American Journal of Analytical Chemistry
Vol.4 No.10A(2013), Article ID:37311,10 pages DOI:10.4236/ajac.2013.410A1003
Spectral Evidence for the One-Step Three-Electron Oxidation of Phenylsufinylacetic Acid and Oxalic Acid by Cr(VI)
1Research Department of Chemistry, Aditanar College of Arts and Science, Tiruchendur, India
2Department of Chemistry, Govindammal Aditanar College for Women, Tiruchendur, India
Email: *subramaniam.perumal@gmail.com
Copyright © 2013 Perumal Subramaniam, Natesan Thamil Selvi. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 23, 2013; revised July 25, 2013; accepted August 20, 2013
Keywords: Phenylsulfinylacetic Acid; Oxalic Acid; Co-Oxidation; Nucleophilic Attack of Sulfur; Three-Electron Transfer; Substituent Effect
ABSTRACT
The co-oxidation of a mixture of phenylsulfinylacetic acid (PSAA) and oxalic acid (OxH2) by Cr(VI) in 20% acetonitrile-80% water (v/v) medium follows third order kinetics, first order, each with respect to PSAA, OxH2 and Cr(VI). The reaction involves nucleophilic attack of sulfur atom of PSAA on chromium of the oxidizing species, Cr(VI)-OxH2 to form a ternary complex, Cr(VI)-OxH2-PSAA followed by a one-step three-electron reduction of Cr(VI) to Cr(III) and simultaneous oxidation of both the substrates. The reaction is catalysed by Mn2+ ion while retarded by Al3+ ion. Electron releasing substituents in the metaand para-positions of the phenyl ring of PSAA enhance the rate of co-oxidation while electron withdrawing substituents retards the reaction. The Hammett plots at different temperatures exhibit excellent correlation with negative ρ values. The reaction series obey isokinetic relationship and the observed isokinetic temperature is lying below the experimental range of temperature.
1. Introduction
Oxalic acid acts as an efficient catalyst and enhances the rate of Cr(VI) oxidation of many inorganic [1-3] and organic compounds [4-10]. The oxalic acid catalysed processes involve the formation of Cr(IV)/Cr(V) intermediate species depending on the substrate, nature of Cr(VI) oxidizing reagent and experimental conditions. However, in the Cr(VI) co-oxidation of alcohol and oxalic acid, Rocek and co-workers [11] observed a direct reduction of Cr(VI) to Cr(III) without the formation of these intermediate species of chromium. In co-oxidation, oxalic acid undergoes oxidation in a stoichiometric ratio yielding CO2 along with the other reductants with tremendous rate acceleration. Such type of rate enhancement with oxalic acid by Cr(VI) is observed with diverse substrates like formic acid [12], aromatic anils [13], anilides [14], cycloalkanones [15] and cinnamic acids [16].
Since the co-oxidation process offers the possibility of several synthetic applications, Cr(VI) co-oxidation studies are of striking significance. Srinivasan et al. [17] reported the co-oxidation of phenylmethyl sulfoxides and oxalic acid with Cr(VI) and designed the mechanism for the reaction which also highlights the substituent effect. Das et al. [18] monitored the micellar effect on the cooxidation of dimethyl sulfoxide and oxalic acid by Cr(VI) considering it as a probe for three-electron transfer in a single step. In the case of co-oxidation of S-phenylmercaptoacetic acid and oxalic acid with pyridinium chlorochromate [19], two different mechanisms, based on substrate concentration, have been suggested. The significant utility of phenylsulfinylacetic acid (PSAA) in various synthetic routes [20-26] prompted the present investtigation. Detailed literature scanning also reveals that there is no systematic kinetic report on PSAA and so, the present study of Cr(VI) co-oxidation of PSAA with oxalic acid (OxH2) is undertaken and its salient features along with the substituent effects are discussed .
2. Experimental
The substrate, phenylsulfinylacetic acid and several metaand para-substituted PSAAs were synthesized from the corresponding phenylmercaptoacetic acids by controlled oxidation with H2O2 [27], recrystallised from suitable solvents and their melting points were checked with the literature values [28]. The purity was further verified by LCMS. Potassium dichromate (BDH), sodium perchlorate (BDH), perchloric acid (Merck), oxalic acid (Merck), aluminium nitrate (Merck) and all other reagents were of AnalaR grade and they were used as such for the preparation of solutions of desired concentration for the kinetic study using double distilled water. Literature procedures were adopted for the purification of solvents, acetonitrile and water.
2.1. Kinetic Measurements
The kinetic study was performed under pseudo-first order conditions by maintaining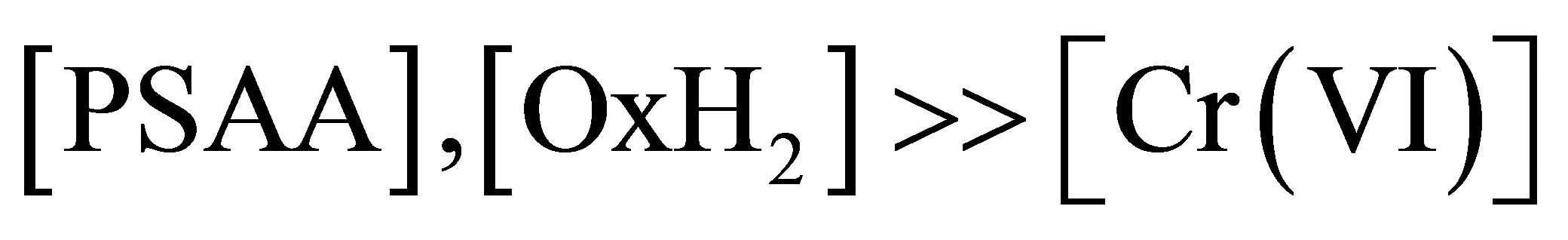 , in 20% acetonitrile-80% water mixture (v/v) and the progress of the reaction was observed by monitoring the decay of Cr(VI) at 351 nm at different time intervals with an ELICO Double Beam UV-vis Bio-spectrophotometer with an inbuilt thermostat. Figure 1 shows the spectral changes recorded during the reaction.
, in 20% acetonitrile-80% water mixture (v/v) and the progress of the reaction was observed by monitoring the decay of Cr(VI) at 351 nm at different time intervals with an ELICO Double Beam UV-vis Bio-spectrophotometer with an inbuilt thermostat. Figure 1 shows the spectral changes recorded during the reaction.
The pseudo-first order rate constant (k1) for each kinetic run was evaluated from the slope of the linear plot of log OD vs time by the method of least squares. The second order rate constant (k2) was calculated from the relation: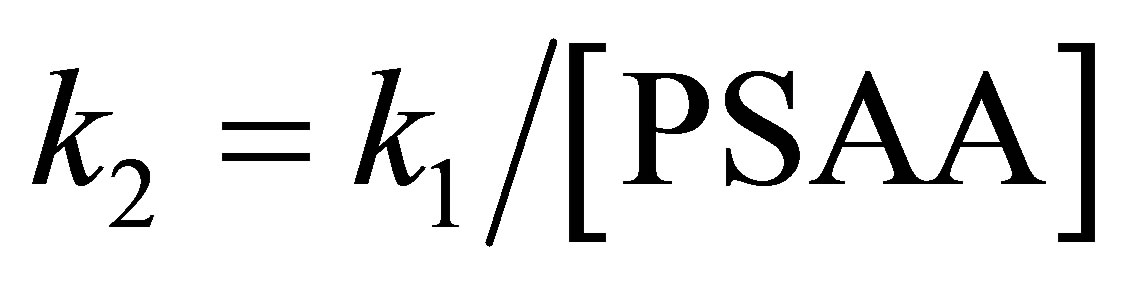 . The precision of the k values is given in terms of 95% confidence limits of Students’ t test.
. The precision of the k values is given in terms of 95% confidence limits of Students’ t test.
2.2. Product Analysis
To characterize the products of the reaction, the reaction mixture containing excess of Cr(VI) over PSAA and OxH2 was kept for 48 hours for completion of the reaction and then extracted with ether. The ether extract was
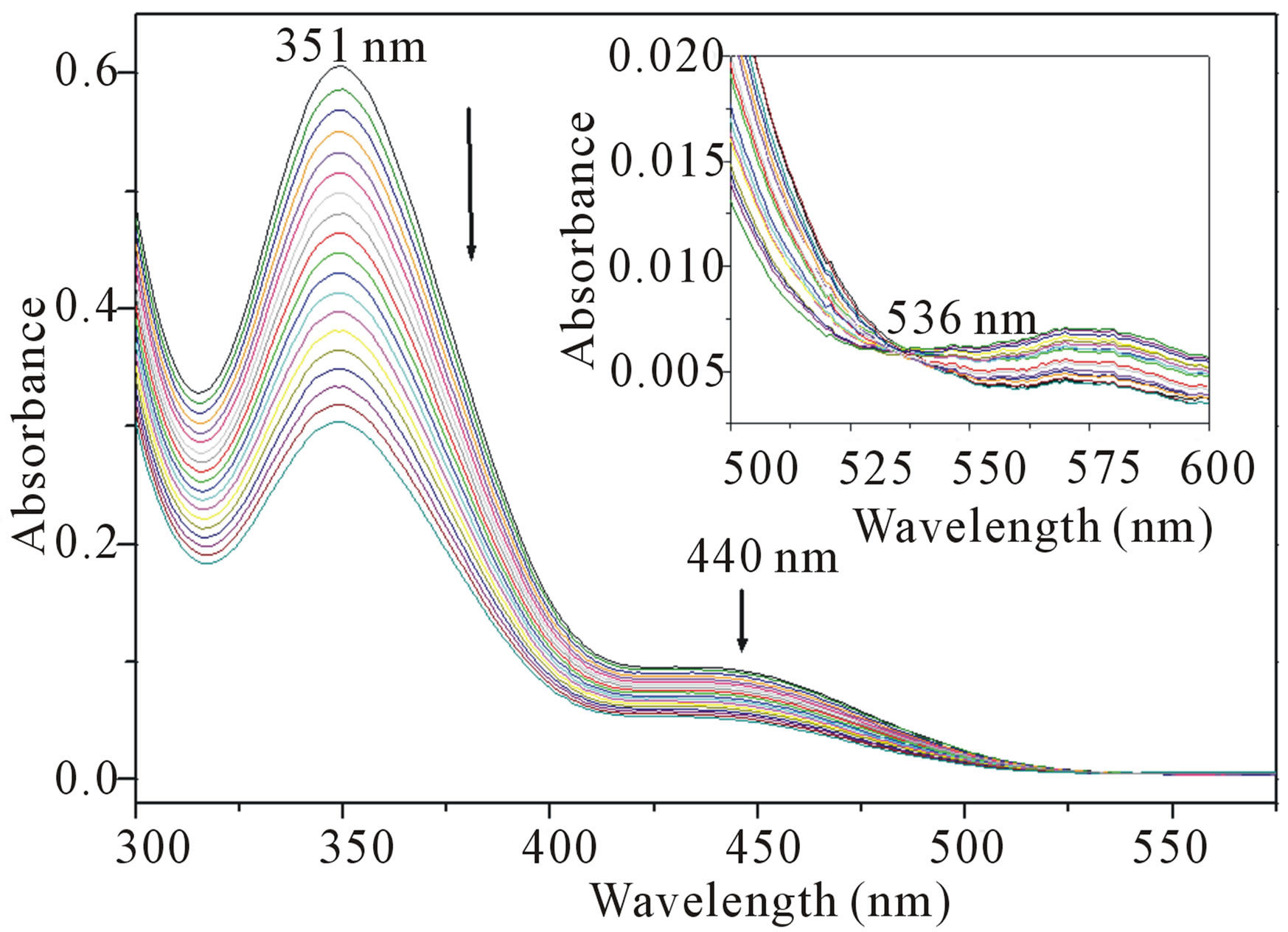
Figure 1. UV-visible spectra at different time intervals for the reaction of Cr(VI) with PSAA and OxH2 in 20% acetonitrile-80% H2O (v/v) [PSAA] = 3.0 × 10−2 mol∙dm−3; [OxH2] = 1.0 × 10−2 mol∙dm−3; [Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [H+] = 7.5 × 10−1 mol∙dm−3.
