Computational Molecular Bioscience
Vol.3 No.2(2013), Article ID:32857,5 pages DOI:10.4236/cmb.2013.32004
BEAR, a Molecular Docking Refinement and Rescoring Method
Dipartimento di Scienze della Vita, Università di Modena e Reggio Emilia, Modena, Italy
Email: *giulio.rastelli@unimore.it
Copyright © 2013 Andrew Anighoro, Giulio Rastelli. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 5, 2013; revised May 5, 2013; accepted May 15, 2013
Keywords: Virtual Screening; Bear; Structure-Based Drug Design
ABSTRACT
BEAR (Binding Estimation After Refinement) is a computational method for structure-based virtual screening. It was set up as a post-docking processing tool for the refinement of ligand binding modes predicted by molecular docking programs and the accurate evaluation of free energies of binding. BEAR has been validated in a number of computational drug discovery applications. It performed well in discriminating active ligands with respect to molecular decoys of biological targets belonging to different protein families as well as in discovering biologically active hits. Recently, it has also been validated in the emerging field of G-protein coupled receptors structure based virtual screening.
1. Introduction
Early stage drug discovery aims at identifying new active compounds against biological targets involved in a given pathology. A widely used method for the discovery of biologically-active hits is High-Throughput Screening (HTS). However, the high costs and relatively low hit rates of HTS make computational tools for drug design an attractive and straightforward alternative [1]. Notably, virtual screening represents an efficient computational approach for the inexpensive and efficient assessment of large libraries of small molecules [2]. When structural information for a target of interest is available, StructureBased Virtual Screening (SBVS) techniques such as molecular docking may be applied [3,4]. Molecular docking programs focus on the prediction of suitable binding modes for small molecules in the binding site of the selected target and the subsequent scoring for the assessment of the binding strength [5]. Despite some valuable improvements in the accuracy and efficiency of the algorithms used for molecular docking, there are still considerable drawbacks and limitations to face [3,6]. Among these, a major hurdle to overcome is the reliable simulation of the flexibility of both ligands and receptor, which play a central role in molecular interaction establishment and binding strength. Furthermore, different scoring functions have been implemented in currently used molecular docking programs, but obtaining a fair correlation between docking scores and experimental activity data remains a difficult task. These limitations are responsible for the occurrence of false-positive and false-negative hits in the ranked lists resulting from the screenings performed with standard docking methods. Hence, it is a general opinion that molecular docking results may be improved by post-processing with more accurate tools, gaining higher accuracy in both binding modes prediction and energy scoring.
For this purpose, we developed Binding Estimation After Refinement (BEAR) [7], an automated post-docking tool consisting of a conformational refinement of docking binding modes with molecular dynamics followed by a prediction of binding free energy performed with MM-PBSA and MM-GBSA algorithms [8-12]. Recently, BEAR has been validated and applied in a number of drug discovery applications. In this review we will give an overview of the BEAR workflow and the results obtained so far.
2. The BEAR Workflow
The BEAR program consists of an initial pre-processing step followed by an automated three steps procedure for structural refinement of the ligand-receptor complexes. Then, a final step is made for computing the binding free energy of the ligand in the refined complex. The procedure is iterative and able to automatically screen compounds in a database. The workflow is graphically represented in Figure 1. We implemented for these purposes modules available in the AMBER suite (Leap, Antechamber, Sander, pbsa) [13,14].
During the pre-processing step, hydrogen atoms are added to the receptor, atomic charges (AM1-BCC) are calculated for the small molecules, missing force-field parameters are assigned with parmcheck, and topologies are built for the ligand, receptor and the complex. Then, the following iterative three steps procedure is based on Molecular Mechanics (MM) and Molecular Dynamics (MD) cycles. In particular, an initial MM energy minimization of the whole protein-ligand complex is performed, followed by a short MD simulation where the ligand is allowed to move, and a final re-minimization of the entire complex. Finally, the binding free energy of the refined complexes is calculated by MM-PBSA and MM-GBSA methods. Further details about energy minimization, MD, and binding free energy calculations can be found in reference [7].
3. Validation Studies
BEAR has been extensively validated as a useful tool for post-processing molecular docking results in different case studies [7,15-19]. In these works, proteins spanning different classes have been selected as targets. In general, BEAR proved to be able to predict free energies of binding that correlate with experimental activities and to recognize true actives seeded within larger datasets of molecular decoys.
