Journal of Biosciences and Medicines
Vol.05 No.07(2017), Article ID:77710,16 pages
10.4236/jbm.2017.57003
Exploration of Activated Pathways for Improving Antifungal Agent FR901469 Productivity in Fungal Species No.11243 Using Comprehensive Pathway Model
Itaru Takeda1,2*, Hiroya Itoh3*, Makoto Matsui3, Takashi Shibata3, Masayuki Machida1,4, Sachiyo Aburatani2,5#
1Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, Tokyo, Japan
2Biotechnology Research Institute for Drug Discovery, National Institute of Advanced Industrial Science and Technology (AIST), Tokyo, Japan
3Astellas Pharma Inc., Tsukuba Biotechnology Research Center, Ibaraki, Japan
4Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Sapporo, Japan
5Computational Bio Big-Data Open Innovation Laboratory (CBBD-OIL), Tokyo, Japan

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution-NonCommercial International License (CC BY-NC 4.0).
http://creativecommons.org/licenses/by-nc/4.0/



Received: May 25, 2017; Accepted: July 15, 2017; Published: July 18, 2017
ABSTRACT
Secondary metabolites are important for various industrial applications. The production of secondary metabolites is often improved by the activation of substrate supply pathways for biosynthesis. However, many important pathways have remained unclear. In this study, we explored possible pathways related to substrate supply for the biosynthesis of the antifungal agent FR901469 which is a nonribosomal peptide and a fungal secondary metabolite. To clarify the unknown activated pathways, we utilized the Comprehensive Pathway Model (CPM) which was developed in our previous study. We verified that the overexpression of the hypothetical beta-alanine-aminotransferase (BAL-AT), which was included in the explored pathways, improved the FR901469 productivity. The genes encoding the BAL metabolic enzymes are considered to be important for improving the FR901469 productivity.
Keywords:
Bioinformatics, Metabolic Pathway Model, Fungi, Secondary Metabolites

1. Introduction
The improvement of secondary metabolite productivity is a crucial task for various industries. Many secondary metabolites have bioactivities and are important resources as lead compounds for new drugs, active constituents of cosmetics, etc. [1] [2] [3] . Many fungal and bacterial species produce secondary metabolites [4] . In order to improve the productivity of secondary metabolites, some traditional methods, such as culture optimization and high-producing strain development, have been used [5] [6] [7] . Other approaches, in which the secondary metabolite productivity was improved by genetic technology, have been reported recently. For example, the productivity of a secondary metabolite has been enhanced by the overexpression of a transcription factor in a gene cluster [8] . In addition, laeA, which is known as a global regulator of secondary metabolism, has been overexpressed [9] . However, many important metabolic pathways for the substrate supply have remained unclear. Secondary metabolites are typically synthesized from primary metabolites, such as amino acids and acetyl-CoA [10] . The biosynthetic pathways of these primary metabolites are considered to be important for improving the productivities of secondary metabolites. Thus, the pathways that are activated according to the production of a secondary metabolite should be explored.
In order to explore all pathways related to the biosynthesis of useful compounds, we developed an algorithm for the construction of a Comprehensive Pathway Model (CPM) in our previous study [11] . Since a cell is regarded as a closed space, the by-products of each enzymatic reaction are used in other enzymatic reactions. The fluctuation of the by-product is considered to affect other enzymatic reactions as well as the main product. Consequently, we constructed the CPM as a metabolic pathway model in which the chemical transformations of the main products and the by-products were considered. The CPM can reveal the pathways hidden behind the main pathways. In our previous study, we explored the by-pass of galactose metabolism in Saccharomyces cerevisiae [11] , and found some possible pathways related to galactose metabolism. Some GAL genes are induced and repressed by the addition of galactose and glucose, respectively [12] [13] [14] . Our explored pathways were composed not only of GAL genes but also other genes, which are induced and repressed in the same manner as the known GAL genes.
In this study, we applied the CPM to an industrial organism. Recently, the whole genome of fungal species No.11243 (No.11243 hereafter) was determined [15] . The wild type strain No.11243 was isolated from a decayed leaf sample, which was picked up at Ayabe, Kyoto, Japan [15] . This organism produces the antifungal agent FR901469 [16] [17] [18] . FR901469 is water-soluble antifungal lipopeptidolactone [16] . FR901469 is an inhibitor of 1,3-beta-glucan synthase [17] . Antifungal activity of this compound was varified against some funagl species [17] . This compound is cyclic peptide [18] . In the industrial process development of FR901469, some mutant strains with high productivity were obtained from the wild-type strain by UV irradiation [6] . Part of the genealogy is shown in Figure 1(a). The productivities of three mutant strains, #10-10, #1769, and #2889, were measured in the previous study [19] . Their relative FR901469 productivities are shown in Figure 1(b). However, the mechanisms underlying the improved productivity in the mutant strains remained unclear. In this study, we explored the activated pathways that are assumed to be responsible for substrate supply. In order to find an important key gene for improving FR901469 productivity, we selected an enzymatic gene from the explored pathways. We verified that the expression of this enzymatic gene affected the productivity of FR901469.
2. Materials and Methods
2.1. Cultivation
We used special grade chemicals from Wako, unless otherwise specified. Seed medium (20 mL), containing 4% soluble starch, 2% cottonseed flour, 1% soybean flour, 1% KH2PO4, and 0.2% CaCO3, was poured into 100-mL Erlenmeyer flasks and sterilized at 121˚C for 20 min. A quadrangular (5 mm × 5 mm) plate culture was inoculated into each flask and cultured under shaking conditions (220 rpm) at 25˚C for 5 days.
Production medium (20 mL), consisting of 7% soluble starch, 1% glucose, 4% cottonseed flour, 6% corn steep liquor (Sigma-Aldrich), 1% (NH4)2SO4, 0.5% BactoTM Peptone (Becton, Dickinson and Company), 0.44% K2HPO4, 1.0% KH2PO4, 0.1% antiform PE-L (Wako), and 0.1% antiform SI (Wako), was adjusted to pH 6.0 by adding 1 M NaOH, poured into each 100-mL Erlenmeyer flask, and sterilized at 121˚C for 20 min. The resultant seed culture (2 mL) was transferred to the production medium and cultured under shaking conditions (220 rpm) at 25˚C for 6 days.
In this work, we cultured one mutant No.11243 strain [6] , which was obtained from the wild type strain (FERM BP-3373), and one transformant obtained in this study: #10-10 (Figure 1(a)) and #10-10+ANO11243_080900.

