American Journal of Analytical Chemistry
Vol.06 No.02(2015), Article ID:53326,11 pages
10.4236/ajac.2015.62010
Application of Total Error Strategy in Validation of Affordable and Accessible UV-Visible Spectrophotometric Methods for Quality Control of Poor Medicines
Jérémie Kindenge Mbinze1,2*, Jean Nsangu Mpasi2*, Elza Maghe2, Sébastien Kobo2, Rodrigue Mwanda2, Guy Mulumba2, Jacquie Bolavie Bolande3, Trésor Menanzambi Bayebila2, Maguy Borive Amani4, Philippe Hubert1, Roland Djang’eing’a Marini1,4#
1University of Liege (ULg), Department of Pharmacy, CIRM, Laboratory of Analytical Chemistry, Liège, Belgium
2Laboratory of Drug Analysis, Faculty of Pharmaceutical Sciences, University of Kinshasa, Kinshasa, Democratic Republic of Congo
3Laboratory Service, Institute of Research in Science of Health, Kinshasa, Democratic Republic of Congo
4Faculty of Medicine and Pharmacy, University of Kisangani, Kisangani, Democratic Republic of Congo
Email: #rmarini@ulg.ac.be
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 2 December 2014; accepted 12 January 2015; published 19 January 2015
ABSTRACT
In the framework of fighting against the poor quality medicines sold in developing countries using classical analytical methods easily accessible in those countries, four UV-Visible spectrophotometric methods for one antimalarial (quinine) and two antibiotics (amoxicillin and metronidazole) have been developed and validated according to the total error strategy using the accuracy profiles as a decision tool. The dosing range was 2 - 10 µg/mL (for quinine sulfate in tablet), 4 - 12 µg/mL (for quinine bichlorhydrate in oral drop-metronidazole benzaote in oral suspension) and 15 - 35 µg/mL (for amoxicillin trihydrate in capsule). The validated methods were then applied in determining the content of some analogous medicines sold in the Democratic Republic of Congo. Thus, the proposed UV-Visible spectrophotometric methods are simple and suitable to quantify quinine, amoxicillin and metronidazole in different pharmaceutical forms.
Keywords:
Poor Quality Medicines, UV-Visible Spectrophotometric Methods, Quinine, Amoxicillin, Metronidazole, Validation, Total Error, Accuracy Profiles

1. Introduction
Nowadays, malaria remains the most dramatic tropical parasitic disease in the world. It is responsible of more than one million deaths annually. Nearly 60% of the world population lives in areas at risk, namely in Africa, Asia, Central America and South America [1] .
Used a longtime ago in the treatment of uncomplicated malaria, monotherapy drug treatment have shown increasing cases of resistance mainly of Plasmodium falciparum [2] . To avoid this phenomenon towards recent antimalarial drugs, World Health Organization (WHO) recommended the use of combination therapies such as the Artemisinin-based combination [3] - [6] . Hence, quinine is still recommended alone in the treatment of severe malaria attacks as well as for chloroquine-resistant falciparum malaria [7] .
Malaria disease is often accompanied with symptoms such as fever, diarrhea, vomiting and some infections, which justifies the medical prescription of other drugs that can relieve these symptoms and treat these infections besides of antimalarial medicine. Amoxicillin and metronidazole are among the most used antibiotics and antiparasitic to treat certain infections. These kinds of medicines are very often bought in self-medication in many developing countries.
Besides the high risk of failure treatment due to plasmodial resistance to monotherapy, the spread of illicit distribution and the marketing of poor quality medicines worldwide and particularly in developing countries are seriously affecting the success of this treatment. Several other consequences towards the public health can be noticed such as adverse reactions, increase of morbidity and of mortality as well as loss of public confidence and waste of scarce resources, etc. [8] . Poor quality medicines can be classified into three main categories: Counter- feit, substandard and degraded medicines [9] . WHO reported that 6% of drugs worldwide are counterfeit while Food and Drug Administration (FDA, USA) estimated this proportion to 10% [10] . In fact, the true proportion cannot be estimated since values vary from one region to another and are related to a certain period. In 2003, P. Newton et al. published the results of a survey conducted in Southeast Asia on the medicines containing artesunate that is used against malaria chemo-resistant. In 40% of cases, these medicines were found to be falsified [11] . Therefore, to guarantee an access of populations to safe and sure medicines, it is important to set up appropriate measures that will allow evaluating and preserving the quality of those medicines, for example: having appropriate analytical method, strong quality assurance and regulatory, etc.
