American Journal of Analytical Chemistry
Vol.4 No.6(2013), Article ID:33425,7 pages DOI:10.4236/ajac.2013.46036
A Validated Stability-Indicating UHPLC Method for Determination of Naproxen and Its Related Compounds in Bulk Drug Samples
1Analytical Research, Product and Technology Development, Ecologic Technologies Limited, Hyderabad, India
2Department of Chemistry, Gitam Institute of Science, GITAM University, Visakhapatnam, India
Email: *tirumalwithu@gmail.com
Copyright © 2013 K. Tirumala Rao, L. Vaikunta Rao. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 25, 2013; revised April 25, 2013; accepted May 15, 2013
Keywords: Naproxen; Stability-Indicating; Related Substances; Assay; Validation; UHPLC
ABSTRACT
A simple, rapid, precise, accurate, rugged and robust stability-indicating ultra-fast high performance liquid chromatographic (UHPLC) method has been developed for the estimation of related compounds (imp-A, imp-B, imp-C, imp-D and imp-E) in Naproxen and also the assay of Naproxen from bulk drug samples. The stability indicating capability of the method was proven by subjecting the samples to stress conditions such as acid, base, oxidation, photolysis and thermal degradation. The efficient chromatographic separation was achieved using mobile phase solution A prepared as buffer solution 10 mM monobasic potassium phosphate pH 4.0 ± 0.05 adjusted with diluted ortho phosphoric acid solution and solution B acetonitrile with linear gradient elution on poroshell 120 EC-C18 shot column (50 mm × 4.6 mm, 2.7 µm) and UV detection at 235 nm at a flow rate 1.0 mL/min, column oven temperature was set to 25˚C. The above are all known impurities and degradation impurities are well resolved with Naproxen peak and these are eluted within a 10 min runtime of HPLC. The photo diode array detector was used for peak homogeneity testing during stress study experiments and the overall mass balance was found to be 99.2% to 100.2% in all stress conditions. The linear calibration range was found to be 0.05 µg/mL to 0.75 µg/mL for related compounds and 50 µg/mL to 150 µg/mL for Naproxen and the accuracy of the method was found to be 91.5% to 98.5% recovery for the related substance method and 95.4% to 97.4% recovery for the assay method. The Naproxen and related compounds were found to be stable up to 48 hours and the method validation data show excellent results for precision, linearity, specificity, limit of detection, limit of quantitation and robustness. The present method can be successfully used for routine QC and stability studies and it will help to reduce the analysis cost, time and effluent load compared to conventional HPLC methods.
1. Introduction
Naproxen is chemically, (S)-6-methoxy-α-methyl-2-naphthaleneacetic acid as sodium salt [1]. The molecule is a non-steroidal anti-inflammatory drug commonly used for the reduction of moderate to severe pain, fever, inflammation and stiffness. It works by inhibiting both COX-1 and COX-2 enzymes, like other NSAIDs, Naproxen is capable of producing disturbances in the gastrointestinal tract. Regulatory requirements for the identification, qualification and control of impurities in drug substances and their formulated products are now being explicitly defined, particularly through the ICH. It is also recommended by ICH that all routine impurities at or above 0.10% level should be identified through appropriate analytical methods [2- 4]. Naproxen is cited in the European Pharmacopoeia to have contamination by compounds A, B, D, E [5,6] and Imp-C is likely to be present in the preparation of Naproxen by using asymmetric hydrogenation step process [7]. A survey on literature reveals that many chromatographic methods have been reported for the determination of Naproxen in raw material [8], tablets [9,10], plasma [11-13], urine [14], intestinal perfusion samples [15] and pharmaceutical preparations [16]. However, no stability indicating UHPLC method is reported for estimation of related compounds and assay of naproxen from bulk drug samples.