collected, dried over anhydrous sodium sulfate and the solvent was removed by evaporation. The residue obtained from the ether extract was dried and subjected to GC-MS (Figure 2) and LC-MS (Supplementary Material: Figure F1) analysis. The parent peak at 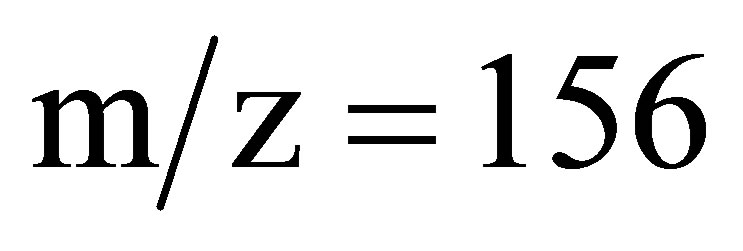 in GC-MS and the peak eluted in LC-MS at a retention time of 1.87 min ionizes in APCI (+) mode with an area of 86% corresponding to the mass 157 indicate that methylphenyl sulfone is the sole product formed. The presence of strong IR bands (Supplementary Material: Figure F2) at 1148 cm−1 and 1290 cm−1 characteristic of symmetric and asymmetric stretching respectively of >SO2 group further supports the formation of the product, methylphenyl sulfone.
in GC-MS and the peak eluted in LC-MS at a retention time of 1.87 min ionizes in APCI (+) mode with an area of 86% corresponding to the mass 157 indicate that methylphenyl sulfone is the sole product formed. The presence of strong IR bands (Supplementary Material: Figure F2) at 1148 cm−1 and 1290 cm−1 characteristic of symmetric and asymmetric stretching respectively of >SO2 group further supports the formation of the product, methylphenyl sulfone.
The final fate of chromium (VI) can be predicted from the UV-visible spectroscopy by scanning the aqueous extract of the product mixture and comparing the spectral changes observed with that of authentic sample of Cr(III). The two UV-vis peaks of Cr(III) at 421 nm and 592 nm (Figure 3(a)) which were assigned to its octahedral transitions, 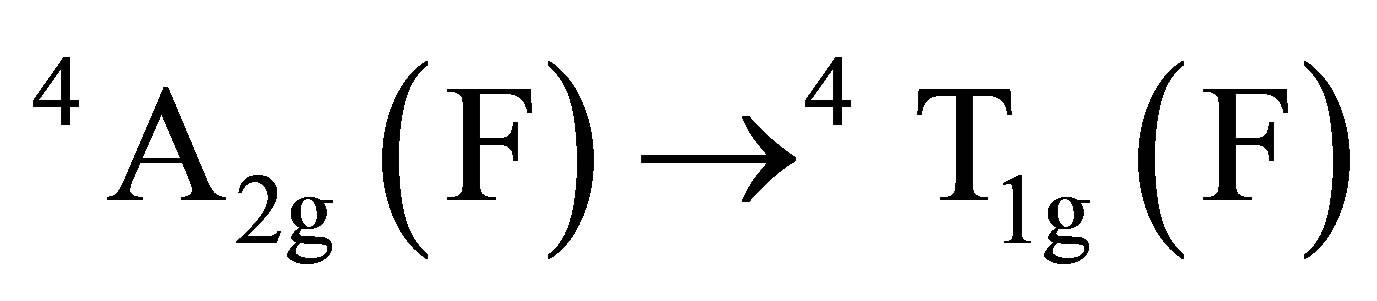 and
and 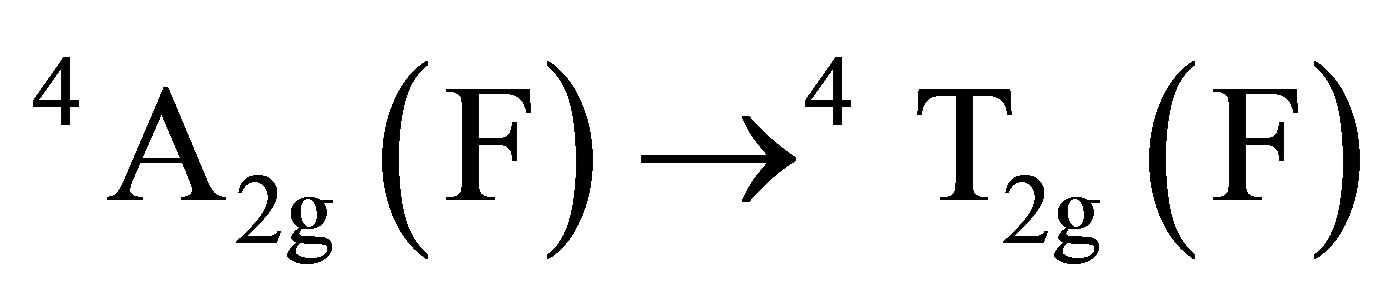 [29,30] were found to shift to lower wavelength regions, 414 nm and 562 nm for the product mixture (Figure 3(b)). This observation clearly indicates the existence of chromium as Cr(III) in the form of complex species probably with oxalic acid as observed by other researchers [31,32] and not exist as free ion.
[29,30] were found to shift to lower wavelength regions, 414 nm and 562 nm for the product mixture (Figure 3(b)). This observation clearly indicates the existence of chromium as Cr(III) in the form of complex species probably with oxalic acid as observed by other researchers [31,32] and not exist as free ion.
3. Results and Discussion
The order of the reaction with respect to Cr(VI) is unity under pseudo-first order conditions of
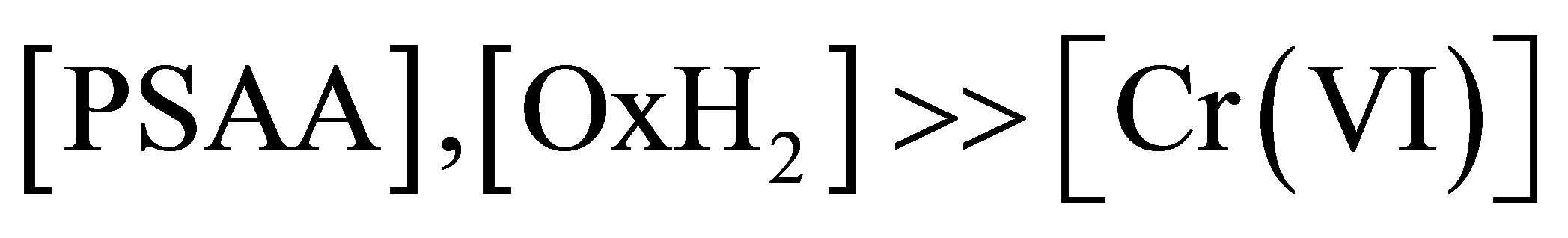
which is clear from the linear plots of logOD vs time even upto 75% completion of reaction and constant pseudofirst order rate constant values. However, it is observed that in the lower regions of [Cr(VI)], the pseudo-first order rate constant increases with decrease in [Cr(VI)]
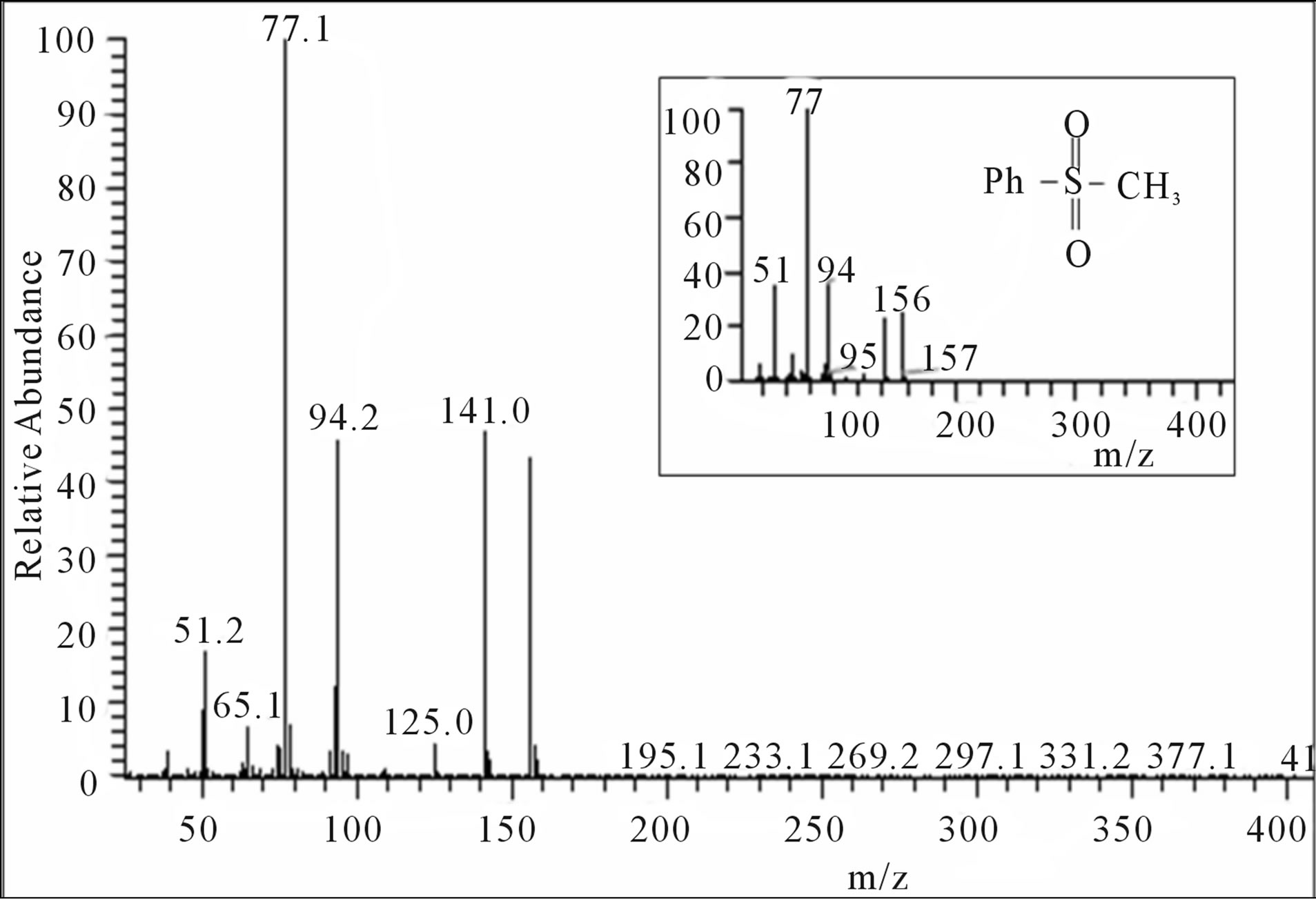
Figure 2. GC-MS spectrogram of the product.