However, several laboratories in the developing countries do not yet dispose new analytical technology capable of satisfying to these requirements (HPLC, GC etc.). They still apply the classic methods such as titration or those described in pharmacopoeia for raw material analysis that they adapt and the UV-Visible spectrophotometric methods. Hence, these methods are affordable, available and technically feasible. Therefore, it is mandatory to verify if this adaptation fits well with the new purpose namely the assay of the active ingredient in new pharmaceutical formulations usually not described qualitatively of quantitatively.
In this study, we were interested to verify by means of validation four UV-Visible spectrophotometric methods currently used in several quality control laboratories in Kinshasa in Democratic Republic of Congo (DRC): method for the determination of quinine 20% in oral drops and of quinine tablets 500 mg, method for the determination of amoxicillin trihydrate (3H2O) 500 mg in capsule and method for the determination of metronidazole 125 mg/5mL in oral suspension. The validation of analytical methods is an obligation concept that is relatively recent in main sub-Saharan countries however that is increasing in requirement by legal health authorities. For the preparation of validation standards, four matrix formulations were provided by the Indian manufacturing laboratories legally authorized in the DRC. Finally, the validated method was used to analyze drugs marketed in DRC. The chemical structures of the concerned active ingredients are shown in Figure 1.
2. Method Validation Strategy
Validation of an assay is a set of operations carried out to demonstrate that a procedure is sufficiently accurate and reliable to have confidence in the results provided for the intended use (of the assay) [12] - [16] . Several strategies have been developed for this purpose [12] [17] - [22] . From a statistical point of view, they have certain insufficiency including the lack of consideration of decision making based on acceptance limits defined a priori, lack of use of the risk related to the future use of the method. A new validation strategy based on the accuracy profile was described [23] [24] . It is in perfect agreement with the objective of an analytical method, namely its ability to quantify as accurately as possible each of the unknown quantities a laboratory will determine in the future [24] . This new validation strategy combines two basic and fundamental criteria, bias and precision to the

Figure 1. The chemical structures of quinine, amoxicillin and metronidazole.
final result of a measurement, and therefore reflect the total measurement error, systematic error and random error, respectively [23] . The principle of this validation strategy can be translated by Equation (1) which states that the difference between a value of measurement (x) and its true value (μ) must be less than an acceptance limit (λ).
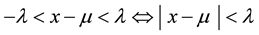 (1)
(1)
From Equation (1), one can notice that the consideration of acceptance limits introduces a first concept for the analyst to take decisions based on the objective of the analytical method and the limitation or control of error propagation. Commonly, the acceptance limit is fixed at 1% or at 2% for the assay of active ingredients in a raw material, at 5% for dosage forms [23] [24] . In other referential, it is set at 10% for pharmaceutical dosage forms [25] . Always from the Equation (1), one can notice a second concept the accuracy profile that is constructed from estimates of the tolerance interval of β-expectation measurements at each concentration level. Another im- portant concept derived from that Equation (1) is the set of “good analytical procedure” with a known risk which may result in the following equation:
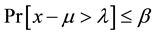 (2)
(2)
With β the proportion of measurements within the limits of acceptance, and λ the quantity defining the acceptance limits fixed a priori according to the constraints of the industry. The risk of a procedure is evaluated by the proportion of measurements (Pr) that could be out of the acceptance limits [23] [26] . Thus, the accuracy profile can be considered as a decision tool based on the risk associated with the method. The concept of risk is related to the guarantee for the future analysis of unknown samples while applying the validated method. As a decision tool, the accuracy profile can be used to accept or reject an analytical method according to the expected usage. Thus decision tool can be exploited as a diagnostic tool, for example, to select the most suitable regres- sion model for calibration, to determine the limits of upper and lower quantification that will define the dosing interval [27] .
3. Experimentation
3.1. Chemical and Reagents
Amoxicillin trihydrate (99.1%) and metronidazole benzoate (99.9%) were purchased from Fagron N. V. (Waregem, Belgium), quinine sulfate (96.9%) from Sigma Aldrich (Antwerp, Belgium) and quinine bichlorhydrate (100.8%) from Molekula Limited (Dorset, UK). Sodium hydroxide was supplied from Merck (Darmstadt, Germany) while the following excipients aerosil, avicel, benzoic acid, gelatin, lactose, magnesium stearate, propylene glycol, talc, starch, sodium benzoate, sorbitol, sodium saccharin, sodium carboxymethyl cellulose (CMC), tween 80, xanthine gum were obtained at New Cesamex Laboratory (Kinshasa, DRC).