3.1. Aldose Reductase
As a first attempt to set up and validate BEAR, the workflow was applied to a series of aldose reductase inhibitors [15]. In particular, 28 inhibitors spanning five orders of magnitude of biological activity and characterized by significant chemical diversity were selected. Their binding modes were taken from the available X-ray crystal structures. Then, the combined MM/MD protocol followed by binding free energy scoring was applied and tested for the ability to score the ligands in agreement with their known biological activity. The results obtained in such a challenging task showed good correlations (r2 = 0.80 by MM-PBSA, r2 = 0.73 by MM-GBSA) between computed and experimental free energies of binding. This was a first important indication that our refinement and rescoring method was able to provide results in significant agreement with experiment. It can be applied for an accurate rescoring of structurally unrelated molecules in a given binding site, and the computational times needed to refine and rescore the docked complexes are compatible with virtual screenings.
3.2. Pf-DHFR
Three independent studies tested the ability of BEAR to recognize Plasodium falciparium dihydrofolate reductase (Pf-DHFR) active molecules from larger datasets of
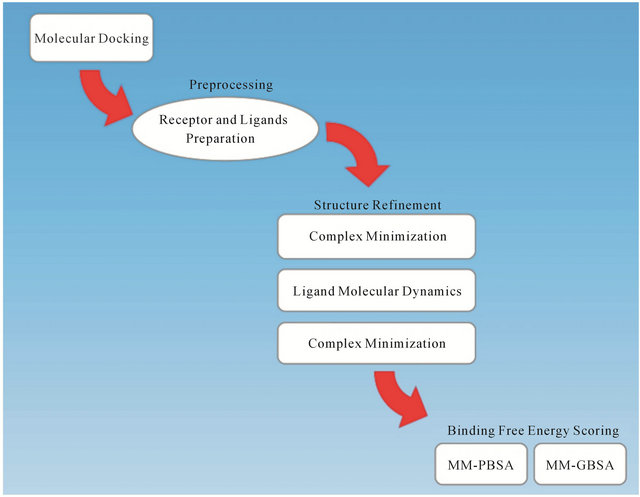
Figure 1. The BEAR workflow.
molecular decoys or molecules without known activities for this target [7,16,17].
In the first study, BEAR was able to significantly enrich 14 known ligands seeded into the National Cancer Institute (NCI) diversity database [20], containing 1720 compounds with unknown activities on Pf-DHFR [7]. The BEAR performance was clearly superior with respect to that of the docking method used, i.e. AutoDock [21]. In the second case, BEAR performance was evaluated using 201 known ligands and 7150 decoys contained in the DHFR data set taken from the directory of useful decoys (DUD) [16,22]. In the third case, the same 201 known ligands were seeded in 1.5 million compounds contained at that time in the lead-like subset of the ZINC database [17,23]. Here, the objective was to test the ability of BEAR to discriminate a very limited number of true binders from a very large set (1.5 million) of compounds, a situation typical of virtual screening campaigns. The results obtained after docking and BEAR post-processing were compared by analysing the enrichment factors. In all three cases, including the more challenging one, BEAR was able to perform strikingly better compared to standard docking. Therefore, BEAR performed well in virtual screenings against a biological target with known structure and different database of compounds. In this regard, BEAR demonstrated to be a reliable computational tool to be implemented in drug discovery.
3.3. Estrogen Receptor, Thymidine Kinase, Factor Xa, Adenosine Deaminase, Aldose Reductase, and Enoyl ACP Reductase
In a later investigation, we tested the performance of BEAR on several targets with different binding sites, using both a single and a multiple conformation approach. The use of a collection of multiple protein conformations is considered a valuable approach to account for protein flexibility in virtual screenings. This procedure is usually referred to as ensemble docking. Recently, BEAR has been applied for post-processing docking results of six targets spanning different protein families, namely, estrogen receptor, thymidine kinase, factor Xa, adenosine deaminase, aldose reductase, and enoyl ACP reductase [18]. In this work, we used two docking programs, LibDock [24] and Autodock, and multiple conformations for each target.