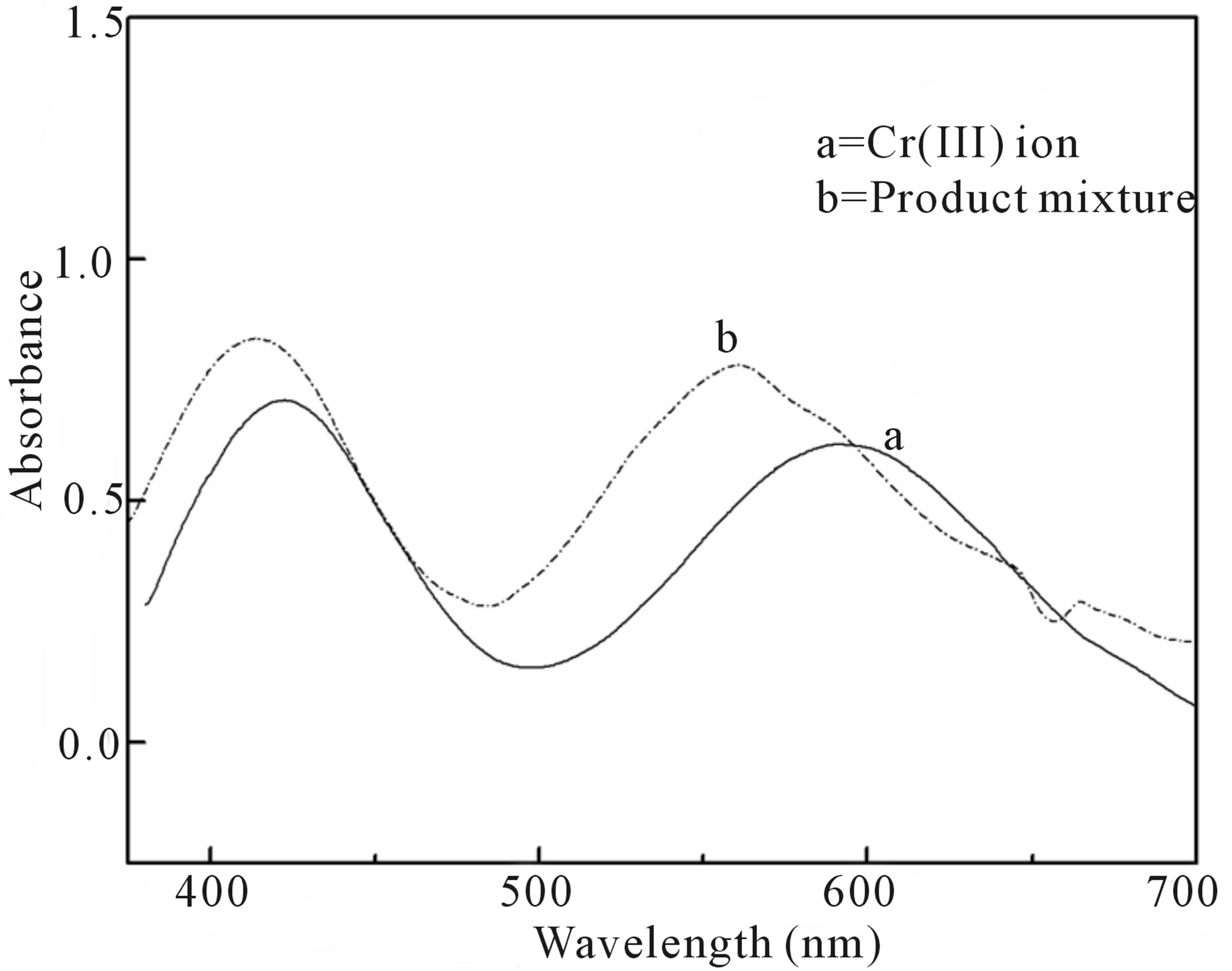
Figure 3. UV-vis spectra of the product mixture.
(Table 1). Such type of decrease in rate with increase in [Cr(VI)] has been observed by several researchers [33- 36]. However, the rate constants remain constant at higher concentration range of Cr(VI). The decrease in rate with increase in Cr(VI) in lower concentration region may be due to the dimerization of Cr(VI) followed by decrease in concentration of active species [33-35] or due to the existence of some sort of weak association between monomeric Cr(VI) species [36].
The reaction is first order each in PSAA and oxalic acid, which are evidenced from the constant values obtained by dividing k1 by [PSAA] or [OxH2] and also from the unit slope of their respective log-log plots of k1 vs [PSAA] and [OxH2]. The co-oxidation rate increases linearly with increase in [H+] (Supplementary material: Table T1) and shows a first order dependence with respect to [H+]. This shows the protonation of Cr(VI) species in the experimental conditions which enhances the electrophilic activity of the oxidant. Solvent variation of the medium from 10% to 80% acetonitrile shows an accelerating effect with decrease in dielectric constant of the medium (Supplementary material: Table T1).
The effect of Mn2+ on rate (Supplementary material: Table T2) indicates that the rate of co-oxidation increases linearly with increase in [Mn2+] which is contrary to the retardation effect of Mn2+ on Cr(VI) oxidation due to the removal of Cr(IV) intermediate by Mn2+. The observed acceleration effect not only excludes the involvement of Cr(IV) intermediate in this reaction but also indicates that Mn2+ acts as a catalyst. Such type of Mn2+ catalysis during co-oxidation process is witnessed by several authors [14,15,17]. Suppression of rate by the addition of aluminium nitrate (Supplementary material: Table T2) may be visualized due to the removal of OxH2 by Al3+.
3.1. Substituent Effect
The effect of substituents on the rate of co-oxidation is examined with several metaand para-substituted PSAAs at three different temperatures viz., 15˚C, 20˚C and 30˚C. The second order rate constants and the activation parameters calculated from the Eyring’s plot are enumerated in Table 2. To have a better insight into the contribution of substituent effect on the rate and mechanism, the rate constants were correlated with Hammett σ constants and found that an excellent correlation is obtained with negative ρ values (Supplementary material: Figure F3).
An analysis of the data in respect of linear free energy relationships reveals that the reaction series obeys isokinetic relationship as per the equation,
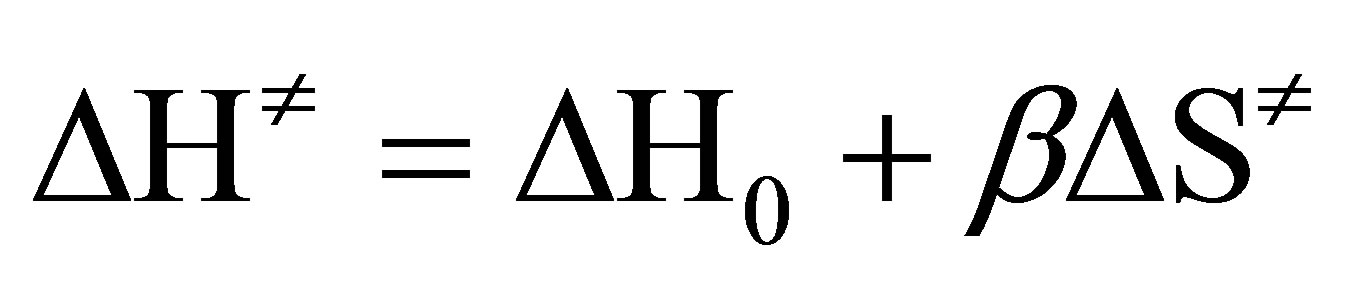
where, β is the isokinetic temperature, at which all the substituents in a given series have the same reactivity. Though the Petersen’s [37] error criteria (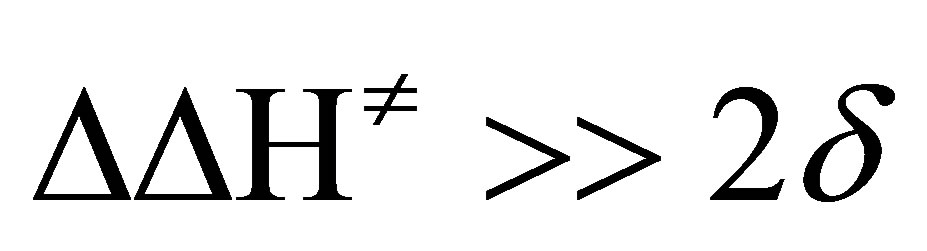 ; δ is the maximum possible error in ∆H≠) is satisfied, the plot of ∆H≠ vs ∆S≠ gives only a bad correlation (r = 0.861). However, Exner’s plot [38] of logk2 at 35˚C against logk2 at 20˚C correlate excellently (r = 0.998) and the slope of the plot (b) affords the isokinetic temperature, β.
; δ is the maximum possible error in ∆H≠) is satisfied, the plot of ∆H≠ vs ∆S≠ gives only a bad correlation (r = 0.861). However, Exner’s plot [38] of logk2 at 35˚C against logk2 at 20˚C correlate excellently (r = 0.998) and the slope of the plot (b) affords the isokinetic temperature, β.
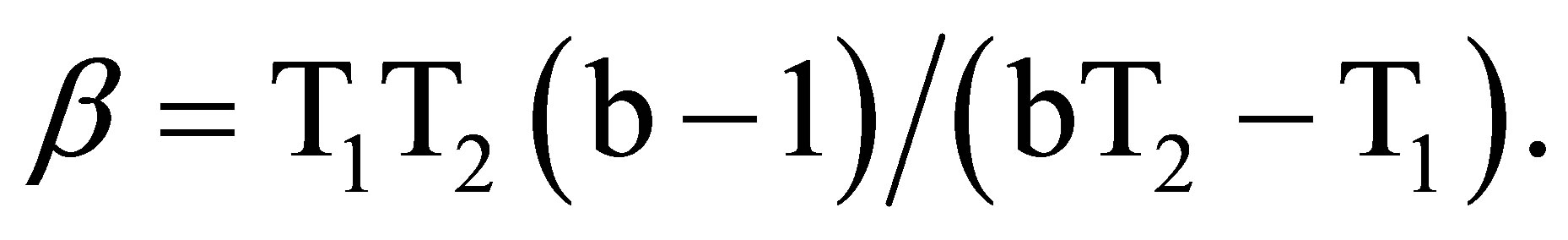
The isokinetic temperature computed from the above equation, 169.5 K is below the experimental temperature showing that the reaction is of entropy controlled. The linearity of Hammett plot and Exner’s plot, evidence the involvement of same mechanism in all substituted PSAAs.