3.2. Materials
The spectrophotometer used was brand HP/Agilent 8453 with Chemstation software from Agilent Technologies (Gyeonggi-do, South Korea) and an electronic analytical balance SHIMADZU AUW 220 D (Kyoto, Japan). Prior to their use, the material were qualified according to an internal qualification procedure. The wavelengths used were 229 nm for amoxicillin, 235 nm for quinine and for metronidazole. Ultrapure water was obtained from Aqua MaxTM―Basic 360 series, purification system from YL Instruments (Gyeonggi-do, South Korea).
For statistical data treatment, validation treatment and graph drawing, the Enoval® software v3.0 (Arlenda, Liege, Belgium) was used.
3.3. Standard Sample Preparations
Four different dissolutions solvent were used for the dissolution of the active ingredients: ultra pure water for quinine dihydrochloride, methanol for quinine sulfate and metronidazole benzoate, 0.1 N sodium hydroxide for amoxicillin trihydrate. The same solvent was used as blank. The stock solutions were prepared by dissolving 10 mg of each of the active ingredients in 100 mL of solvent corresponding to dissolution media.
For the calibration standards (CS), dilutions were performed in different dissolutions solvent in order to obtain solutions at three concentration levels (m = 3), as mentioned in Table 1.
For validation standards (VS), independent stock solutions of the active ingredients were prepared in the same way as described for the CS. The excipients of each pharmaceutical formulation were added in stock solutions.
Subsequent dilutions were carried out in dissolutions solvent to obtain solutions at five different concentration levels (m = 5), as mentioned in Table 2.
The VS were independently prepared in the matrix, in such a way to simulate as much as possible the corres- ponding formulation and its routine analysis. For each concentration level and each standard, three independent repetitions (n = 3) were performed daily for three days (p = 3).

Table 1. Content of each targeted active ingredient for the calibration standards and the validation standards according to pharmaceutical formulations.
Table 2. Qualitative and quantitative composition of the different matrices.
4. Results and Discussion
4.1. Validation
4.1.1. Consideration of Different Limits
As a first step in the validation of the four UV-Visible spectrophotometric methods evaluated in this study, the acceptance limit was settled at 10% that corresponds to the assay specification of 90.0% - 110.0% for the pharmaceutical formulations concerned. Unless specified, we have considered a risk of 5%, meaning that the assay method should give guarantee of having 95% of future measurements within the 10% acceptance limits.
4.1.2. Evaluation of Quality of Results
After having run the practical experiences of validation according to the experimental design, several tools were used to evaluate the quality of the results collected for each analytical method. At first, by means of calibration standards data several regression models were computed and used to evaluate the fitting of the validation standards. For each regression model, several indexes were computed, namely, the precision, the trueness and the dosing range. An index value closer to 1 indicated a good suitability of the validation parameters, e.g., good precision, good trueness according to the 10% acceptance limits, whereas a dosing range index closer to 1 relates to the ability of the assay method to cover the maximum of the tested range for quantification. In Table 3 we reported only the most adequate and appropriate regression models to easy use in routine. The accuracy index values were also computed as the geometric mean of the three other indexes. As can be seen, the accuracy indexes for quinine sulfate in tablet, quinine bichlorhydrate in oral drop, metronidazole benzoate in oral suspension and amoxicillin trihydrate in capsule were all above 0.82 indicating very good method accuracy with the ability to quantifiy quinine sulfate, quinine bichlorhydrate, metronidazole benzoate and amoxicillin trihydrate all over the tested dosing range (index of 1).
4.2. Validation Parameters
The following models were selected to evaluate the other validation parameters: linear regression for quinine bichlorhydrate in oral drop and for metronidazole benzoate in oral suspension, linear regression through 0 fitted using the highest concentration level only for quinine sulfate in tablet and for amoxicillin trihydrate in suspension. The risks were set at 5% in all cases.
Table 3. Evaluation of quality of fit for the selected regression model for quinine sulfate, Quinine bichlorhydrate, amoxicillin trihydrate, and metronidazole benzoate.