Enrichment factor plots were built for each target and the percentage of known inhibitors, taken from the DUD data set, retrieved at different early stages of the ranked databases were annotated. A comparative analysis of the enrichment factors showed that the scoring functions implemented in BEAR are generally able to yield higher enrichments of known ligands compared to the standard docking scoring functions. However, rather poor enrichments of known ligands have been obtained with both docking and BEAR in case of particularly challenging targets for docking studies, such as adenosine deaminase and enoyl ACP reductase. This was mainly due to difficulties in retrieving correct binding modes for these targets.
The use of multiple protein conformations combined with the application of BEAR showed potential benefits in virtual screening context yielding to higher enrichment factors. Nevertheless, different strategies may be implemented for the compound selection (best scoring docking solution within multiple structures, top scoring compounds in each structure).
3.4. G-Protein Coupled Receptors
Recently, we have applied BEAR to G-protein coupled receptors (GPCRs) SBVS [19]. These proteins are highly relevant targets for drug discovery, and advances in GPCRs structural determination by X-ray crystallography are opening unprecedented opportunities to discover new biologically active hits using SBVS [25]. Therefore, we wanted to test BEAR on GPCRs with known crystal structure.
In our work, four GPCRs have been studied, namely β2-adrenergic (β2), adenosine A2a (A2a), dopamine D3 (D3), and histamine H1 (H1). Receptor flexibility and induced fit are known to play a major role in the binding and potency of their antagonists, hence it was investigated whether a structural refinement of the docking complexes with molecular dynamics could be particularly useful. Furthermore, we hypothesized that a more accurate description of desolvation free energies performed by MM-PBSA could improve the binding affinity predictions of the many protonated biogenic amines found among the known antagonists.
For each of the four targets analysed, the ability to recognize a small number of known antagonists (between 20 and 56) in a database of 1000 drug-like decoys have been assessed through the analysis of enrichment factor plots and binding modes assessment. In most cases, BEAR gave better performances compared to the docking program used, i.e. AutoDock. A strikingly better performance was obtained in the case of the β2-adrenergic receptor, where AutoDock gave enrichment factors close to random while BEAR significantly enriched the hit list with active antagonists. Moreover, we showed that the results could be improved for the A2a receptor by using a suitable combination of structural waters that participate in hydrogen bonding network with the antagonists. Finally, for the first time, BEAR’s implications on multitarget drug screening were put forward. In particular, we found that five known antagonists in common between the H1 and D3 receptors were scored and ranked favourably in each of the two target screenings. The rational design of multi-target (polypharmacology) ligands is becoming increasingly important in drug discovery [26], and our results suggest that BEAR may be useful in discovering multi-target hits shared by different GPCRs.
3.5. Plasmepsin II
Plasmepsin II, an aspartic protease with a key role in the metabolism of Plasmodium falciparum, is a validated target in the therapy of malaria. In a recent successful virtual screening campaign, BEAR was applied for the discovery of new Plasmepsin inhibitors [27].
In this work, 5000 top-scoring compounds obtained from a previous docking screening [28] conducted with FlexX [29] have been selected and post-processed with BEAR. The post-processed compounds have been ranked according to the estimated free energy of binding and the top fraction of the ranked lists has been analysed and then biologically tested on the purified protein. Among the best energy scoring compounds BEAR identified four promising chemical classes, namely N-alkoxyamidines, guanidines, amides, and ureas and thioureas. Visual inspection of the binding modes of the top-ranking derivatives belonging to these four classes confirmed that these molecules were able to establish key interactions with the receptor binding site. After this filtering, thirty bestscoring compounds were selected as representatives of the four classes and then biologically tested on the purified protein. Remarkably, 26 compounds out of the 30 tested showed IC50 values ranging from 4.3 nM to 1.8 μM. This was a fairly exceptional hit rate for a virtual screening.
4. Final Remarks
Our studies have shown that BEAR performed well in a number of validation studies conducted on proteins of different families and ligands and databases of different nature. Moreover, we have shown the potential of BEAR in both single and multiple receptor conformation approaches as well as its utility in screening transmembrane GPCRs. Finally, we provided the first evidence that BEAR may be suited to recognize multi-target active compounds. Using BEAR, biologically active compounds were discovered with a very good hit rate.
All these evidence testifies that BEAR is a useful and reliable computational tool to be implemented in structure-based virtual screenings.
