American Journal of Analytical Chemistry
Vol. 3 No. 7 (2012) , Article ID: 21042 , 8 pages DOI:10.4236/ajac.2012.37062
Development and Validation of Stability Indicating LC Method for 10-Hydroxycamptothecin
1Analytical Development Laboratory, Avra Laboratories Private Limited, Hyderabad, India
2Institute of Science & Technology, Jawaharlal Nehru Technological University, Hyderabad, India
Email: *Venkateshwarlu@avralab.com
Received April 15, 2012; revised May 21, 2012; accepted June 2, 2012
Keywords: 10-Hydroxycamptothecin; HPLC Method; Validation; Degradation; Anti-Cancer Activity and Related Substances
ABSTRACT
The present method gives a detailed description for the development and validation of a simple stability indicating reverse phase liquid chromatographic method for 10-hydroxycamptothecin(10-HCTN) in the presence of its impurities namely Imp A and Imp B along with degradation products generated from forced degradation studies. The drug was subjected to stress conditions of hydrolysis (acid, base and neutral), oxidative, photolytic and thermal stress degradation. Degradation was observed when subjected to treatment with peroxides or under conditions normally used for typical acid and base hydrolysis. The drug was found to be stable under other stress conditions attempted such as photolytic and thermal. Successful separation and isolation of the drug from related impurities and degradation products formed under stress conditions was achieved on an Inertsil ODS-3V (250 mm × 4.6 mm, 5 µm) column using a phosphate buffer, acetonitrile, methanol and Nanopure water. The developed HPLC method was validated with respect to specificity, linearity, accuracy, precision, sensitivity, robustness and solution stability. The assay method was found to be linear in the range of 0.16 mg/mL to 0.24 mg/mL with a correlation coefficient of 0.999 and the linearity of the impurities was established from 0.02% (LOQ) to 0.3%. Recoveries of assay and impurities were found between 99.4% and 100.3%. The developed HPLC method can be used to determine the related substances and assay determinations of 10-HCTN and also to evaluate the quality and long term stability of production samples.
1. Introduction
10-Hydroxycamptothecin((4S)-4-Ethyl-4,9-dihydroxy-1 H-pyrano[3’,4’:6,7]indolizino[1,2-b]quinoli-ne-3,14(4H,1 2H)-dione) (10-HCTN) (Figure 1) is a potent DNA topoisomerase I inhibitor. It induces apoptosis in human breast cancer cells. It inhibits the activity of topoisomerase I and has a broad spectrum of anti-cancer activity in vitro and in vivo. 10-HCTN is a single agent delivered by oral administration in the treatment of human colon cancer. 10-HCTN significantly repressed the proliferation of colon 205 cells at a relatively low concentration (5 - 20 nM). HCPT-induced ultra-structural changes in nuclei and nuclear matrix were similar to those typically associated with lesions of DNA replication or RNA transcription [1-4]. 10-HCTN is obtained by a total synthesis involving 13 steps. The presence of residual impurities also referred as related substances that are obtained in trace amounts during the synthesis of 10-HCTN, can have a significant impact on the quality and safety of the final drug. In order to optimize a process that provides the required Active Pharmaceutical Ingredient (API) with minimal amounts of impurities, it is important that these substances are identified by suitable and reliable analytic methods. Such studies provide a deeper insight into the impurity profile of the API and allow researchers to identify synthetic or purification methods to lower and control their formation during the manufacturing of the drug product. As per the International Conference on Harmonization (ICH) guidelines, all unknown impurities which are forming at a level greater than 0.1% with respect to drug substance or API, they should be identified, synthesized and characterized thoroughly [5-7].
To the best of our knowledge, no HPLC methods are available in the literature for the analysis of 10-HCTN [8]. A RP-HPLC-PDA method was used for the analysis of 10-HCTN. The same method was used to determine the 10-HCTN when exposed to forced degradation conditions. A literature survey revealed that there is no HPLC method for performing a stability analysis that allows for the determination of related substances along with the quantitative estimation of 10-HCTN. An ideal

Figure 1. Chemical structures and labels of 10-HCTN and its impurities.
stability indicating chromatographic method should estimate the required substance whilst resolving and detecting all the impurities and degradation products that may be present.