3.2. Mechanism
The existence of active Cr(VI) species in the reaction mixture depends mostly on the concentration of H+ and Cr(VI). In aqueous acidic solution, the various possible forms of Cr(VI) species are H2CrO4, 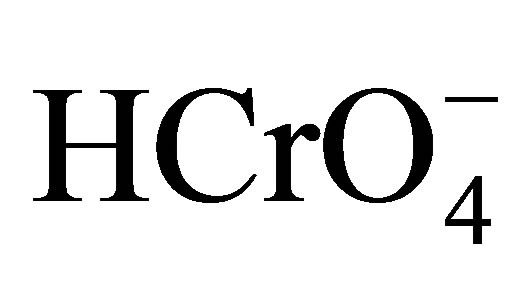 and
and 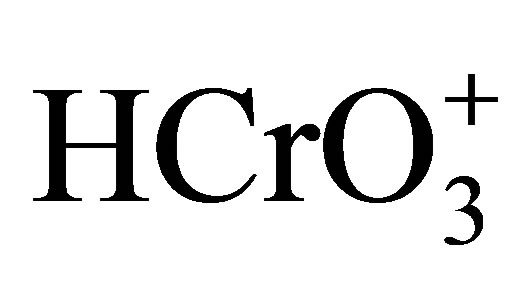 [39,40]. The species,
[39,40]. The species, 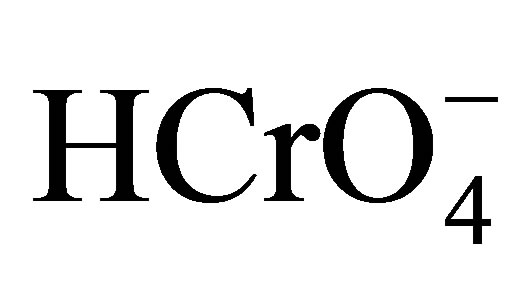 exists only at very low concentration of H+ [41] and hence under the maintained experimental conditions of H+ the participation of
exists only at very low concentration of H+ [41] and hence under the maintained experimental conditions of H+ the participation of 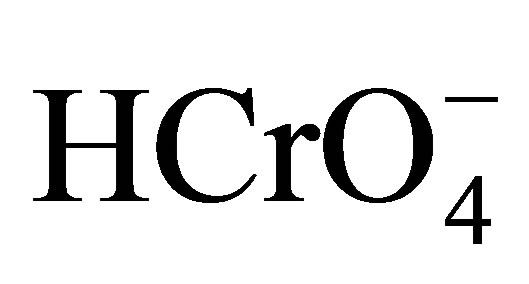 is excluded. At higher concentrations of H+, the existence of Cr(VI) in the form
is excluded. At higher concentrations of H+, the existence of Cr(VI) in the form 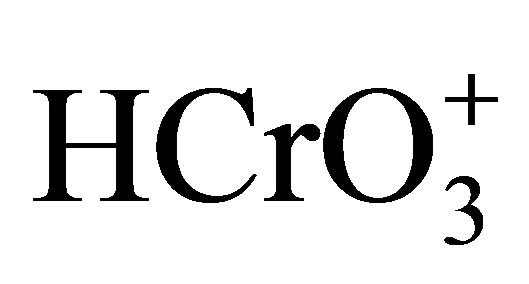 has been proved by several researchers [35,42,43] and on this basis its participation in the present co-oxidation process is envisaged. The positive slope obtained from the linear plot of logk1 vs 1/D along with first order dependence on [H+] supplement the involvement of a positively charged Cr(VI) species in the rate determining step.
has been proved by several researchers [35,42,43] and on this basis its participation in the present co-oxidation process is envisaged. The positive slope obtained from the linear plot of logk1 vs 1/D along with first order dependence on [H+] supplement the involvement of a positively charged Cr(VI) species in the rate determining step.
The kinetic results revealed that the co-oxidation of phenylsulfinylacetic acid and oxalic acid by Cr(VI) follows the rate law,

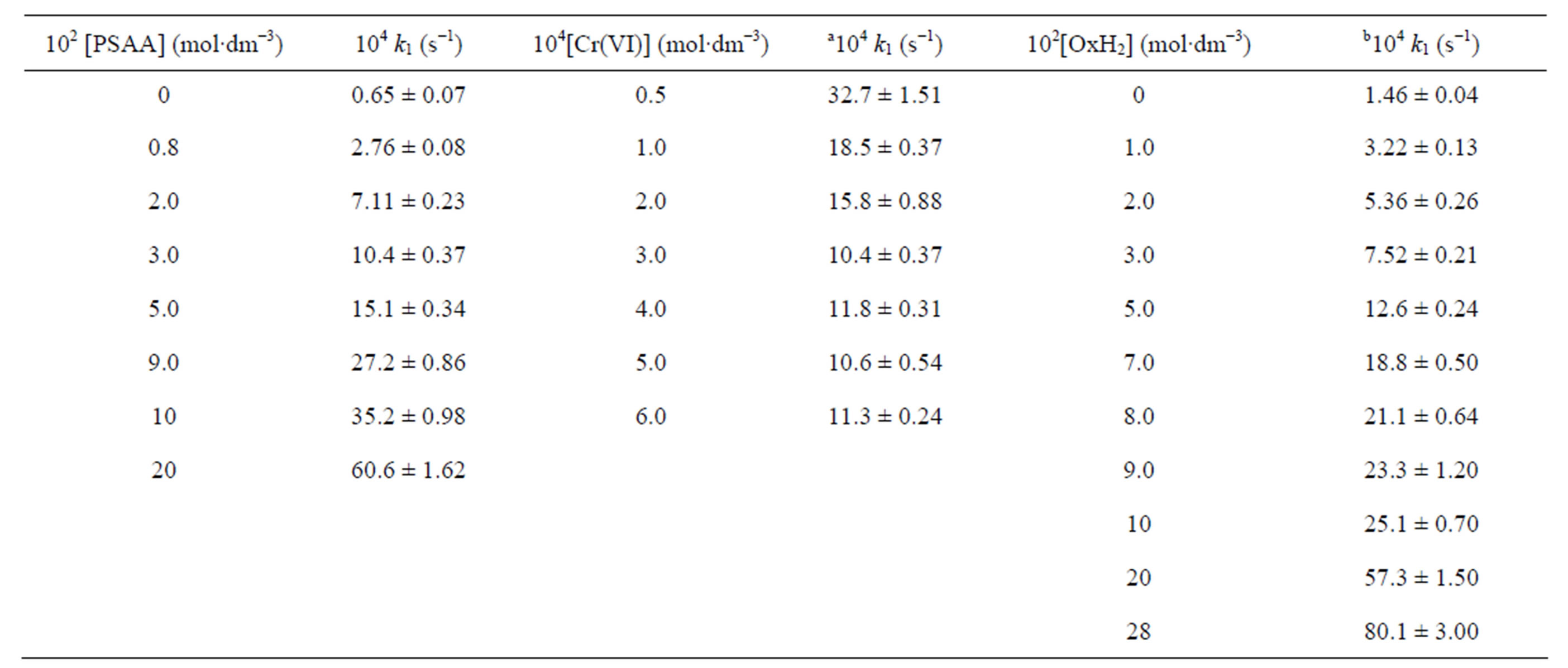
Table 1. Rate constants for the co-oxidation of PSAA and OxH2 by Cr(VI).
[Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [OxH2] = 1.0 × 10−2 mol∙dm−3; [H+] = 0.75 mol∙dm−3; I = 0.8 mol∙dm−3; solvent = 20% CH3CN-80% H2O (v/v); a[PSAA] = 3.0 × 10−2 mol∙dm−3; b[PSAA] = 1.0 × 10−2 mol∙dm−3.
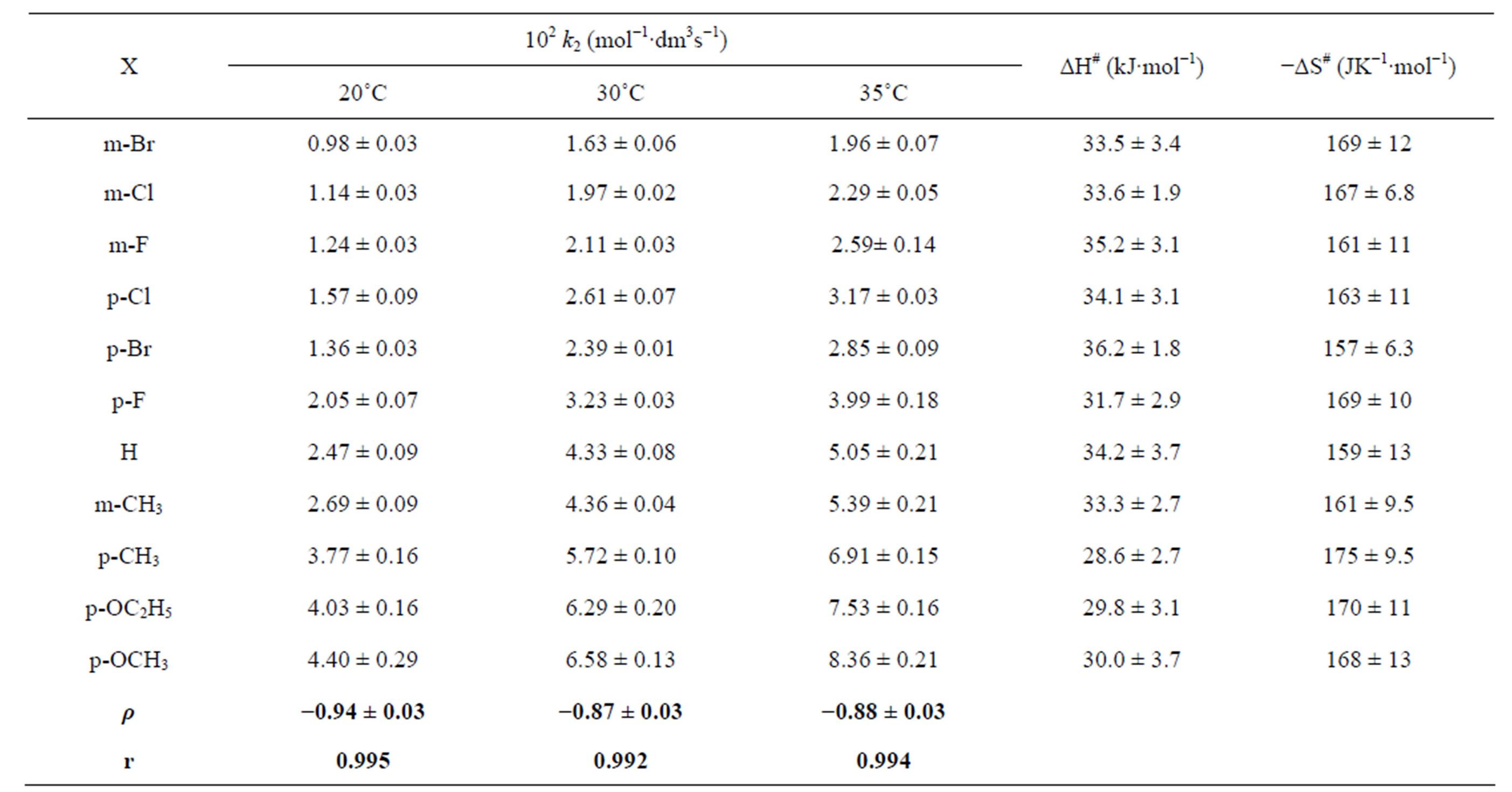
Table 2. Second-order rate constants, enthalpy and entropy of activations for the co-oxidation of X-C6H4SOCH2COOH and OxH2 by Cr(VI).