In the present investigation, current chromatographic technology (UHPLC) method has been developed and described for the simultaneous estimation of related compounds and Naproxen in bulk drug samples and the samples were subjected to stress studies. The present validated [17] stability indicating method can be used as an alternative method for routine quality control and stability study of bulk drug manufacturing samples.
2. Material and Methods
2.1. Materials
Working standard of Naproxen and related impurities were synthesized and further confirmation was performed by using spectral techniques (NMR, MASS, IR & UV) and bulk drug samples are obtained from Ecologic Technologies Limited (Hyderabad, India) for this entire study. A chemical structure of Naproxen and related compounds are shown in Figure 1. Gradient grade acetonitrile, potassium dihydrogen orthophosphate, orthophosphoric acid HPLC grade (Merck, India) was used and HPLC grade water was obtained by Millipore water purification system.
2.2. Instrumentation
The ultra-high performance liquid chromatographic (UHPLC) system was used model of Agilent 1260 Infinity series with a diode array detector (quaternary pump: G1311B, column thermostat: G1316B, Auto sampler with cooler: G1329B & G1330B and detector: G4212B) for method development, validation and stress study experiments, the chromatographic data and peak purity of the Naproxen was tested by using Chemstation DAD software (Agilent Technologies, Clara, US). A column poroshell 120 EC C18 UHPLC column was manufactured by Agilent
(Agilent Technologies, Clara, US).
2.3. Methods
2.3.1. Chromatographic Conditions
The chromatographic separation was achieved on poroshell 120 EC-C18 UHPLC column (50 mm × 4.6 mm, 2.7 µm) using solvent A as 10 mM potassium dihydrogen orthophosphate buffer pH 4.0 ± 0.05 adjusted with diluted ortho phosphoric acid and solvent B as acetonitrile with a linear gradient programme: time (min)/% B is 0/25, 8.0/ 75, 8.5/25 and 10/25 at flow rate of 1.0 mL/min with UV detection of 235 nm, column temperature was maintained at 25˚C using 5.0 µL injection volume.
2.3.2. Standard Solution
Standard solution was prepared by dissolving Naproxen working standard equivalent to 50 mg in 60 mL of acetonitrile and then diluted to 100 mL to get a solution of concentration 500 µg/mL with the same solvent.
The above solution was further diluted to get a solution of concentration 100 µg/mL of Naproxen used for assay determination and mass balance study during stress study experiments.
Similarly stock solutions of related compounds were prepared individually to get a solution of concentration 100 µg/mL with acetonitrile solvent. The stock solutions were further diluted and used for related substances determination.
2.3.3. Estimation of Naproxen and Related Compound from Bulk Samples
The composite sample of Naproxen from bulk material taken an amount of 50 mg was transferred to dried 100 mL volumetric flask. Dissolved and diluted to 100 mL with acetonitrile related substances determination.
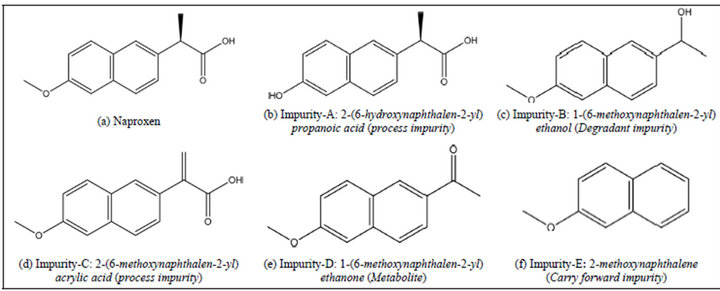
Figure 1. Chemical structure of Naproxen and its related substances.
During stress studies experiments, the sample solution was further diluted to get a solution of concentration 100 µg/mL for assay and peak purity measurements.
2.4. Validation Parameters
2.4.1. Specificity/Selectivity
Specificity of the method was evaluated by injecting the blank, individual related compound, sample solution was prepared by spiking related compounds at 0.1% level of test concentration (i.e., 500 µg/mL) and injected as system suitability test (SST) into UHPLC.