The current stability test guidelines Q1A(R2) issued by the ICH recommend that the stress study should be carried on a substance to establish its inherent stability characteristics, leading to separation of degradation products and hence supporting the stability of the proposed analytical procedures. It is also imperative that the analytical test procedures for such studies should be stability indicating and fully validated [9,10]. Hence, an attempt has been made to develop an accurate, rapid, specific and reproducible method for the determination of the purity of 10-HCTN and the level of impurities present in 10- HCTN samples. The stability studies were performed on 10-HCTN and the analytical methods were validated as per ICH guidelines.
2. Experimental
2.1. Chemicals and Reagents
10-HCTNwas synthesized by a total synthesis. Impurity A (Imp A) and Impurity B (Imp B) were prepared as depicted in the synthetic Schemes 1 and 2. HPLC grade acetonitrile (ACN) was purchased from Rankem (New Delhi, India). Sodium dihydrogen phosphate monohydrate (AR grade) was obtained from spectrochem (Mumbai, India). High purity water was prepared by using a Nanopure diamond water purification system (Barnstead, Thermo Fisher Scientific, Mumbai, India).
2.2. Instrumentation
High Performance Liquid Chromatography (HPLC)
A Shimadzu HPLC system LC-2010 CHT with a photo diode array detector (PDA) was used for method development, validation and forced degradation studies. The output signal was monitored and processed using LCsolution software.

Scheme 1. Synthetic route of Imp A.
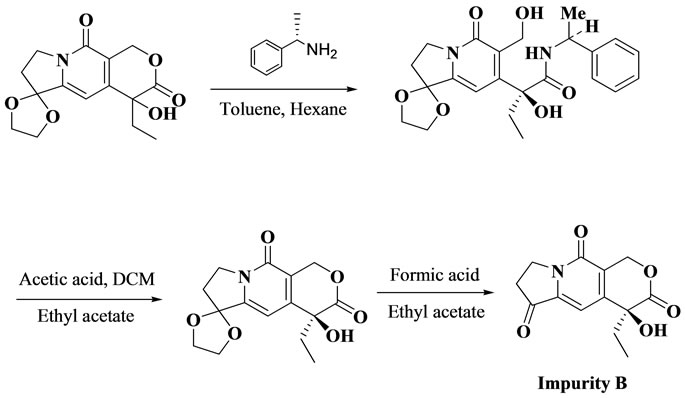
Scheme 2. Synthetic route of Imp B.
2.3. Chromatographic Conditions
An Inertsil ODS C18, (250 mm × 4.6 mm, 5.0 µm) column was used for this analysis and the mobile phase consists of solvent A (0.01 M phosphate buffer; pH adjusted to 3.0) and solvent B (acetonitrile: methanol; v/v 70:30). The flow rate of the mobile phase was 1.0 mL/min. The optimized gradient elution programme is given in the Table 1. A wave length of 220 nm was found to be suitable for this analysis and the column temperature was maintained at 40˚C. The injection volume was 10 μL.
2.4. Preparation of Solutions
2.4.1. Preparation of Standard Solutions
A stock solution of 10-HCTN (0.2 mg/mL) was prepared by dissolving an appropriate amount of 10-HCTN in diluent. A stock solution of Imp A (0.2 mg/mL) and Imp B (0.2 mg/mL) were prepared by dissolving an appropriate amount of respective impurities in diluent. Working solutions were made from above stock solutions for related substances determination and assay determination, respectively.
2.4.2. Preparation of Sample Solution
A 10 mg of sample was weighed and transferred into a 50 mL volumetric flask. 35 mL of solvent B was added
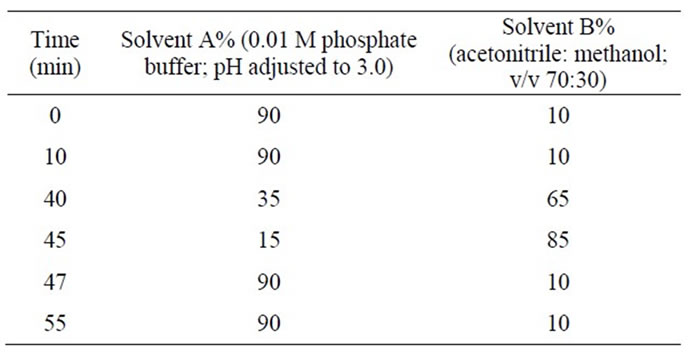
Table 1. The optimized gradient elution program for 10- HCTN.
to dissolve the sample and the mixture was sonicated for 5 min after which it was further diluted with solvent A to make up the volume up to the mark.