[X-C6H4SOCH2COOH] = 3.0 × 10−2 mol∙dm−3; [OxH2] = 0.01 mol∙dm−3; [Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [H+] = 0.75 mol∙dm−3; I = 0.75 mol∙dm−3; solvent = 40% CH3CN-60% H2O ( v/v).
It has been reported that in the well-documented Cr(VI) oxidation of several substrates in the presence of oxalic acid, the cyclic form of an intermediate formed between Cr(VI) and oxalic acid is assumed to be the oxidizing species. Such complex formation between the oxidant and oxalic acid is indispensable for co-oxidation to occur [17]. In the present case too, addition of oxalic acid to Cr(VI) leads to a substantial hyperchromic shift at 263 nm and 351 nm in the UV-vis spectra of Cr(VI) (Figure 4(b)) which indicates the involvement of Cr(VI) in complex formation. Based on the above facts, it has been proposed that  forms a cyclic complex (C1, Scheme 1) with oxalic acid in the first step of the mechanism. The rate acceleration is almost cancelled by the addition of Al3+ ion which is a direct evidence for the difficulty in the formation of Cr(VI)-OxH2 species as Al3+ removes OxH2. Further, broadening of the peak at 263 nm which merges with the peak at 351 nm (Figure 4(d)) is perceived during the addition of Al3+ ion to the reaction mixture.
forms a cyclic complex (C1, Scheme 1) with oxalic acid in the first step of the mechanism. The rate acceleration is almost cancelled by the addition of Al3+ ion which is a direct evidence for the difficulty in the formation of Cr(VI)-OxH2 species as Al3+ removes OxH2. Further, broadening of the peak at 263 nm which merges with the peak at 351 nm (Figure 4(d)) is perceived during the addition of Al3+ ion to the reaction mixture.
In order to comprehend the subsequent steps in the mechanism of co-oxidation and the binding nature of PSAA with the oxidizing species, the UV-vis spectrum was recorded for the reaction mixture. The change in the shape as well as broadening of the peak at 263 nm in the presence of PSAA (Figure 4(c)) clearly reveals the formation of a new species probably termolecular intermediate complex, C2. The formation of this intermediate, (Cr(VI)-OxH2-PSAA) as a result of nucleophilic attack of sulfur of PSAA on chromium atom of the Cr(VI)- OxH2 complex is the rate determining step. The importance of this intermediate, C2 during co-oxidation is known from the significant rate acceleration observed when both PSAA and OxH2 co-exists than their individual rate constants with Cr(VI) under the same experimental conditions. The high negative entropy of activation for this co-oxidation system (−158.88 ± 12.8 JK−1∙mol−1) when compared to that of the reaction without oxalic acid (−24.49 ± 0.09 JK−1∙mol−1) suggests that there exists a highly ordered transition state in the rate determining step.
As a result of electron transfer from sulfur of PSAA to chromium, a positive charge is developed on the sulfur center of the complex C2. The negative ρ value observed in the substituent effect study also confirms the existence
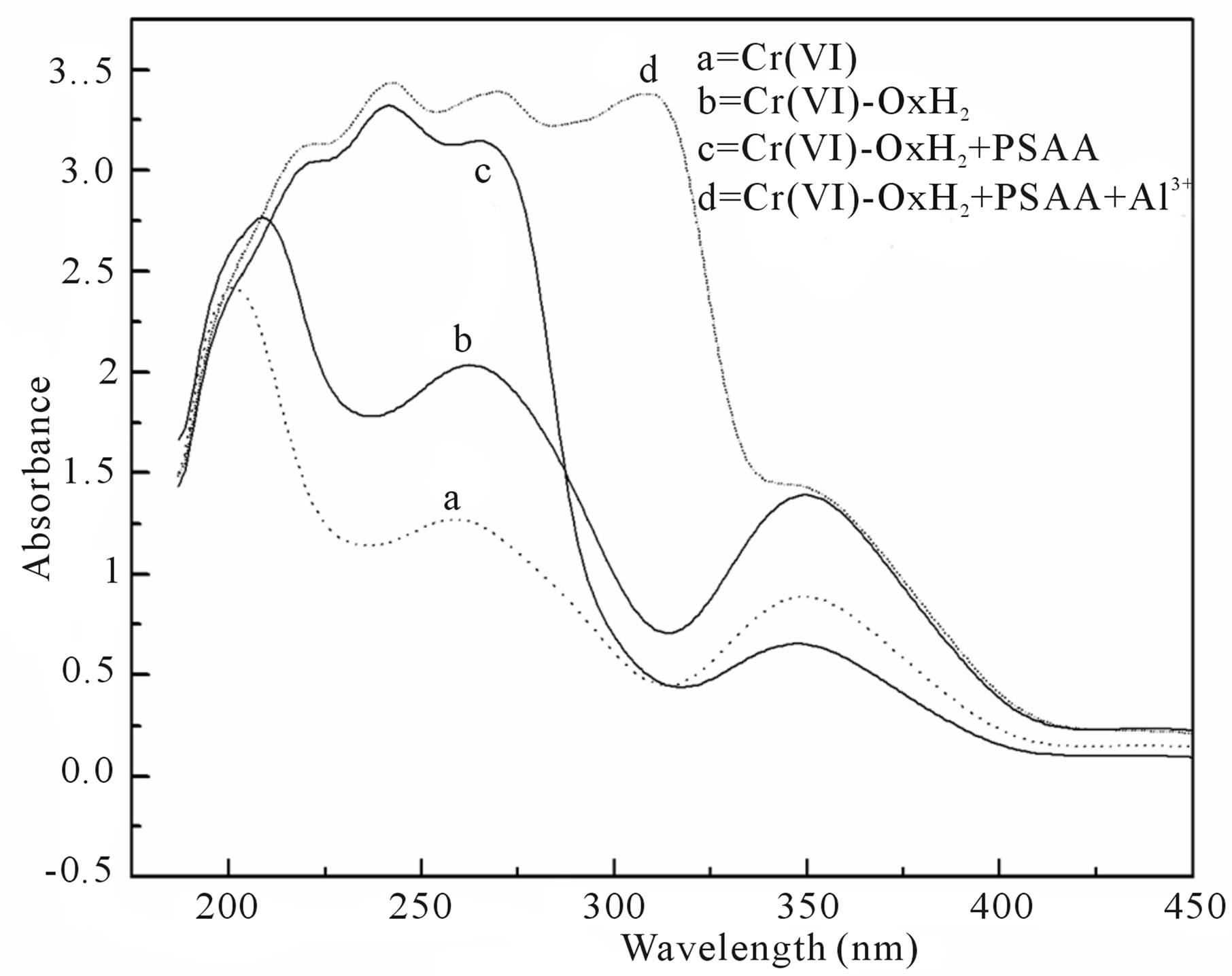
Figure 4. UV-vis spectra of (a) Cr(VI) alone (b) Cr(VI) and OxH2; (c) Cr(VI), OxH2 and PSAA and (d) Cr(VI), OxH2, PSAA and Al3+ [Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [PSAA] = 3.0 × 10 −2 mol∙dm−3; [OxH2] =1.0 × 10−2 mol∙dm−3; [HClO4] = 7.5 × 10−1 mol∙dm−3; solvent = 20% CH3CN-80% H2O (v/v).
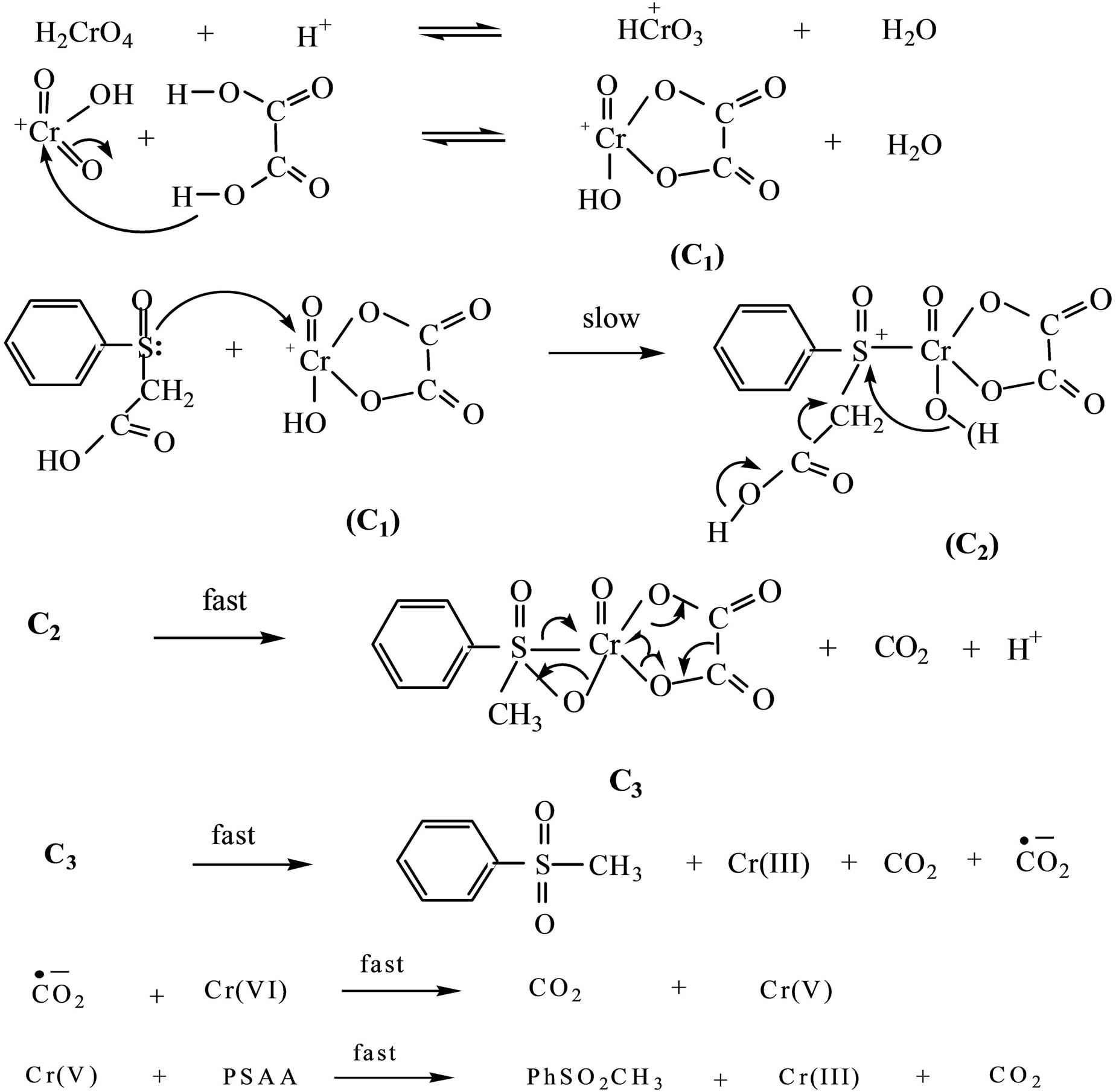
Scheme 1. Co-oxidation of PSAA and oxalic acid by Cr(VI).
of sulfonium ion intermediate in the rate determining step. The presence of electron releasing groups in the PSAA increase the electron density on sulfur atom of PSAA which makes PSAA a more efficient nucleophile for binding with the positively charged oxidizing species, Cr(VI)-OxH2. Besides, electron releasing substituents stabilize the intermediate, C2 while electron withdrawing substituents in PSAA destabilize it. The rapid oxidation of PSAA-oxalic acid mixture by Cr(VI) supports that the reduction of Cr(VI) through a termolecular complex must be much more favourable and hence faster than their individual oxidation reactions.