REFERENCES
- B. C. Duffy, L. Zhu, H. Decornez and D. B. Kitchen, “Early phase Drug Discovery: Cheminformatics and Computational Techniques in Identifying Lead Series,” Bioorganic Medicinal Chemistry, Vol. 20, No. 18, 2012, pp. 5324-5342. doi:10.1016/j.bmc.2012.04.062
- P. Ripphausen, B. Nisius, L. Peltason and J. Bajorath, “Quo Vadis, Virtual Screening? A Comprehensive Survey of Prospective Applications,” Journal of Medicinal Chemistry, Vol. 53, No. 24, 2010, pp. 8461-8467. doi:10.1021/jm101020z
- T. Cheng, Q. Li, Z. Zhou, Y. Wang and S. H. Bryant, “Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review,” The AAPS Journal, Vol. 14, No. 1, 2012, pp. 133-141. doi:10.1208/s12248-012-9322-0
- G. Rastelli, “Emerging Topics in Structure-Based Virtual Screening,” Pharmaceutical Research, Vol. 30, No. 5, 2013, pp. 1458-1463. doi:10.1007/s11095-013-1012-9
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath. “Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications,” Nature Reviews. Drug Discovery, Vol. 3, No. 11, 2004, pp. 935-949. doi:10.1038/nrd1549
- G. Schneider, “Virtual Screening: An Endless Staircase?” Nature Reviews. Drug Discovery, Vol. 9, No. 4, 2010, pp. 273-276. doi:10.1038/nrd3139
- G. Rastelli, G. Degliesposti, A. Del Rio and M. Sgobba, “Binding Estimation after Refinement, a New Automated Procedure for the Refinement and Rescoring of Docked Ligands in Virtual Screening,” Chemical Biology & Drug Design, Vol. 73, No. 3, 2009, pp. 283-286. doi:10.1111/j.1747-0285.2009.00780.x
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, III, “Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models,” Accounts of Chemical Research, Vol. 33, No. 12, 2000, pp. 889-897. doi:10.1021/ar000033j
- J. Wang, P. Morin, W. Wang and P. A. Kollman, “Use of MM-PBSA in Reproducing the Binding Free Energies to HIV-1 RT of TIBO Derivatives and Predicting the Binding Mode to HIV-1 RT of Efavirenz by Docking and MM-PBSA,” Journal of the American Chemical Society, Vol. 123, No. 22, 2001, pp. 5221-5230. doi:10.1021/ja003834q
- B. Kuhn, P. Gerber, T. Schulz-Gasch and M. Stahl, “Validation and Use of the MM-PBSA Approach for Drug Discovery,” Journal of Medicinal Chemistry, Vol. 48, No. 12, 2005, pp. 4040-4048. doi:10.1021/jm049081q
- P. D. Lyne, M. L. Lamb and J. C. Saeh, “Accurate Prediction of the Relative Potencies of Members of a Series of Kinase Inhibitors Using Molecular Docking and MMGBSA Scoring,” Journal of Medicinal Chemistry, Vol. 10, No. 49, 2006, pp. 4805-4808. doi:10.1021/jm060522a
- A. Weis, K. Katebzadeh, P. Soderhjelm, I. Nilsson and U. Ryed, “Ligand Affinities Predicted with the MM/PBSA Method: Dependence on the Simulation Method and the Force Field,” Journal of Medicinal Chemistry, Vol. 49, No. 22, 1995, pp. 6596-6606. doi:10.1021/jm0608210
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, III, S. DeBolt, D. Ferguson, G. Seibel and P. Kollman, “AMBER, a Package of Computer Programs for Applying Molecular Mechanics, Normal Mode Analysis, Molecular Dynamics and Free Energy Calculations to Simulate the Structural and Energetic Properties of Molecules,” Computer Physics Communications, Vol. 91, No. 1-3, pp. 1-41. doi:10.1016/0010-4655(95)00041-D
- D. A. Case, T. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods, “The Amber Biomolecular Simulation Programs,” Journal of Computational Chemistry, Vol. 26, No. 16, 2005, pp. 1668-1688. doi:10.1002/jcc.20290
- A. M. Ferrari, G. Degliesposti, M. Sgobba and G. Rastelli, “Validation of an Automated Procedure for the Prediction of Relative Free Energies of Binding on a Set of Aldose Reductase Inhibitors,” Bioorganic & Medicinal Chemistry, Vol. 15, No. 24, 2007, pp. 7865-7877. doi:10.1016/j.bmc.2007.08.019
- G. Rastelli, A. Del Rio, G. Degliesposti and M. Sgobba, “Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA,” Journal of Computational Chemistry, Vol. 31, No. 4, 2010, pp. 797-810.