2.5. Analytical Method Validation
The developed chromatographic method was validated with respect to specificity, sensitivity, linearity, accuracy, precision, robustness, solution stability and system suitability.
2.5.1. Specificity
Specificity is the ability of the method to measure the analyte response in the presence of its impurities. Stress testing of the drug substance can help to identify the likely degradation products. These studies allow us to understand the degradation pathways and intrinsic stability of the molecule and validate the stability indicating capability of the analytical procedures developed and utilized. The specificity of 10-HCTN in the presence of its impurities namely Imp A and Imp B along with other degradation products was determined by the developed RP HPLC method. Forced degradation studies were also performed on 10-HCTN to offer an indication of the stability indicating property and specificity of the method. The stress conditions opted for the degradation study comprises of light (carried out as per ICH Q1B), heat (60˚C), acid hydrolysis (1N HCl), base hydrolysis (1N NaOH) and oxidation (30% H2O2). For light and thermal stress studies, the study period was 7 days, whereas the acid, base and peroxide related stability study was carried out for a period of 24 h at 60˚C. Peak purity of stressed samples of 10-HCTN was checked by using a LC-2010 CHT with PDA detector of Shimadzu Corporation, Japan. Assay studies were carried out for stress samples against qualified reference standards and the mass balance (% of assay + % of impurities + % of degradation products) was calculated.
2.5.2. Sensitivity
The sensitivity of the method was determined by establishing the limit of detection (LOD) and limit of quantification (LOQ) for Imp A and Imp B, which was estimated at a signal-to-noise ratio of 3:1 and 10:1 respectively, by injecting dilute solutions of a known concentration. The precision study was carried out at the LOQ level by injecting six individual preparations of Imp A and Imp B and the %RSD for LOQ level for the areas of each analytical profile was calculated.
2.5.3. Linearity and Range
Linearity test solutions for the assay method were prepared from stock solution at five concentration levels from 80% to 120% of assay analyte concentrations (0.16, 0.18, 0.20, 0.22 and 0.24 mg/mL).
A linearity test solution for the related substances method was prepared by diluting the impurity stock solution to the required concentrations. The solutions were prepared at five concentration levels. From 10% (LOQ) to 150% of the permitted maximum level of the impurity (i.e. LOQ (0.02%), 0.10%, 0.15%, 0.20% and 0.30%) was subjected to linear regression analysis with the least squares method. The calibration equation obtained from regression analysis was used to calculate the corresponding predicted responses. The residual and sum of the residual squares were calculated from the corresponding predicted responses. Linearity was checked for both assay and related substances method and the %RSD value of the slope and Y-intercept of the calibration curve were calculated. The upper and lower levels of the range were also established.
2.5.4. Accuracy
The accuracy of the assay method was assessed in triplicate at three concentration levels i.e. 80% (0.16 mg/mL), 100% (0.2 mg/mL) and 120% (0.24 mg/mL). At each concentration, three sets were prepared and injected in triplicate. The percentage of recovery was calculated at each level.
The 10-HCTN sample shows no detection of Imp A and Imp B (limit: not more than 0.20% of known, 0.10% for unknown impurities and 0.50% for total impurities). The study was carried out for related substances in triplicate at 0.10%, 0.20% and 0.30% of analyte concentration (0.2 mg/mL).
2.5.5. Precision
The precision of the related substances method was checked by injecting six individual preparations (0.2 mg/mL) of 10-HCTN where each sample was spiked with 0.2% impurities. The %RSD for percentage of each impurity was calculated. The intermediate precision (ruggedness) of the method was also evaluated by different analyst, different column and different instrument in the same laboratory. Assay method precision was assessed by carrying out six independent assays of test sample of 10-HCTN against qualified reference standard. The %RSD of six assay values obtained was calculated. The intermediate precision of related substances method and assay method was evaluated by different analysts, different column and by using a different instrument from the same laboratory.
2.5.6. Robustness
By deliberate changes in experimental conditions the resolution between 10-HCTN, Imp A and Imp B was evaluated. To study the effect of flow rate on the resolution, 0.1 units were changed i.e. 0.9 and 1.1 mL/min. The effect of pH on resolution of impurities was studied by varying ±0.05 pH units (i.e. buffer pH altered from 3.0 to 2.95 and 3.05). The effect of column temperature on resolution was studied at 38˚C and 42˚C instead of 40˚C. In all the above varied conditions, the constituents of the mobile phase were held constant.