The intermediate, C2 then undergoes several fast steps including ligand coupling and decarboxylation from PSAA moiety leading to the formation of the products. During this process, Cr(VI) is directly reduced to Cr(III) by a one-step three-electron transfer which involves a two electron transfer from PSAA and a one electron transfer from oxalic acid. The direct evidence for the one-step three electron transfer, i.e., reduction of Cr(VI) to Cr(III) arises from the existence of isobestic point at λmax 540 nm (Figure 1) in the overlay of the reaction mixture.
The involvement of OxH2 as co-reductant and not as a catalyst in the reaction can be arrived from the amount of CO2 liberated from the reaction. The number of moles of CO2 liberated in the reactions with and without oxalic acid is determined quantitatively by the method described by Crossno et al. [44]. In the present reaction it has been observed that the amount of CO2 liberated is more than one mole and this shows that CO2 is liberated from oxalic acid too besides PSAA. The  thus formed may then undergo further rapid oxidation with Cr(VI) producing CO2 and Cr(V). Then, Cr(V) reacts with another molecule of PSAA forming products.
thus formed may then undergo further rapid oxidation with Cr(VI) producing CO2 and Cr(V). Then, Cr(V) reacts with another molecule of PSAA forming products.
4. Conclusion
The co-oxidation of phenylsulfinylacetic acid and oxalic acid by Cr(VI) follows three electron transfer mechanisms. The reduction of Cr(VI) to Cr(III) in a concerted step is assumed to be effective by the active oxidising species, Cr(VI)-OxH2 via a termolecular complex, Cr(VI)-OxH2- PSAA. The product analysis is also in support of the proposed mechanism.
REFERENCES
- G. F. Vandegrift and J. Rocek, “Catalysis in Oxidation Reactions. II. The Oxalic Acid Catalyzed Oxidation of Iodide,” Journal of American Chemical Society, Vol. 98, No. 6, 1976, pp. 1371-1376. http://dx.doi.org/10.1021/ja00422a015
- R. F. Hintze and J. Rocek, “Catalysis in Oxidation Reactions. 3. The Oxalic Acid Catalyzed Chromic Acid Oxidation of Tris(1,10-phenanthroline)iron(II),” Journal of American Chemical Society, Vol. 99, No. 1, 1977, pp. 132-137. http://dx.doi.org/10.1021/ja00443a025
- T. Satyanarayana, N. R. Anipindi, V. Subbiah and M. W. Pandit, “The Oxalic Acid Catalysed Oxidation of Bis(2,4, 6-tripyridyl-1,3,5-triazine)-iron(II) by Chromium(VI) in Acetate Buffer,” Transition Metal Chemistry, Vol. 18, No. 1, 1993, pp. 93-95. http://dx.doi.org/10.1007/BF00136060
- S. P. Meenakshisundaram, M. Gopalakrishnan, S. Nagarajan and N. Sarathi, “Oxalic Acid Catalyzed Chromium(VI) Oxidation of Some 2-Amino-4,6-diarylpyrimidines,” Catalysis Communications, Vol. 8, No. 4, 2007, pp. 713- 718. http://dx.doi.org/10.1016/j.catcom.2006.08.033
- G. Rajarajan, N. Jayachandramani, S. Manivarman, J. Jayabharathi and V. Thanikachalam, “Kinetics and Mechanism of the Oxidation of Some Substituted Aldonitrones by Quinolinium Cholorochromate in Aqueous DMF Medium in the Absence and Presence of Oxalic Acid,” Journal of the Serbian Chemical Society, Vol. 74, No. 2, 2009, pp. 171-182. http://dx.doi.org/10.2298/JSC0902171R
- D. C. Gaswick and J. H. Krueger, “Kinetics and Mechanism of the Chromium(VI)-iodide Reaction,” Journal of American Chemical Society, Vol. 91, No. 9, 1969, pp. 2240-2244. http://dx.doi.org/10.1021/ja01037a010
- I. Baldea and G. Niac, “Reaction between Chromate and Thiosulfate. II. Kinetics of Tetrathionate Formation,” Inorganic Chemistry, Vol. 9, No. 1, 1970, pp. 110-114. http://dx.doi.org/10.1021/ic50083a023
- K. A. Muirhead and G. P. Haight, “Kinetics and Mechanism of the Oxidation of Thiocyanate Ion by Chromium (VI),” Inorganic Chemistry, Vol. 12, No. 5, 1973, pp. 1116- 1120. http://dx.doi.org/10.1021/ic50123a028
- M. A. Olatunji and A. McAuly, “Metal-Ion Oxidations in Solution. Part XII. Oxidation of Thiourea and NN′-ethylenethiourea by Chromium(VI) in Perchlorate Media,” Journal of the Chemical Society, Dalton Transactions, 1975, No. 8, pp. 682-688. http://dx.doi.org/10.1039/dt9750000682
- L. F. Sala, C. Palopoli, V. Alba and S. Signorella, “A Secondary Reaction in the Oxidation of L-Methionine by Chromium(VI) in Acidic Medium,” Polyhedron, Vol. 12, No. 18, 1993, pp. 2227-2234. http://dx.doi.org/10.1016/S0277-5387(00)88261-3
- F. Hasan and J. Rocek, “Cooxidation of isopropyl alcohol and oxalic acid by chromic acid, One-step three-electron oxidation,” Journal of American Chemical Society, Vol. 94, No. 9, 1972, pp. 3181-3187.
- A. K. Das, A. Roy, B. Saha and M. Das, “Cooxidation of Formic Acid and Oxalic Acid by Chromium(VI) in Aqueous Acid Media: A Kinetic Study,” Journal of Chemical Research, No. 8, 2001, pp. 334-335.
- R. Srinivasan and A. N. M. Kasim, “Cooxidation of Aromatic Anils and Oxalic Acid by Chromic Acid in Aqueous Acetic Acid Medium,” Asian Journal of Chemistry, Vol. 21, No. 9, 2009, pp. 6909-6914.
- K. A. Basheer Ahamed, “Cooxidation of Anilides and Oxalic Acid by Chromic Acid: A One-Step, Three-Electron Oxidation,” International Journal of Chemical Kinetics, Vol. 33, No. 1, 2001, pp. 21-28. http://dx.doi.org/10.1002/1097-4601(20010101)33:1<21::AID-KIN3>3.0.CO;2-R
- S. Meenakshisundaram and R. Sockalingam, “Enhanced Reactivity in the Quinolinium Fluorochromate Cooxidation of Some Cycloalkanones and Oxalic Acid,” Bulletin of the Chemical Society of Japan, Vol. 74, No. 6, 2001, pp. 1043-1049. http://dx.doi.org/10.1246/bcsj.74.1043
- G. Vanangamudi and S.Srinivasan, “Kinetic Studies on the Oxidation of Some Para and Meta-Substituted Cinnamic Acids by Pyridinium Bromochromate in the Presence of Oxalic Acid (A Co-Oxidation Study),” E-Journal of Chemistry, Vol. 6, No. 3, 2009, pp. 920-927. http://dx.doi.org/10.1155/2009/242743
- C. Srinivasan, P. Pandarakutty Jegatheesan, S. Rajagopal and N. Arumugam, “Substituent Effects in Cooxidation: Cr(VI)-Oxalic Acid-Sulfoxides Systems,” Canadian Journal of Chemistry, Vol. 65, No. 10, 1987, pp. 2421-2424. http://dx.doi.org/10.1139/v87-403
- A. K. Das, B. Saha, and M. Islam, “Micellar Effect on the Catalytic Cooxidation of Dimethyl Sulfoxide and Oxalic Acid by Chromium(VI) in Aqueous Acid Media: A Kinetic Study,” Progress in Reaction Kinetics and Mechanism, Vol. 30, No. 3, 2005, pp. 215-226. http://dx.doi.org/10.3184/007967405779134047
- G. Mangalam and S. Meenakshisundaram, “Cooxidation of S-Phenylmercaptoacetic Acid and Oxalic Acid with Pyridinium Chlorochromate,” Polish Journal of Chemistry, Vol. 72, No. 3, 1998, pp. 582-586.
- V. Sumangala, B. Poojary, N. Chidananda, T. Arulmoli and S. Shenoy, “Synthesis, Characterization, Antimicrobial and Antioxidant Activity of Some Disubstituted [1,3, 4]-Oxadiazoles Carrying 4-(Methylsulfonyl/sulfinyl)benzyl Moieties,” Journal of Chemical and Pharmaceutical Research, Vol. 4, No. 3, 2012, pp. 1661-1669.
- N. J. Gogan, M. J. Newlands and B.-Y. Tan, “Ambident Ligands.1. Phenylsulfinylacetic Acid,” Canadian Journal of Chemistry, Vol. 50, No. 19, 1972, pp. 3202-3206. http://dx.doi.org/10.1139/v72-510
- M. Janczewski, T. Najda and T. Jablonska Pikus, “Effect of Molecular Structure on Optical Properties of Sulfoxide Systems. [Phenyland (m-bromophenyl)-sulfinyl]acetic Acids and Some of Their Derivatives. Part I,” Polish Journal of Chemistry, Vol. 56, 1982, pp. 1297-1312.
- A. A. Jaxa-Chamiec, P. G. Sammes and P. D. Kennewell, “A New Route to 5-Substituted Resorcinols and Related Systems,” Journal of the Chemical Society, Perkin Transactions 1, 1980, pp. 170-175. http://dx.doi.org/10.1039/p19800000170
- Q. B. Cass, A. A. Jaxa-Chamiec and P. G. Sammes, “Preparation and Rearrangement of Some Conjugated Phenylsulfinylacetate Derivatives,” Journal of the Chemical Society, Chemical Communications, Vol. 24, 1981, pp. 1248-1250. http://dx.doi.org/10.1039/c39810001248
- T. Allmendinger, “Ethyl Phenylsulfinyl Fluoroacetate, a New and Versatile Reagent for the Preparation of α-Fluoro-α,β-Unsaturated Carboxylic Acid Esters,” Tetrahedron, Vol. 47, No. 27, 1991, pp. 4905-4914. http://dx.doi.org/10.1016/S0040-4020(01)80956-X
- K.-S. Lee, “Tert-Butyl 3-Oxo-4-(phenylsulfinyl)-2-(triphenyl-λ5-phosphanylidene)butanoate: A New Reagent for the Efficient Synthesis of Triphenylphosphorane Ylide Precursors to Vicinal Tricarbonyls from Alkyl Halides,” Bulletin of the Korean Chemical Society, Vol. 32, No. 9, 2011, pp. 3477-3479. http://dx.doi.org/10.5012/bkcs.2011.32.9.3477
- D. Walker and J. Leib, “The Acid-Catalysed Cleavage of Phenylsulfinylacetic Acid,” Canadian Journal of Chemistry, Vol. 40, No. 7, 1962, pp. 1242-1248. http://dx.doi.org/10.1139/v62-192
- W. J .Kenney, J. A. Walsh and D. A. Davenport, “An Acid-Catalyzed Cleavage of Sulfoxides,” Journal of American Chemical Society, Vol. 83, No. 19, 1961, pp 4019- 4022. http://dx.doi.org/10.1021/ja01480a016
- A. B. P. Lever, “Inorganic Electronic Spectroscopy,” 2nd Edition, Elsevier, Amsterdam, 1984.