2.4.2. Forced Degradation (Stress Testing)
To further evaluate the stability indicating power of the analytical method, Naproxen was subjected to stress testing as per ICH recommended test conditions. The sample was subjected to base hydrolysis using 0.5 N sodium hydroxide solution and acid hydrolysis by using 0.5 N hydrochloric acid; oxidation by using 5% v/v solution of hydrogen peroxide; thermal and photolysis.
The objective of the stress study was to generate degradation products under various stress conditions. For acid, base and oxidation degradation study period was up to 6 h, thermal and photolysis study period was 7 days and 11 days respectively. The spectral purity of the main peak was evaluated using photodiode array detector and overall mass balance was calculated in each stress study experiment.
2.4.3. Precision
Six solutions were prepared containing Naproxen (500 µg/mL) with related compounds at 0.1% level of test concentration (i.e., 500 µg/mL) and injected into chromatographic system. The value of %RSD was calculated considering peak area of Naproxen and each related compound. Similarly, intermediate precision of the method was also evaluated by another analyst, on a different day in the same laboratory.
2.4.4. Limit of Detection and Limit of Quantitation
The limit of detection (LOD) and limit of quantitation (LOQ) for Naproxen and related compounds were determined. Precision study was also carried out at the LOQ level by injecting six (n = 6) individual preparations and calculated %RSD for Naproxen and each related compound.
2.4.5. Linearity
Linearity test solutions for the assay method were performed from 25% to 150% (i.e., 25, 50, 75, 100 and 150 µg/mL). The responses were measured as peak areas and plotted against concentration. Similarly linearity test solutions for the related substances method were performed from LOQ to 150% at impurity level (i.e., LOQ, 0.025%, 0.05%, 0.075%, 0.10%, 0.125% and 0.15%). The calibration curve was drawn by plotting the each impurity peak area versus its corresponding concentration. The correlation co-efficient, slope and Y-intercept was measured for Naproxen and also each impurity.
2.4.6. Accuracy
The accuracy of the assay method was evaluated in triplicate (n = 3) at the concentration levels of naproxen 50, 100 and 150 µg/mL (50%, 100% and 150%) and the % recovery was calculated at each level. Similarly accuracy of the related substances method was evaluated in triplicate (n = 3) at the concentration levels of each related compound 0.25, 0.5 and 0.75 µg/mL (50%, 100% and 150%) and the % recovery was calculated for each related compound.
2.4.7. Robustness
The robustness of the method was determined as a measure of the analytical method capability to be unaffected by small variation in method parameters. The different variations such as variation in flow rate by ± 0.1 mL/min, variation in column temperature by ± 5˚C; variation in composition of mobile phase ± 10% (in terms of organic component) and variation in pH of buffer by ± 0.1 units. At these changed conditions the system suitability was evaluated at each condition.
2.4.8. Stability of the Solution
A sample solution of assay method and related substance method was checked at different time intervals up to 48 h by keeping the solution at room temperature and calculated cumulative %RSD for the peak area of naproxen and its related compounds.
3. Results and Discussion
3.1. Method Development
The main objective of the method is to develop a common chromatographic method for the estimation of naproxen and related compounds from bulk drug samples. As naproxen is containing acidic functional group (-COOH), so pH of the buffer solution was chosen in acidic medium as phosphate buffer and various trials were attempted in different buffer pH conditions (i.e., 2.0, 3.0, 3.5, 4.0 and 4.5) on various columns. At these chromatographic conditions, the resolution between impurity-C and naproxen is more than 2.0 was observed at buffer pH 4.0 condition. Further fine-tuned and downscale the method into UHPLC method by using chromatography conversion tool. Efficient chromatographic separation was achieved on above described method section. A typical blank and system suitability chromatogram is given in Figures 2(a) and (b).