2.5.7. Solution and Mobile Phase Stability
The solution stability of 10-HCTN in the assay method was carried out by leaving the test solutions of sample in tightly capped volumetric flasks at room temperature for 48 h. The same sample solutions were assayed in 6 h intervals against freshly prepared standard solutions. The mobile phase stability was also carried out by performing an assay with the freshly prepared sample solutions against freshly prepared reference standard solutions at six hours intervals. The mobile phase prepared was kept constant during the study period. The %RSD of assay of 10-HCTN was calculated for the study period during the mobile phase and solution stability experiments.
The solution stability of 10-HCTN and its impurities in the related substances method was carried out by leaving both spiked and un-spiked sample solutions in tightly capped volumetric flasks at room temperature for 48 h. Content of Imp A and Imp B were determined at 6 h intervals during the study period. The mobile phase stability studies were also carried out for 48 h by injecting the freshly prepared sample solutions during 6 h intervals. The contents Imp A and Imp B were verified in the test solutions. The mobile phase prepared was kept constant during the study period.
3. Results and Discussion
3.1. Method Development and Optimization
The objective was to devise a HPLC method that would allow for the separation of Imp A and Imp B. Initial experiments, using a KH2PO4 buffer (10 mM) with pH 3.0 as solvent A and acetonitrile as solvent B on a Gemini Phenomenex C18 column (250 mm × 4.6 mm, 5 μm) did not result in any noticeable separation. In this experiment, the flow rate was set to 1.0 mL/min and a 10-HCTN sample was spiked with the above two impurities and was injected, the resolution (Rs) between both impurities was <0.5. In this attempt less resolution was observed between Imp A and Imp B.
Another attempt was made by changing the composition of solvent A and solvent B. KH2PO4 buffer was replaced with Na2HPO4 (10 mM) buffer in solvent A. Mixture of acetonitrile: methanol (70:30) was used in place of acetonitrile in solvent B, the gradient program was altered and the spiked sample was injected. In this attempt there was some improved resolution (Rs = 1.2) and the impurities peak shapes. Following on this lead, another experiment was carried out by using the same mobile phase and Inertsil ODS column (250 mm × 4.6 mm, 5 μm) in place of Gemini Phenomenex, with a column temperature at 40˚C. Under these conditions both the impurities were well resolved. The retention time peak of 10-HCTN was observed at 29 min and the resolution between the impurities and 10-HCTN was >2.0. Thus, satisfactory resolution was observed between the impurities and a uniform and symmetrical peak of 10-HCTN was obtained using this method.
3.1.1. Forced Degradation Studies
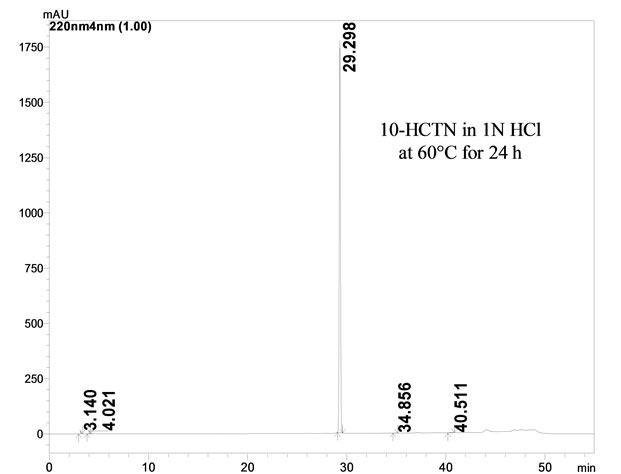
10-HCTN samples were separately exposed to 1N HCl, 1N NaOH and 30% H2O2 at 60˚C. 10-HCTN has shown significant sensitivity towards treatment of 1N HCl, 1N NaOH and 30% H2O2 (Figures 2(a)-(c)). Gradually, the drug undergoes degradation with time in 1N HCl, 1N NaOH and 30% H2O2. No major degradation products were observed when 10-HCTN is subjected to stress in photolytic and thermal (heat at 60˚C) conditions for 7 days (Figures 2(d)-(e)). From the degradation studies using a PDA detector, it was confirmed that the 10- HCTN peak was homogeneous and pure in all the analyzed stress samples. The average mass balance of stressed sample was close to 99.6%. No degradation products were observed after 45 min in the extended runtime of 60 min of all the 10-HCTN samples. The developed HPLC method was found to be specific in the presence of Imp A and Imp B and their degradation products confirm the stability indicating power of the developed method. HPLC studies on 10-HCTN under different stress conditions suggested the following degradation behavior.