- G. Degliesposti, C. Portioli, M. D. Parenti and G. Rastelli, “BEAR, a Novel Virtual Screening Methodology for Drug Discovery,” Journal of Biomolecular Screening, Vol. 16, No. 1, 2011, pp. 129-133. doi:10.1177/1087057110388276
- M. Sgobba, F. Caporuscio, A. Anighoro, C. Portioli and G. Rastelli, “Application of a Post-Docking Procedure Based on MM-PBSA and MM-GBSA on Single and Multiple Protein Conformations,” European Journal of Medicinal Chemistry, Vol. 58, 2012, pp. 431-440. doi:10.1016/j.ejmech.2012.10.024
- A. Anighoro and G. Rastelli, “Enrichment Factor Analyses on G-Protein Coupled Receptors with Known Crystal Structure,” Journal of Chemical Information and Modeling, Vol. 53, No. 4, 2013, pp. 739-743. doi:10.1021/ci4000745
- NCI Diversity Set Information. http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, “A Semiempirical Free Energy Force Field with ChargeBased Desolvation,” Journal of Computational Chemistry, Vol. 28, No. 6, 2007, pp. 1145-1152. doi:10.1002/jcc.20634
- N. Huang, B. K. Shoichet and J. J. Irwin, “Benchmarking Sets for Molecular Docking,” Journal of Computational Chemistry, Vol. 49, No. 23, pp. 6789-6801.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, “ZINC: A Free Tool to Discover Chemistry for Biology,” Journal of Chemical Information and Modelling, Vol. 52, No. 7, pp. 1757-1768. doi:10.1021/ci3001277
- D. J. Diller and K. M. Merz Jr., “High throughput Docking for library Design and Library Prioritization,” Proteins, Vol. 43, No. 2, 2001, pp. 113-124. doi:10.1002/1097-0134(20010501)43:2<113::AID-PROT1023>3.0.CO;2-T
- R. C. Stevens, V. Cherezov, V. Katritch, R. Abagyan, P. Kuhn, H. Rosen and K. Wüthrich, “The GPCR Network: A Large-Scale Collaboration to Determine Human GPCR Structure and Function,” Nature Reviews. Drug Discovery, Vol. 12, No. 1, 2013, pp. 25-34. doi:10.1038/nrd3859
- X. H. Ma, Z. Shi, C. Tan, Y. Jiang, M. L. Go, B. C. Low, Y. Z. and Chen, “In-Silico Approaches to Multi-Target Drug Discovery,” Pharmaceutical Research, Vol. 27, No. 5, 2010, pp. 739-749. doi:10.1007/s11095-010-0065-2
- G. Degliesposti, V. Kasam, A. Da Costa, H. K. Kang, N. Kim, D. W. Kim, V. Breton, D. Kim and G. Rastelli, “Design and Discovery of Plasmepsin II Inhibitors Using an Automated Workflow on Large-Scale Grids,” ChemMedChem, Vol. 4, No. 7, 2009, pp. 1164-1173. doi:10.1002/cmdc.200900111
- V. Kasam, M. Zimmermann, A. Maass, H. Schwichtenberg, A. Wolf, N. Jacq, V. Breton and M. HofmannApitius, “Design of New Plasmepsin Inhibitors: A Virtual High throughput Screening Approach on the EGEE Grid,” Journal of Chemical Information and Modelling, Vol. 47, No. 5, 2007, pp. 1818-1828. doi:10.1021/ci600451t
- M. Rarey, B. Kramer, T. Lengauer and G. Klebe, “A Fast Flexible Docking Method Using an Incremental Construction Algorithm,” Journal of Molecular Biology, Vol. 261, No. 3, 1996, pp. 470-489. doi:10.1006/jmbi.1996.0477
NOTES
*Corresponding author.