3.2. Specificity/Selectivity
There was no interference from the blank and also from the related compounds at the retention time of naproxen peak. The peak purity data shows the peaks are pure and there was no co elution found at the retention time of naproxen and related compound peaks. The obtained SST results are given in Table 1.
3.3. Forced Degradation (Stress Testing)
The percentage of total degradation in acid hydrolysis, oxidation is observed 0.50 & 0.24 respectively and there was no degradation found in other stress conditions during the study period. Peak purity was passing in all the stressed samples and also well separation was observed degradation products from the naproxen. The results of stress studies are tabulated in Table 2.
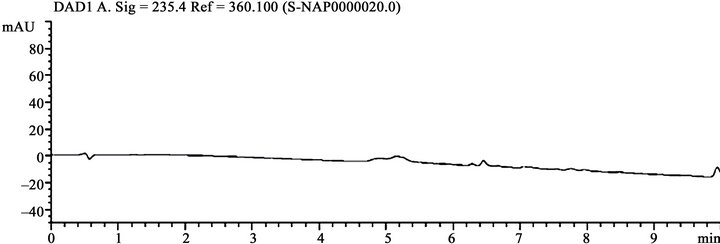 (a)
(a)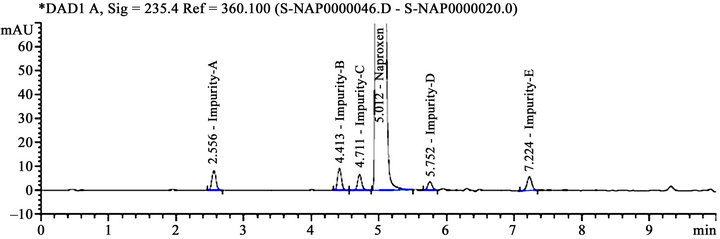 (b)
(b)
Figure 2. (a) A typical blank chromatogram; (b) A system suitability chromatogram.
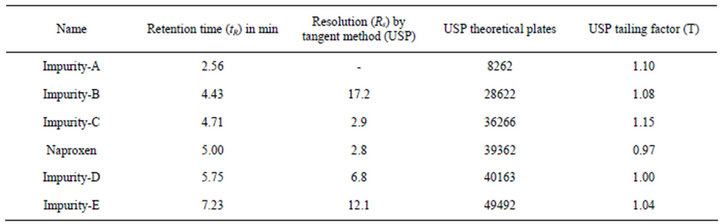
Table 1. System Suitability Test (SST) results.
3.4. Precision
The %RSD of assay of naproxen during assay method precision study was well within 1.0 and %RSD of content of related compound during related substance method precision study was below 3.5. The %RSD of assay of naproxen results is obtained in the intermediate precision less than 1.0 and the related substance method was found to be less than 3.1. Precision results are given in Table 3.
3.5. Limit of Detection and Limit of Quantitation
The LOD and LOQ for naproxen and related compounds were found to be less than 0.03 µg/mL. LOQ & LOD results are given in Table 3.
3.6. Linearity
The obtained calibration curve by least square regression analysis between peak areas versus concentration is showed linear relationship with correlation coefficient of 0.9998 for naproxen and ≥0.9975 respectively for related compounds.
The linear calibration range was found to be 25 to 150 µg/mL for naproxen and LOQ to 0.75 µg/mL for each related compound. The results are demonstrating that an excellent correlation existed between the peak area and concentration, linear regression least square curve obtained results are given in Table 3. The calibration curve was shown in Figure 3.