3.1.2. Degradation in Acidic Solution
10-HCTNwas exposed to 1N HCl at 60˚C temperature after 24 h, minor degradation products were observed at RRT 0.11 and 0.14.
3.1.3. Degradation in Basic Solution
10-HCTNwas exposed to1N NaOH solution at 60˚C temperature for 24 h. Major degradation products were observed under these basic conditions at RRT of 0.81, 0.83 and 0.95.
 (a)
(a) (b)
(b) (c)
(c)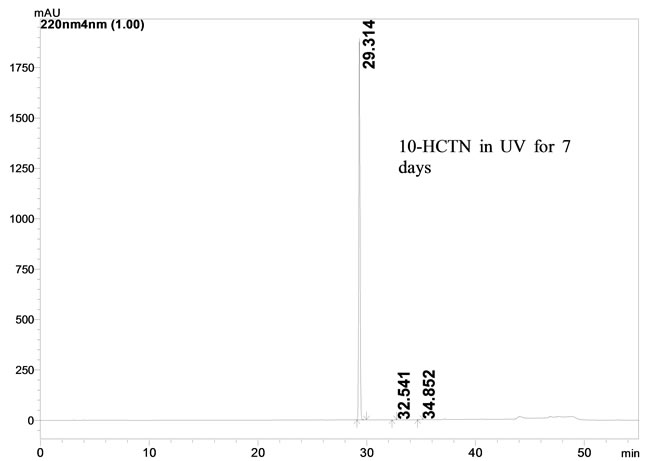 (d)
(d) (e)
(e)
Figure 2. Typical HPLC chromatograms of 10-HCTN under stress conditions. (a) Acid hydrolysis; (b) Base hydrolysis; (c) Peroxide hydrolysis; (d) UV and (e) Heat.
3.1.4. Oxidative Hydrolysis
The drug was exposed to 30% H2O2 at 60˚C temperature for 24 h. The drug gradually underwent degradation with time and significant degradation was observed. Three major degradation products were observed at RRT of 0.25, 0.26, 0.64 and 0.90.
3.1.5. Photolytic and Thermal Conditions
When the 10-HCTN sample was exposed to photolytic and thermal conditions for 7 days, no degradation was observed. The mass balance of stressed samples was close to 99.6%. The assay of 10-HCTN is unaffected in the presences of Imp A and Imp B and its degradation products confirm the stability indicating power of the developed method.
3.2. Method Validation
Under the optimized conditions, 10-HCTN, Imp A and Imp B were well separated with a resolution greater than 4 and typical retention times of Imp A, Imp B and 10- HCTN were about 12.6, 17.1 and 29.3 respectively (Figure 3). The system suitability results are given in Table 2. The resolution is greater than 4 between any two peaks. The tailing factor for all the peaks was about 1.06 and theoretical plates were greater than 10,000.
3.2.1. Sensitivity
The limit of detection of Imp A and Imp B were 0.013% and 0.013%, and limit of quantification was 0.02% and 0.02% (analyte concentration, i.e. 0.2 mg/mL) respectively with 10 μL injection volume. The %RSD of the precision at LOQ concentration for Imp A and Imp B was below 2.0%.
3.2.2. Linearity
Linear calibration plot for the assay method was obtained over the calibration range tested, i.e. 0.16 mg/mL to 0.24 mg/mL and the correlation coefficient obtained was greater than 0.999. The results indicated an excellent correlation between the peak area and concentration of the analyte. Linear calibration plot for the related substance method was obtained over the calibration range tested i.e. LOQ to 0.3% for Imp A and Imp B. The correlation coefficient obtained was greater than 0.999 for all two impurities. The results indicated an excellent cor relation between the peak area and concentration of Imp A and Imp B. The linear equation obtained for Imp A was y = 59906342.2359x + 41.9908; Imp B was y = 28286919.3623x – 6.5578 and drug was y = 73472224.975x + 476002.867 (Figures 4(a)-(c)).
3.2.3. Accuracy
The percentage recovery of 10-HCTN in the samples ranged from 99.7% to 100.3% (Table 3). The percentage recovery of Imp A and Imp B in the samples ranged from 99.4% to 99.9% (Table 4). HPLC chromatogram of the spiked samples with the two impurities in 10-HCTN are shown in Figure 3.