4.2.1. Method Selectivity
As a next step, the selectivity criteria was evaluated for each UV-Visible spectrophotometric method using the matrix blank containing only the excipients of drug as mentioned in Table 2 with the compound not targeted by the method. No interference was observed as analytical response.
4.2.2. Trueness
The results of trueness illustrate the mean of bias observed between the series of measurements and the reference concentrations. Using the calibration curve of each analyte and the concentrations of the validation standards (VS) was back-calculated and expressed in terms of absolute bias (µg/mL) and relative bias (%). As shown in Table 4, the trueness of the developed methods was found acceptable since the relative biases were below 2.78%.
4.2.3. Precision
Precision refers to the ability of the methods to provide proximate results obtained from multiple measurements of the same samples, under the same conditions. It was expressed in terms of relative standard deviation (RSD, %) for repeatability (intraday variations) and intermediate precision (inter-day variations) at each concentration level. As shown in Table 4, precision was found acceptable, as the RSD values for repeatability and intermediate precision were below 2.37% and 2.62%, respectively.
Table 4. Summary of the validation criteria for quinine sulfate, quinine bichlorhydrate, amoxicillin trihydrate, and metronidazole benzoate.
4.2.4. Accuracy
The accuracy was determined by the 95% β-expectation tolerance interval in order to observe the closeness of agreement of every single concentration measured by the method, and the assumed true value of this concentration. It combines the uncertainties of trueness and precision and is expressed as actual values and as a percentage of the targeted concentration (Table 4). As shown in Figure 2, all methods were found accurate all over the tested concentration levels since the lower and upper tolerance intervals were included within the 10% acceptance limits, guaranteeing that 95% of the future quantification results will be accurate in the dosing ranges evaluated. However, the four methods are able to quantify largely the corresponding analytes at the target concentration 100.0% that correspond to 8 µg/mL of quinine sulfate for tablet, 10 µg/mL of quinine bichlorhydrate for oral drop and metronidazole benzoate for oral suspension and to 30 µg/mL of amoxicillin trihydrate for capsule.
In addition, the four methods are also able to quantify largely the corresponding analytes at ±10.0% of the claimed content that can ensure the control during the release of batch production as well quantification of low or high content.
4.2.5. Linearity
The linearity of results was also evaluated for the four tested methods. It expresses the ability of the methods to give results directly proportional to the concentrations. Indication about linearity is given by the parameters of
 Legend: Red = bias (%), Black = acceptance limit (±10%), Blue = 95% β-expectation tolerance interval, Green = individual measures. (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Legend: Red = bias (%), Black = acceptance limit (±10%), Blue = 95% β-expectation tolerance interval, Green = individual measures. (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Figure 2. Accuracy profiles for quantitative methods validation of quinine sulfate in tablet, quinine bichlorhydrate in oral drop, metronidazole benzoate in suspension and amoxicillin trihydrate in capsule.
the linearity equation, namely the coefficient of determination (R2), the slope and the y-intercept. As can be seen in Table 4, the relationship was assumed linear for the four methods as the R2 values and the slope were close to one with acceptable intercepts fairly near to zero. Figure 3 shows that, for each concentration level, the absolute 95% β-expectation tolerance limits were all within the absolute acceptance, confirming the linearity of these four UV-Visible spectrophotometric methods.
4.2.6. Limits of Quantification (LOQ)
As the smallest quantity of the targeted substance in the sample that can be assayed under experimental conditions with a well-defined accuracy, the LOQ was evaluated by calculating the smallest concentration beyond which the accuracy limits or β-expectation tolerance limits go outside the acceptance limits. As the accuracy profile was included inside the acceptance limits (Figure 2), the first concentration level was considered as the lower LOQ for all molecules studied (2 µg/mL for quinine sulfate in tablet, 4 µg/mL for quinine bichlorhydrate in oral drop and for metronidazole benzoate in oral suspension and 15 µg/mL for amoxicillin trihydrate in capsule). On the other hand, the upper LOQ was evaluated as the highest quantity of the targeted substance in the sample that can be assayed under experimental conditions with a well-defined accuracy. It was estimated by calculating the highest concentration above which the accuracy limits or β-expectation tolerance limits go outside the acceptance limits. For all the four tested methods, the upper LOQ were the highest concentration levels, namely 10 µg/mL for quinine sulfate in tablet, 12 µg/mL for quinine bichlorhydrate in oral drop and for metronidazole benzoate in oral suspension and 35 µg/mL for amoxicillin trihydrate in capsule.
