Investigation, 45, 321- 329. http://dx.doi.org/10.1172/JCI105346
- Larsen, B., Johnson, G., Loghem, E., Van Marshall, W.H., Newton, R.M., Pryse-Phillips, W. and Skanes, V. (1980) Immunoglobulin Concentration and Gm Allotypes in a Family with Thirty-Three Cases of Myotonic Dystrophy. Clinical Genetics, 18, 13-19. http://dx.doi.org/10.1111/j.1399-0004.1980.tb01358.x
- Sanders, B.G., Kline, K. and Morton, C.J. (1980) Serum IgG Levels in the Storrs Strain of Hereditary Muscular Dystrophhic Chickens. Biochemical Genetics, 18, 1149-1158.
- Mancini, G., Carbonara, A.O. and Heremans, J.F. (1965) Immunochemical Quantitation of Antigens by Single Radial Immunodiffusion. Immunochemistry, 2, 235-254. http://dx.doi.org/10.1016/0019-2791(65)90004-2
- Al-Hakeim, H.K. (2008) Serum Cortisol, Immunoglobulins and Some Complements among Depressed Patients. Indian Journal of Clinical Biochemistry, 23, 76-80. http://dx.doi.org/10.1007/s12291-008-0018-2
- Olsson, R., Hellner, L., Lindstedt, G., Lundberg, P.A. and Teger-Nilsson, A.C. (1983) Plasma Proteins in Patients on Long Term Anti Epileptic Treatment. Clinical Chemistry, 29, 728-730.
- Riddoch, D. and Thompson, R.A. (1970) Immunoglobulin Levels in the Cerebrospinal Fluid. British Medicine Journal, 1, 396-399. http://dx.doi.org/10.1136/bmj.1.5693.396
- Gholamoli, Y.P., Izadi, S. and Ghaderi, A. (2004) Immunological Correlates of Adults Onset Idiopathic Generalised Tonic-Clanic Epilepsy before and after Sodium Valporate Treatment, Iranion. The Journal of Immunology, 1, 105-110.
- Slavin, B.N., Fenton, G.M., Laundry, M. and Reynolds, E.H. (1974) Serum Immunoglobulin in Epilepsy. Journal of the Neurological Sciences, 23, 353-357. http://dx.doi.org/10.1016/0022-510X(74)90153-1
- Moustafa, S., Osama, A., Saad, A., El-seed, S. and Moustafa, N. (2006) Immunotoxicity of Chronic Phenytoin Administration in Epileptic Patients. Egyptian Journal of Neurology, Psychiatry and Neurosurgery, 43, 435-441.
- Kumar, S. (1989) Medico-Physical Studies on Epilepsy and Other Neurological Disorders. Ph. D. Thesis, University of Delhi, New Delhi.
- Anderson, P. and Moseklide, L. (1977) Immunoglobulin Levels and Autoantibodies in Epileptics on Long-Term Anticonvulsant Therapy. Acta Medica Scandinavica, 201, 69-74.
- Moore, T.L., Ryan Jr., R.E., Pohl, D.A., Roodman, S.T. and Ryan Sr., R.E. (1980) Immunoglobulin, Complement, and Immune Complex Levels during a Migrane Attack. Headache: The Journal of Headache and Face pain, 20, 9-12.
- Lord, G.D.A. and Duchworth, J.W. (1977) Immunoglobulin and Complement Studies in Migrance. Headache: The Journal of Head and Face Pain, 17, 163-168. http://dx.doi.org/10.1111/j.1526-4610.1977.hed1704163.x
- Kumar, S., Kumar, V., Jain, D.C. and Mittal, R. (2013) Immunological Variations in Epileptic Children. Open Journal of Applied Sciences, 3, 71-91.
NOTES

*Corresponding author.