3.2.4. Precision
The %RSD of 10-HCTN during the assay method precision study was within 0.19% and in the intermediate precision study was within 0.56%. The %RSD of area of Imp A and Imp B in the related substances method precision study was within 0.97% and 1.15% respectively, confirming the good precision of the developed analytical method.
3.2.5. Robustness
Close observation of the analytical results from intentionally changed chromatographic conditions (flow rate, pH and column temperature) clearly show that the resolution between eluting impurities namely Imp A and Imp
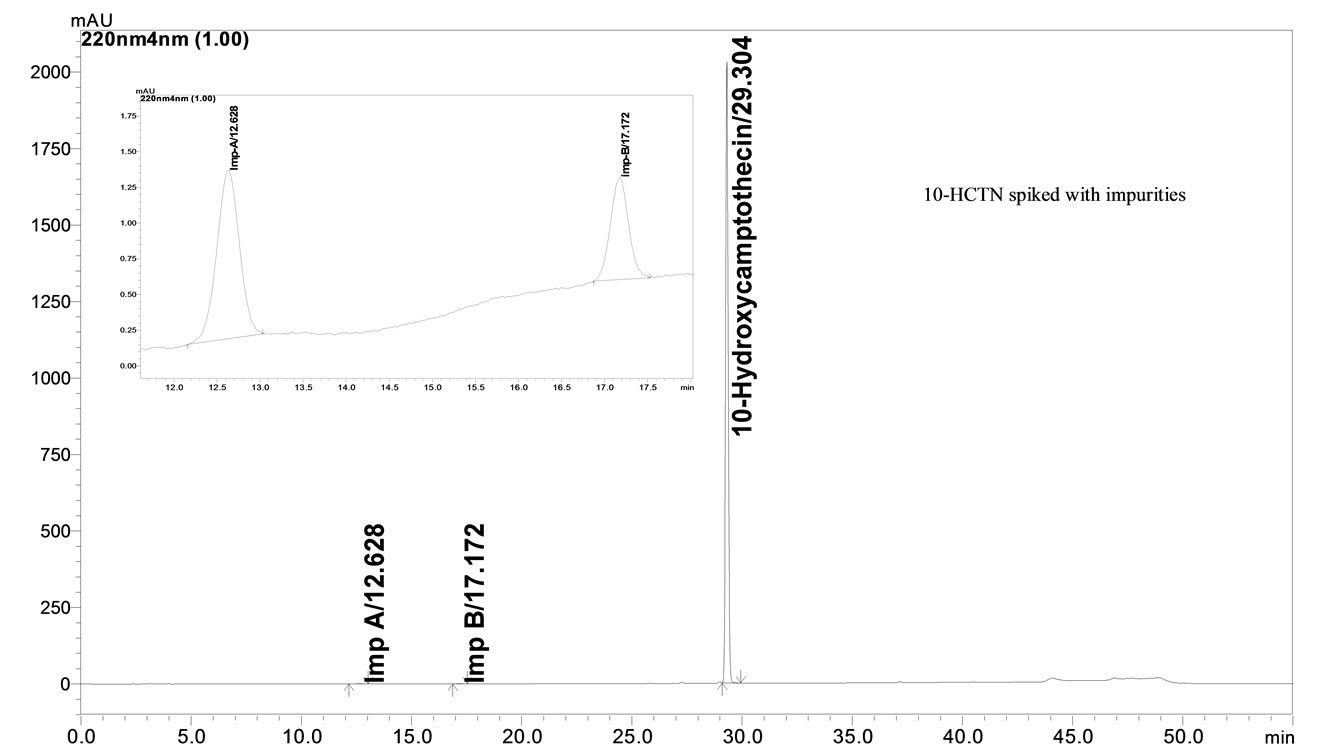
Figure 3. Typical HPLC chromatogram of 10-HCTN spiked with impurities 0.2% specification level.

Table 2. System suitability data.
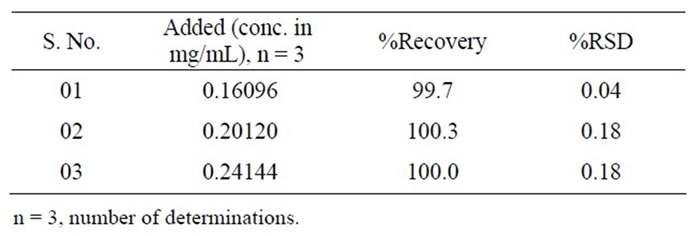
Table 3. Results of accuracy study for 10-HCTN.