 Legend: Gray = identity line, Black = acceptance limit (±10%), Blue = 95% β-expectation tolerance interval, Green = individual measures. (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Legend: Gray = identity line, Black = acceptance limit (±10%), Blue = 95% β-expectation tolerance interval, Green = individual measures. (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Figure 3. Linearity graph for quantitative methods validation of quinine sulfate in tablet, quinine bichlorhydrate in oral drop, metronidazole benzoate in suspension and amoxicillin trihydrate in capsule.
4.3. Other Parameters
4.3.1. Risk Profile
In this study, the risk profile was used to investigate the risk of having future measurements falling outside of the 10% acceptance limits for each concentration level taking into account each analyte with its matrix and the selected regression model. Figure 4 shows that the risks of having future measurements outside the 10% acceptance limits were practically below 1.3% at all over the tested concentration range.
When analyzing of these products in routine, only about 98.7 times out of 100 the future measurements will be included in the 10% acceptance limits.
4.3.2. Uncertainty of Measurements
The uncertainty corresponds to the dispersion of the values that could reasonably be attributed to the measurand. The uncertainty was derived from the variance used to build the β-expectation tolerance limits at each concentration level tested, prior to intermediate precision, between and within-series variances [27] . As shown in Table 4, uncertainties of measurements of the developed methods were found acceptable since the relative expanded uncertainty were below 5.64%, confirming that all the results are within the 10% acceptance limit.
 Legend: (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Legend: (a) Quinine sulfate in tablet; (b) Quinine bichlorhydrate in oral drop; (c) Metronidazole benzoate in suspension; and (d) Amoxicillin trihydrate in capsule.
Figure 4. Graph of the risk profiles with a risk level set a priori at 10%.
4.4. Application
The validated methods were applied for the determination of the four active ingredients (quinine sulfate, quinine bichlorhydrate, amoxicillin, and metronidazole) in several brand pharmaceutical products coded A, B, C, D, and purchased in the DRC. The samples were obtained in non legal Pharmacy located in suburban areas. As it can be noticed in Table 5, all the targeted and claimed active ingredients were quantified and only three products (code C001 and C002 suspension containing metronidazole benzoate and code D001 capsule containing amoxicillin trihydrate) were not compliant with the assay specification 90.0% - 110.0% for the analyzed compound [25] .
5. Conclusion
In the framework of fighting against the phenomenon of poor quality medecines that affects mostly developing countries and ensure that the assay tests results are reliable for UV-Visible spectrophotometric methods, it was necessary to validate these methods and use them for routine analysis in the future. In order to fight this problem, easily accessible classical analytical methods were developed. Four methods were concerned and were validated for the quantitative analysis of active substances present in pharmaceutical formulations suspected to be of poor quality. For these UV-Visible spectrophotometric methods, the assay method of quinine sulfate in tablets was validated in the dosing range of 2 µg/mL to 10 µg/mL, the assay methods for quinine bichlorhydrate in oral drop and metronidazole benzoate in oral suspension were validated in the range of 4 µg/mL to 12 µg/mL and amoxicillin trihydrate in capsule was validated in the range of 15 µg/mL to 35 µg/mL. The strategy of the total error has been used to validate these methods that present advantages of being also simple, rapid and affordable. Finally, the methods have been successfully applied to the determination of quinine, amoxicillin and metronidazole in fifteen pharmaceuticals marketed in DRC.
Table 5. Assay results of fifteen pharmaceuticals marketed in DRC. Results consist in the mean percentage of claimed nominal content and the standard deviation computed on 3 independent samples. Specifications are set to 90% - 110% of the claimed nominal content (500 mg for Quinine sulfate, 3 g/15mL for quinine bichlorhydrate, 125 mg/5mL for metronidazole benzoate and 500 mg for amoxicicilline 3H2O).
Acknowledgements
The authors would like to thank the ARES-CCD (Académie de Recherche et d’Enseignement Supérieur― Com- mission de la Coopération au Développement) of Belgium and Arlenda s.a. respectively for financial support and data processing for validation. The following pharmaceutical production laboratories NEW CESAMEX, AVE PHARMA, LAPHAKI and PHATIKN are also acknowledged for their collaboration.