- S. Signorella, M. Rizzotto, V. Daier, M. I. Franscaroli, C. Palopoli, D. Martino, A. Boussekou and L. F. Sala, “Comparative Study of Oxidation by Chromium(V) and Chromium(VI),” Journal of the Chemical Society, Dalton Transactions, No. 8, 1996, pp. 1607-1611. http://dx.doi.org/10.1039/dt9960001607
- R. E Hamm, R. L. Johnson, R. H. Perkins and R. E. Davis, “Complex Ions of Chromium. VIII. Mechanism of Reaction of Organic Acid Anions with Chromium(III),” Journal of American Chemical Society, Vol. 80, No. 17, 1958, pp. 4469-4471. http://dx.doi.org/10.1021/ja01550a008
- Kabir-ud-Din, K. Hartani and Z. Khan, “Effect of Micelles on the Oxidation of Oxalic Acid by Chromium(VI) in the Presence and Absence of Manganese(II),” Colloids and Surfaces A: Physicochemical and Engineering Aspects, Vol. 193, No. 1-3, 2001, pp. 1-13. http://dx.doi.org/10.1016/S0927-7757(01)00475-7
- A. K. Das, D. Mondal, D. Kar and M. Das, “Micellar Effect on the Reaction of Picolinic Acid Catalysed Chromium(VI) Oxidation of Dimethyl Sulfoxide in Aqueous Acidic Media: A Mechanistic Study,” International Journal of Chemical Kinetics, Vol. 33, No. 3, 2001, pp. 173-181. http://dx.doi.org/10.1002/1097-4601(200103)33:3<173::AID-KIN1011>3.0.CO;2-I
- S. Meenakshisundaram and R. Vinothini, “Kinetics and Mechanism of Oxidation of Methionine by Chromium (VI): Edta Catalysis,” Croatica Chemica Acta, Vol. 76, No. 1, 2003, pp. 75-80.
- K. Pitchumani, V. Subramanian, P. P. Jegatheesan and C. Srinivasan, “Kinetics and Mechanism of Oxidation of N-Substituted Phenothiazines by Chromium(VI),” Proceedings of the Indian Academy of Sciences, Chemical Sciences, Vol. 104, No. 1, 1992, pp. 67-76.
- C. Karunakaran, S. Karuthapandian and S. Suresh, “Kinetic Evidence of a Common Mechanism in the Oxidations of Diethyl Sulfide by Dichromates and Halaochromates of Heterocyclic Bases,” International Journal of Chemical Kinetics, Vol. 35, No. 1, 2003, pp. 1-8.
- R. C. Petersen, J. H. Markgraf and S. D. Ross, “Solvent Effects in the Decomposition of 1,1’-Diphenylazoethane and 2,2’-Azobis-(2-methylpropionitrile),” Journal of American Chemical Society, Vol. 83, No. 18, 1961, pp. 3819- 3823. http://dx.doi.org/10.1021/ja01479a021
- O. Exner, “The Enthalpy-Entropy Relationship,” Progress in Physical Organic Chemistry, Vol. 10, 1973, pp. 411-482. http://dx.doi.org/10.1002/9780470171899.ch6
- K. B. Wiberg, “Oxidation in Organic Chemistry, Part A,” Academic Press, New York, 1965.
- G. Cainelli and G. Cardillo, “Chromium Oxidations in Organic Chemistry,” Springer, Berlin, 1984. http://dx.doi.org/10.1007/978-3-642-69362-5
- N. E. Brasch, D. A. Buckingham, B. A. Evans and C. R. Clark, “17O NMR Study of Chromium(VI) Ions in Water,” Journal of American Chemical Society, Vol. 118, No. 34, 1996, pp. 7969-7980. http://dx.doi.org/10.1021/ja960843j
- L. S. Levitt, “The Common Basis of Organic Oxidations in Acidic Solution,” Journal of Organic Chemistry, Vol. 20, No. 10, 1955, pp. 1297-1310. http://dx.doi.org/10.1021/jo01127a001
- C. Srinivasan, A. Chellamani and S. Rajagopal, “Mechanism of the Oxidation of Alkyl Aryl and Diphenyl Sulfides by Chromium(VI),” Journal of Organic Chemistry, Vol. 50, No. 8, 1985, pp. 1201-1205. http://dx.doi.org/10.1021/jo00208a011
- S. K. Crossno, L. H. Kalbus and G. E. Kalbus, “Determinations of Carbon Dioxide by Titration: New Experiments for General, Physical, and Quantitative Analysis Courses,” Journal of Chemical Education, Vol. 73, No. 2, 1996, pp. 175-176. http://dx.doi.org/10.1021/ed073p175
Supplementary Materials
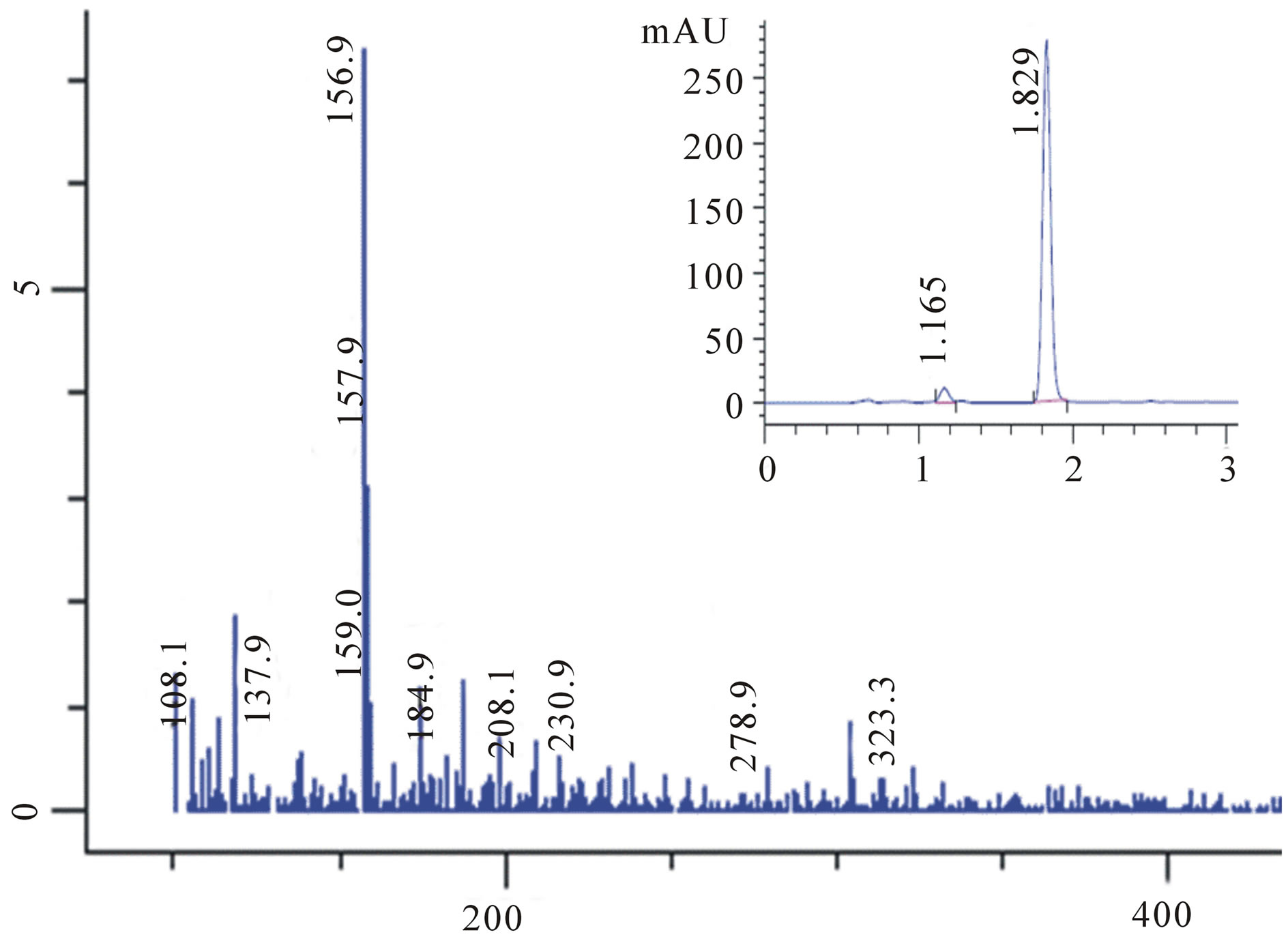
Figure F1. LC-MS analysis of the product.
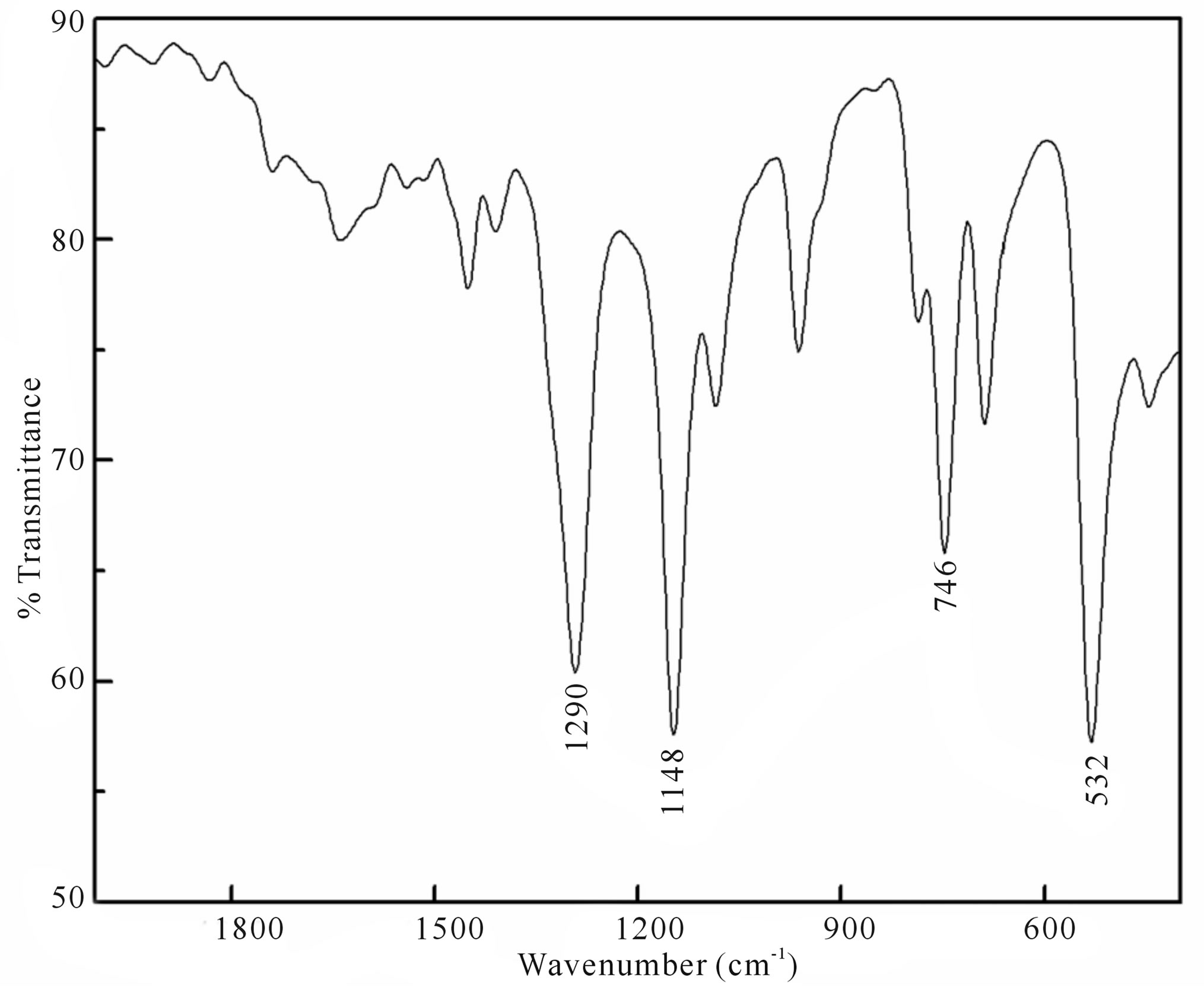
Figure F2. IR spectra of the product.