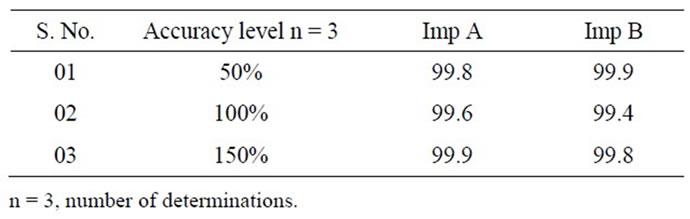
Table 4. Results of accuracy study for impurities.
B was always greater than 4.0, illustrating the robustness of the method.
3.2.6. Solution Stability and Mobile Phase Stability
The %RSD of assay of 10-HCTN during the solution stability and mobile phase stability experiments was within 0.5%. No significant changes were observed in the content of Imp A and Imp B during solution stability and mobile phase stability experiments. The solution stability and mobile phase stability data confirmed that the sample solution and mobile phase used during assay and related substances determination were stable up to the study period was 48 h.
3.2.7. Assay Analysis
Analysis was performed for different batches of 10-HCTN samples (3 batches) ranged from 99.3% to 99.6%.
4. Conclusion
A Precise, accurate and specific HPLC method has been developed and validated for the quantitative determination of all related substances in 10-HCTN. The method was validated with respect to specificity, sensitivity, linearity, accuracy, precision, robustness, solution and mobile
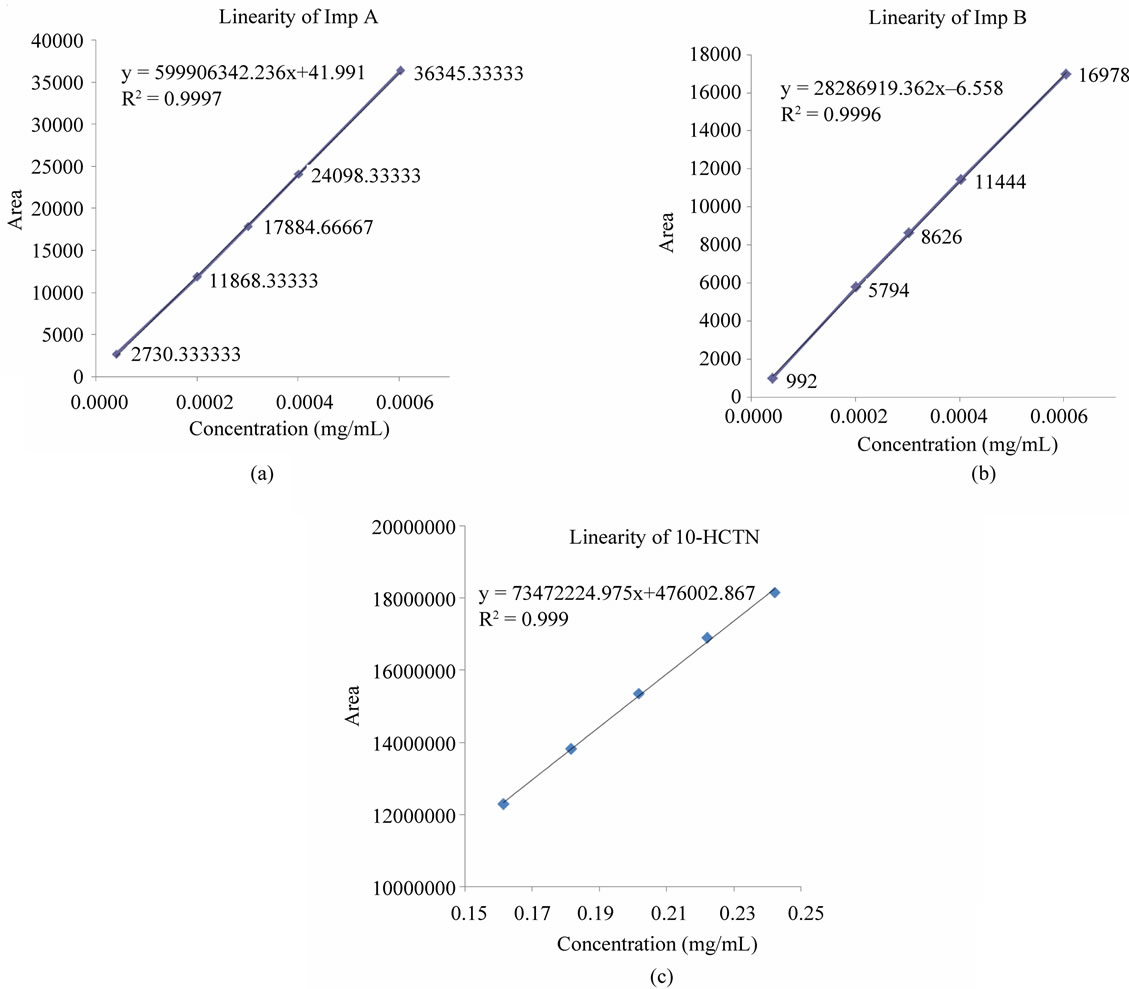
Figure 4. (a) Linearity graph of Imp A; (b) Linearity graph of Imp B; (c) Linearity graph of drug (10-HCTN).