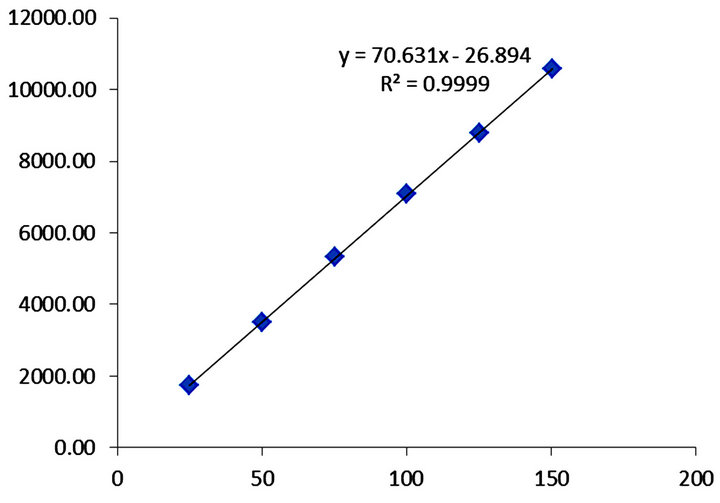
Figure 3. Linearity plot of naproxen.
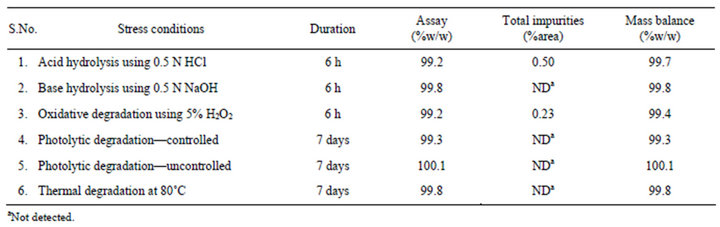
Table 2. Results of stress studies.
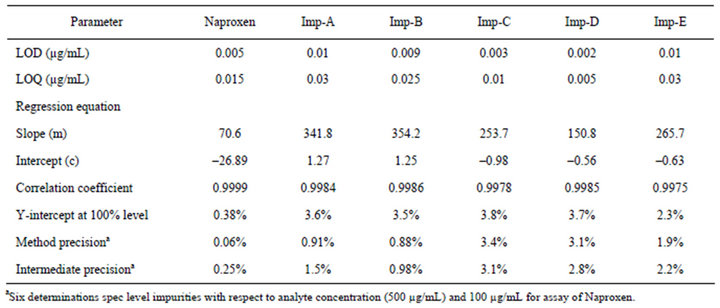
Table 3. LOD, LOQ, regression and precision data.
3.7. Accuracy
The accuracy of the assay method was determined in percentage of recovery of Naproxen from bulk drug samples ranged from 99.5 to 100.3. The % recovery of the five impurities from bulk drug samples is ranged from 91.5% to 98.5% and the obtained results are shown in Table 4.
3.8. Robustness
In all the deliberate varied chromatographic conditions (flow rate, column temperature, pH variation and composition of organic solvent), all analyte and impurity markers were adequately resolved and also elution order remains unchanged. The resolution between critical pair was greater than 2.0 and tailing factor of Naproxen peak was found be less than 1.5. According to robustness experimental results (Table 5), the developed UHPLC method is showing robust for the determination of Naproxen and its related compounds from the bulk drug samples.
3.9. Solution Stability
The RSD percentage of naproxen and its related compound peak areas were found be less than 1% and 5% respectively. The stability of naproxen sample solution by assay method and also related substances method was stable up to 48 h.