References
- Hay, S.I., Guerra, C.A., Tatem, A.J., Noor, A.M. and Snow, R.W. (2004) The Global Distribution and Population at Risk of Malaria: Past, Present and Future. The Lancet Infectious Diseases, 4, 327-336. http://dx.doi.org/10.1016/S1473-3099(04)01043-6
- Ambroise, T. (2000) Traitement du paludisme: Prévenir les résistances par les associations d’antipaludiques. Médecine Tropicale, 60, 219-222.
- Geyer, G. (2001) Les combinaisons thérapeutiques antipaludiques: Rapport d’une consultation technique. Organisation Mondiale de la Santé (World Health Organization), Genève, Suisse, 5-24.
- Mutabingwa, T., Anthony, D., Heller, A., Hallett, R., Ahmed, J., Drakeley, C., Greenwood, B.M. and Whitty, C. (2005) Amo- diaquine Alone, Amodiaquine + Sulfadoxinepyrimethamine, Amodiaquine + Artesunate, and Artemether-Lumefan- trine for Outpatient Treatment of Malaria in Tanzanian Children: A Four-Arm Randomized Effectiveness Trial. The Lancet, 365, 1474-1480. http://dx.doi.org/10.1016/S0140-6736(05)66417-3
- Tall, A., Rabarijaona, L., Robert, V., Bedja, S., Ariey, F. and Randrianarivelojosia, M. (2007) Efficacy of Artesunate plus Amodiaquine, Artesunate plus Sulfadoxine-Pyrimethamine and Chloroquine plus Sulfadoxine-Pyrimethamine in Pa- tients with Uncomplicated Plasmodium Falciparum in the Comoros Union. Acta Tropica, 102, 176-181. http://dx.doi.org/10.1016/j.actatropica.2007.03.004
- Van Vugt, M., Looareesuwan, S., Wilairatana, P., McGready, R., Villegas, L., Gathmann, I., Mull, R., Brockman, A., White, N.J. and Nosten, F. (2000) Artemether-Lumefantrine for the Treatment of Multidrug-Resistant Falciparum Malaria, Transactions of the Royal. Society of Tropical Medicine and Hygiene, 94, 545-548. http://dx.doi.org/10.1016/S0035-9203(00)90082-8
- South East Asian Quinine Artesunate Malaria Trial (SEAQUAMAT) Group (2005) Artesunate versus Quinine for Treat- ment of Severe Falciparum Malaria: A Randomised Trial. Lancet, 366, 717-725. http://dx.doi.org/10.1016/S0140-6736(05)67176-0
- Panusa, A., Multari, G., Incarnato, G. and Gagliardi, L. (2007) High-Performance Liquid Chromatography Analysis of Anti- Inflammatory Pharmaceuticals with Ultraviolet and Electrospray-Mass Spectrometry Detection in Suspected Counter- feit Homeopathic Medicinal Products. Journal of Pharmaceutical and Biomedical Analysis, 43, 1221-1227. http://dx.doi.org/10.1016/j.jpba.2006.10.012
- Newton, P.N., Lee Sue, J., Goodman, C., Fernández Facundo, M., Yeung, S., Phanouvong, S., Kaur, H., Amin, A.A., Whitty, C.J.M., Kokwaro, G.O., Lindegårdh, N., Lukulay, P., White, L.J., Day, N.P.J., Green, M.D. and White, N.J. (2009) Guidelines for Field Surveys of the Quality of Medicines: A Proposal. PloS Medicine, 6, 0252-0257.
- Mazière, M. (2007) Pharmaceutiques, Dossier Contrefaçon. PR Editions, Tour Ariane 5-7 place de la Pyramide 92080 La Défense, Cedex, France, 68-73.
- Newton, P.N., Dondorp, A., Green, M., Mayxay, M. and White, N.J. (2003) Counterfeit Artesunate Antimalarials in Southeast Asia. The Lancet, 362, 169. http://dx.doi.org/10.1016/S0140-6736(03)13872-X
- Caporal-Gauthier, J., Nivet, J.M., Algranti, P., Guilloteau, M., Histe, M., Lallier, M., N’guyen-Huu, J.J. and Russoto, R. (1992) Guide de validation analytique, Rapport d’une commission SFSTP, I Méthodologie. STP Pharma Pratiques, 2, 205-226.