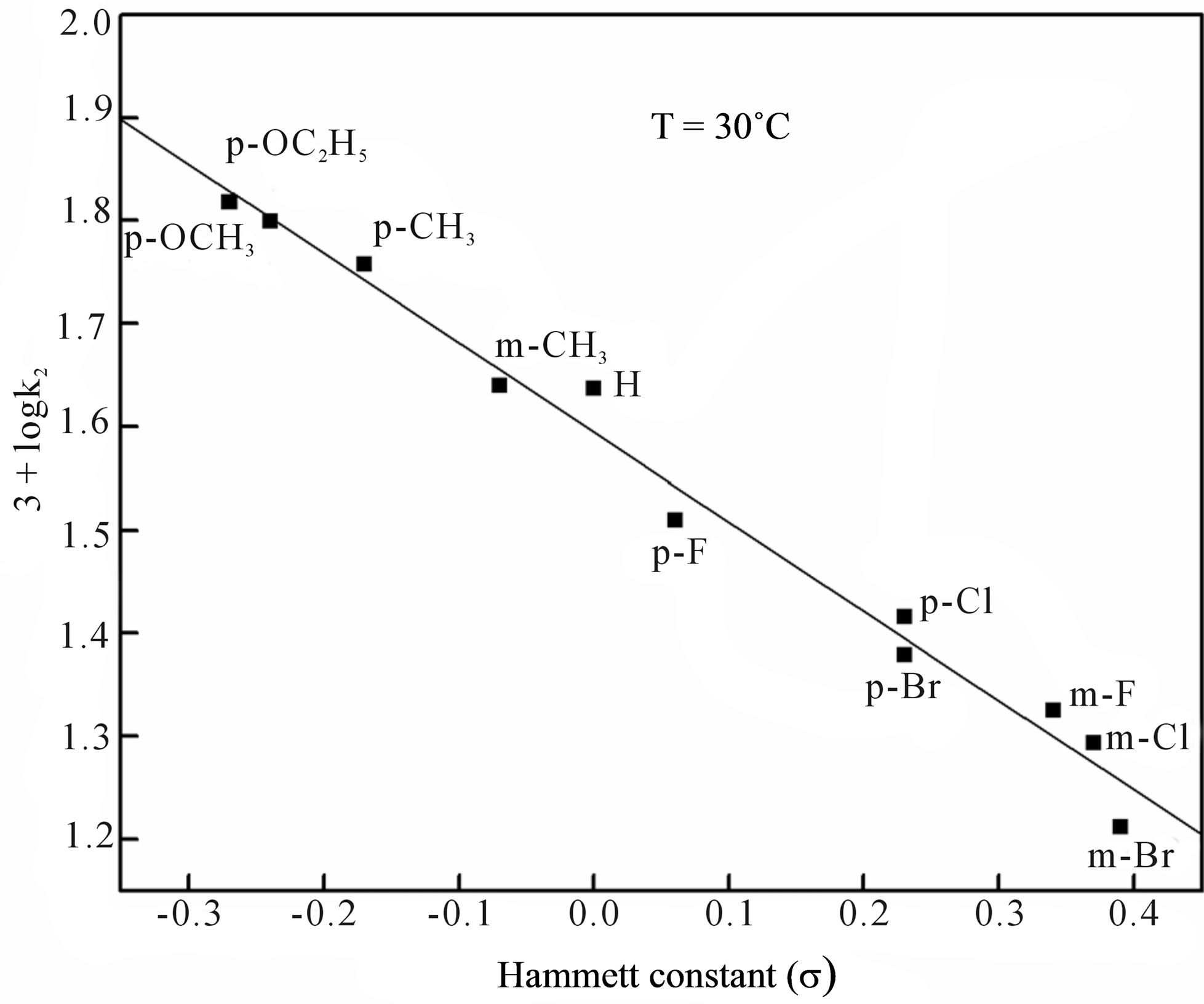
Figure F3. Hammett plot at 30˚C.
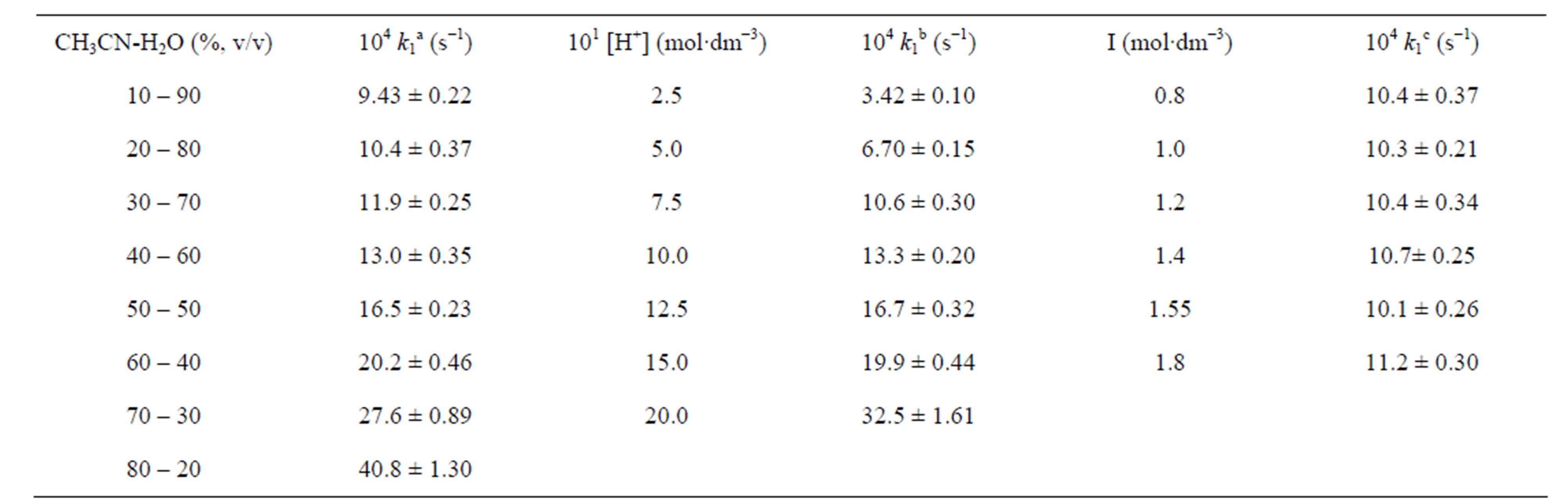
Table T1. Effect of solvent composition, [H+] and ionic strength on rate constant at 30˚C.
[Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [PSAA] =3.0 ×10−2 mol∙dm−3; a,c[H+] =0.75 mol∙dm−3; [OxH2] = 1.0 × 10−2 mol∙dm−3; aI = 0.8 mol∙dm−3; bI = 1.1 mol∙dm−3; b,csolvent = 20% CH3CN-80% H2O (v/v).
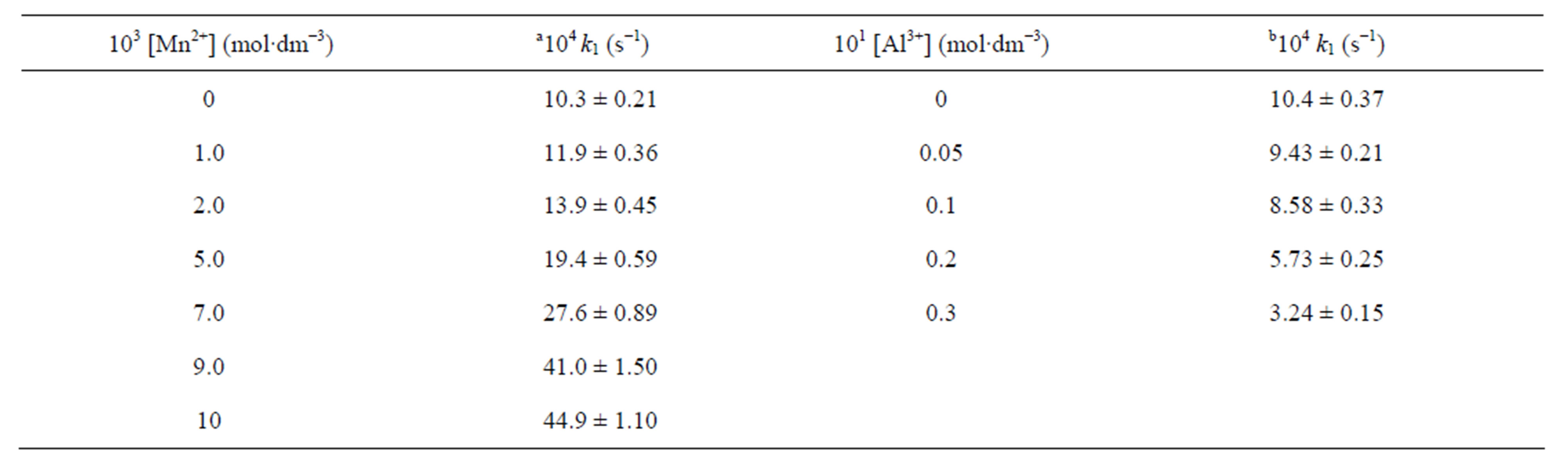
Table T2. Effect of Mn2+ ion and Al3+ ion on reaction rate.
[Cr(VI)] = 3.0 × 10−4 mol∙dm−3; [PSAA] = 3.0 × 10−2 mol∙dm−3; [OxH2] =1.0 × 10−2 mol∙dm−3; [H+] = 7.5 × 10−1 mol∙dm−3; solvent = 20% CH3CN-80% H2O (v/v); aI = 1.0 mol∙dm−3; bI = 2.55 mol∙dm−3.
NOTES
*Corresponding author.