4. Conclusion
The developed analytical UHPLC method is rapid, sim-
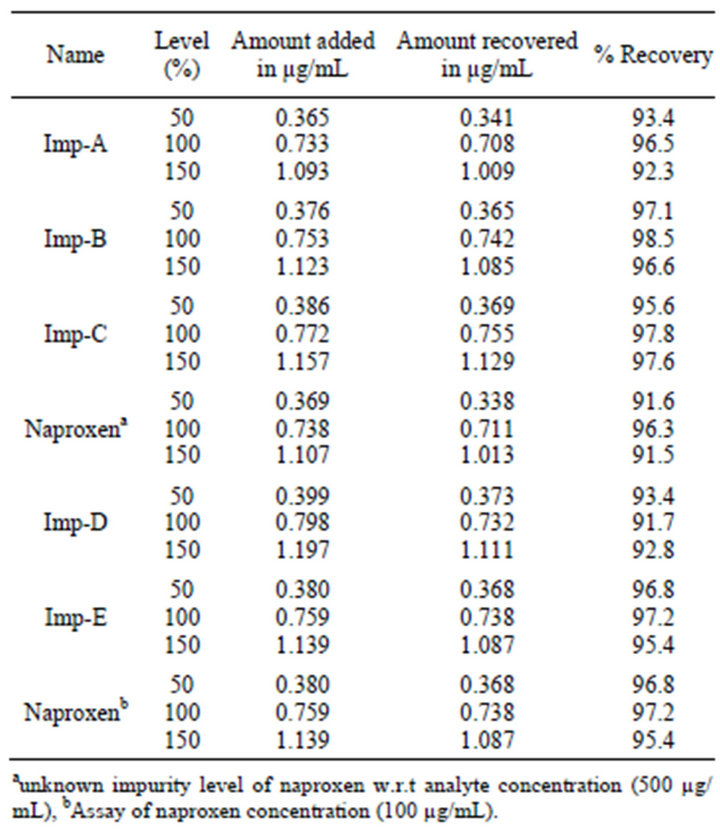
Table 4. Results of accuracy for related substance and assay.
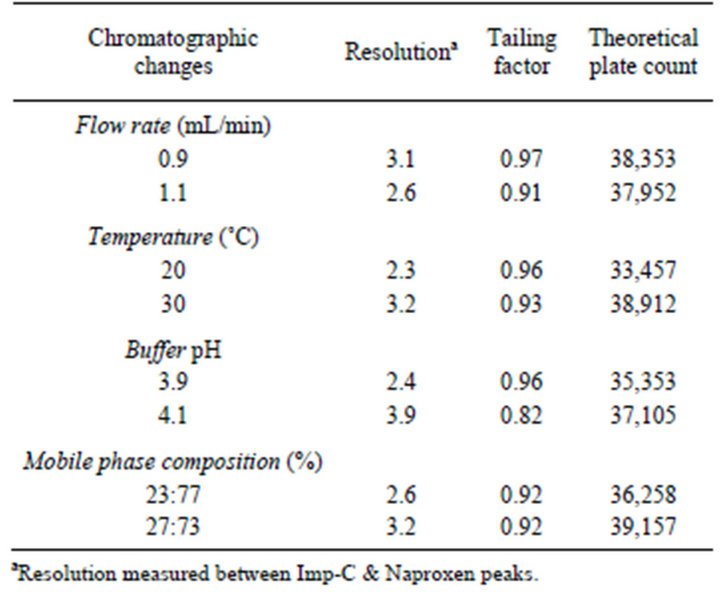
Table 5. Results of robustness evaluation data.
ple, specific, rugged and stability indicating. Hence it can be used as an alternative method for the determination of naproxen and its related compounds from bulk drug samples of routine quality control analysis as well as stability study analysis. This method is more sensitive and superior to the published methods since those are neither stability indicating nor sensitivity levels (i.e., observed LOQ and LOD values are < 0.03 µg/mL) which are estimated to be a low level and also it reduces analysis time, cost and HPLC effluent.
5. Acknowledgements
The authors wish to thank the management of ecoLogic Technologies Limited group supporting this work. We would also thanks to colleagues of Analytical Research of Ecologic Technologies Limited for their support.
REFERENCES
- USP, “Monograph of Naproxen Sodium,” 34th Edition, United States Pharmacopoeial Convention, Rockville, 2011.
- ICH, “Stability Testing of New Drug Substances and Products,” International Conference on Harmonization Guidance Documents, Q1A (R2), 2005.
- ICH, “Impurities in New Drug Substances,” International Conference on Harmonization Guidance Documents, Q3A (R1), 2006.