- Notes explicative III/844/87-FR (1989) Commission des Communautés Européennes. Groupe de travail du Comité des spécialités pharmaceutiques, Bruxelles-Luxembourg, 8.
- Text on Validation of Analytical Procedures: Definitions and Terminology (Q2A) (2005) Tripartite International Conference on Harmonization (ICH) Text. ICH Technical Coordination, Londres.
- Text on Validation of Analytical Procedures: Methodology (Q2B) (2005) Tripartite International Conference on Harmonization (ICH) Text. ICH Technical Coordination, Londres.
- Vessman, J. (1996) Selectivity or Specificity? Validation of Analytical Methods from the Perspective of an Analytical Chemist in the Pharmaceutical Industry. Journal of Pharmaceutical and Biomedical Analysis, 14, 867-869. http://dx.doi.org/10.1016/0731-7085(95)01679-1
- Food and Drug Administration (1995) International Conference on Harmonization: Guideline on Validation of Analytical Procedures: Definitions and Terminology. Federal Register, 60, 11260-11262.
- Food and Drug Administration (1997) International Conference on Harmonization: Guideline on Validation of Analytical Procedures: Methodology. Federal Register, 62, 27463-27467.
- Chapuzet, E., Mercier, N., Bervoas-Martin, S., Boulanger, B., Chevalier, P., Chiap, P., Granjean, D., Hubert, P., La- gorce, P., Lallier, M., Laparra, M.C., Laurentie, M. and Nivet, J.C. (1997) Méthodes chromatographiques de dosage dans les milieux biologiques: Stratégie de validation. STP Pharma Pratiques, 7, 169-194.
- Hubert, Ph., Chiap, P., Crommen, J., Boulanger, B., Chapuzet, E., Mercier, N., Bervoas-Martin, S., Chevalier, P., Grandjean, D., Lagorce, P., Lallier, M., Laparra, M.C., Laurentie, M. and Nivet, J.C. (1999) The SFSTP Guide on the Validation of Chromatographic Methods for Drug Bioanalysis: From the Washington Conference to the Laboratory. Analytica Chimica Acta, 391, 135-148. http://dx.doi.org/10.1016/S0003-2670(99)00106-3
- Feinberg, M. and Raguènès, N. (1999) Development and Application of a Standardized Validation Procedure for Food Chemistry Laboratories. Analytica Chimica Acta, 391, 239-252. http://dx.doi.org/10.1016/S0003-2670(99)00120-8
- Ermer, J. (2001) Validation in Pharmaceutical Analysis. Part I: An Integrated Approach. Journal of Pharmaceutical and Biomedical Analysis, 24, 755-767. http://dx.doi.org/10.1016/S0731-7085(00)00530-6
- Hubert, P., Nguyen-Huu, J.J., Boulanger, B., Chapuzet, E., Chiap, P., Cohen, N., Compagnon, P.A., Dewé, W., Fein- berg, M., Lallier, M., Laurentie, M., Mercier, N., Muzard, G., Nivet, C. and Valat, L. (2003) Validation des procédures analytiques quantitatives: Harmonisation des démarches. STP Pharma Pratiques, 13, 101-138.
- Boulanger, B., Chiap, P., Dewé, W., Crommen, J. and Hubert, Ph. (2003) An Analysis of the SFSTP Guide on Validation of Chromatographic Bioanalytical Methods: Progresses and Limitations. Journal of Pharmaceutical and Biomedical Analysis, 32, 753-765. http://dx.doi.org/10.1016/S0731-7085(03)00182-1
- United States Pharmacopeial Convention (2009) USP Monographs, United States Pharmacopeial Convention 37, National Formulary 32, Rockville, MD, USA.
- Mee, R. (1988) Estimation of the Percentage of a Normal Distribution Lying Outside a Specified Interval. Communications in Statistics: Theory and Methods, 17, 1465-1479.
- Marini, R.D., Chiap, P., Boulanger, B., Rozet, E., Rudaz, S., Crommen, J. and Hubert, Ph. (2006) LC Method for the Determination of R-Timolol in S-Timolol Maleate: Validation of Its Ability to Quantify and Uncertainty Assessment. Talanta, 68, 1166-1175. http://dx.doi.org/10.1016/j.talanta.2005.07.026
NOTES

*These authors have equally contributed to this article.
#Corresponding author.