phase stability as per ICH Q2(R1) guidelines. The data showed satisfactory results which are within the acceptable limits for all the method validation parameters. The developed method is stability indicating and it can be used for the routine analysis of production samples and also used to check the stability of 10-HCTN.
5. Acknowledgements
The authors wish to thank Dr. A. V. Rama Rao and Avra Laboratories, Hyderabad, India for supporting of this work.
REFERENCES
- Y. H. Ping, H. C. Lee, J. Y. Lee, P. H. Wu, L. K. Ho, C. W. Chi, M. F. Lu and J. J. Wang, “Anticancer Effects of Low-Dose 10-Hydroxycamptothecin in Human Colon Cancer,” Oncology Reports, Vol. 15, No. 5, 2006, pp. 1273- 1279.
- R. P. Hertzberg, M. J. Caranfa and S. M. Hecht, “On the Mechanism of Topoisomerase I Inhibition by Camptothecin: Evidence for Binding to an Enzyme-DNA Complex,” Nucleic Acids Research, Vol. 21, No. 3, 1993, pp. 593- 600.
- Y. H. Ling, R. Perez-Soler and M. T. Tseng, “Effect of DNA Topoisomerase I Inhibitor, 10-Hydroxycamptothecin, on the Structure and Function of Nuclei and Nuclear Matrix in Bladder Carcinoma MBT-2 Cells,” Anticancer Research, Vol. 13, No. 5A, 1993, pp. 1613-1617.
- P. I. Yan, K. Jiang, R. Hou, Y. Gong, J. Lin, X. Sun and K. Tang, “Examination of Camptothecin and 10-Hydroxycamptothecin in Camptothecaacuminata Plant and Cell Culture, and the Affected Yields under Several Cell Culture Treatments,” Biocell Official Journal of the Sociedades Latinoamericanas de Microscopia Electronica et al., Vol. 34, No. 3, 2010, pp. 139-143.
- ICH Steering Committee, “Impurities in New Drug Substances,” International Conference on Harmonization of Technical Requirements for Registration of Pharmaceutical for Human Use, Chicago, 25 October 2006.
- M. E. Wall, M. C. Wani, A. W. Nicholas, G. Mani Kumar and C. Tele, “Plant Antitumor Agents. 30. Synthesis and Structure Activity of Novel Camptothecin Analogs,” Journal of Medicinal Chemistry, 1993, Vol. 36, No. 18, pp. 2689-2700. doi:10.1021/jm00070a013
- R. Rao and A. V. Rama Rao, “Method of Synthesizing the Key Intermediates for the Production of Camptothecin Derivatives,” US 0221358 A1, 2008.
- J. Ma, Z. P. Jia, Q. Zhang, J. J. Fan, N. Jiang, R. Wang, H. Xie and J. Wang, “Liquid Chromatography Determination of 10-Hydroxycamptothecin in Human Serum by a Column-Switching System Containing a Pre-Column with Restricted Access Media and Its Application to a Clinical Pharmacokinetic Study,” Journal of Chromatography, Vol. 796, No. 1, 2003, pp. 195-200.
- IFPMA, “Stability Testing of New Drug Substances and Drug Products Q1A(R2),” International Conference on Harmonization, Geneva, 2003.
- ICH Steering Committee, “Validation of Analytical Procedures,” International Conference on Harmonization of Technical Requirements for Registration of Pharmaceutical for Human Use, 27 October 1994.
NOTES
*Corresponding author.