- ICH, “Impurities in New Drug Products,” International Conference on Harmonization Guidance Documents, Q3B (R1), 2006.
- EP, “Monograph of Naproxen Sodium,” 7th Edition, European Pharmacopeia Guideline Document, 2011.
- J. Peter, Harrington and E. Lodewijk, “Twenty Years of Naproxen Technology,” Organic Process Research and Development, Vol. 1, No. 1, 1997, pp. 72-76. doi:10.1021/op960009e
- H. Zhu and M. A. Bo, “Asymmetric Hydrogenation Synthesis of –(S)-(+)-2-(6’-Methoxyl-2-naphthyl) Propionic Acid by Cinchona Modified Pd (0)-α-FeOOH Catalyst,” Chinese Chemical Letters, Vol. 14, 2003, pp. 1101-1104.
- A. Ekpe, J. H. Tong and L. Rodriguez, “High-Performance Liquid Chromatographic Method Development and Validation for the Simultaneous Quantitation of Naproxen Sodium and Pseudoephedrine Hydrochloride Impurities,” Journal of Chromatographic Science, Vol. 39, 2001, p. 81. doi:10.1093/chromsci/39.3.81
- E. Dinc, A. Ozdemir, H. Aksoy, O. Ustundag and D. Baleanu, “Chemometric Determination of Naproxen Sodium and Pseudoephedrine Hydro Chloride in Tablets by HPLC,” Chemical and Pharmaceutical Bulletin, Vol. 54, 2006, p. 415. doi:10.1248/cpb.54.415
- L. Monser and F. Darghouth, “Simultaneous Determination of Naproxen and Related Compounds by HPLC Using Porus Graphitic Carbon Column,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 32, 2003, p. 1087. doi:10.1016/S0731-7085(03)00213-9
- B. M. Tashtoush and B. M. Al-Taani, “HPLC Determination of Naproxen in Plasma,” Die Pharmazie, Vol. 58, 2003, p. 614.
- F. Nielsen-Kudsk, “HPLC Determination of Some AntiInflammatory, Weak Analgesic and Uricosuric Drugs in Human Blood Plasma and Its Application to Pharmacokinetics,” Acta Pharmacol Toxicol, Vol. 47, 1980, p. 267. doi:10.1111/j.1600-0773.1980.tb03653.x
- T. M. Phillips and E. F. Wellner, “Measurement of Naproxen in Human Plasma by Chip-Based Immunoaffinity Capillary Electrophoresis,” Biomedical Chromatography, Vol. 20, 2006, p. 662. doi:10.1002/bmc.673
- E. Mikami, T. Goto, T. Ohno, H. Matsumoto and M. Nishida, “Simultaneous Analysis of Naproxen, Nabumetone and Its Major Metabolite 6-Methoxy-n-Napthyl Acetic Acid in Pharmaceuticals and Human Urine by HPLC,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 23, 2000, p. 917. doi:10.1016/S0731-7085(00)00365-4
- P. Zakeri_Milani, M. Barzegar-Jalali, H. Tajerzadeh, Y. Azrmi and H. Valizadesh, “Simultaneous Determination of Naproxen, Ketoprofen and Phenol Red in Samples from Rat Intestinal Permeability Studies HPLC Method Development and Validation,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 39, 2005, p. 624. doi:10.1016/j.jpba.2005.04.008
- Y. Hsu, Y. Liou, J. Lee, C. Chen and A. Wu, “Assay of Naproxen by High-Performance Liquid Chromatography and Identification of Its Photo Products by LC-ESI/MS,” Biomedical Chromatography, Vol. 20, 2006, p. 787. doi:10.1002/bmc.598
- ICH, “Validation of Analytical Procedures: Text and Methodology,” International Conference on Harmonization Guidance Documents, Q2 (R1), 2005.
NOTES
*Corresponding author.