Figure 1. Mutant strains of fungal sp. No.11243 and relative FR901469 productivities. (a) The genealogy of the mutant strains of No.11243. (b) The relative productivities of the mutant strains. The bar graph represents mean values of three replicate experiments. The error bars are Mean value ± Standard error of three replicate experiments.
2.2. Analysis of FR901469 Productivity
To extract FR901469, 200 μL of liquid medium from 4 and 6 day cultures was placed into 1.5-mL Eppendorf tubes, mixed with 800 μL ethanol, and incubated for 30 min at room temperature. The debris was removed by centrifugation at 15,000 rpm at room temperature for 10 min, and the supernatant was filtered using 0.22-μm pore size filter units (NacalaiTesque). A 2-μL aliquot of the extract was separated on an Acquity UPLC system (Waters, MA, USA) with an Acquity UPLC BEH C18 1.7 μm 2.1 × 50 mm reversed-phase column (Waters), and eluted with a 36% - 46% aqueous acetonitrile (Wako, for LC/MS) gradient containing 0.1% formic acid at a flow rate of 0.6 mL/min. The production of FR901469 was quantified by the peak area of absorbance at 210 nm.
2.3. RNA Sequencing (RNA-Seq)
To isolate RNA, we used the 4 and 6 day production cultures. Mycelia in 2 mL of the production culture were collected by filtration, using Miracloth. The mycelia were washed with Milli-Q water, frozen by liquid nitrogen, and pulverized in a mortar. The total RNA was extracted from the frozen pulverized mycelia using 1.5 mL ISOGEN (Nippon Gene, Tokyo, Japan), and purified with an RNeasy mini kit (Qiagen, Venlo, Netherlands). The Truseq Stranded mRNA and Total RNA Sample Preparation Kit (Illumina, CA, USA) were used for library preparation from the purified RNA, and the libraries were sequenced by MiSeq (Illumina). We registered the RNA-seq data of #10-10 and #10-10+ANO11243_ 080900 in the DDBJ Sequence Read Archive (Accession numbers #10-10: DRX076195-DRX076200, #10-10+ANO11243_080900: DRX076201-DRX076206).
2.4. Sequence Data Processing
In this work, we used five RNA-Seq data sets. For constructing the activated pathway models, the three data sets of mutant strains #10-10, #1769, and #2889 registered in the DDBJ Sequence Read Archive were used (Accession numbers #10-10: DRX040488-DRX040490, #1769:DRX040524-DRX040526, #2889: DRX040566-DRX040568). For evaluating the activated pathways, the two data sets of #10-10 and #10-10+ANO11243_080900 from the 6 day culture periods were used (Accession numbers #10-10: DRX076198-DRX076200, #10-10+ ANO11243_080900: DRX076204-DRX076206). The reference genome was indexed by Bowtie 2 [20] , and high-quality reads were mapped to the No.11243 genome by TopHat 2 [21] . By using Genome Traveler (Insilico Biology), the Reads Per Kilobase of exon model per Million mapped reads (RPKM) values were calculated, as the expression level of each gene. For the #10-10 data set, the mean RPKM value of three replicate experiments was calculated for each gene. In the same way, the mean RPKM values were also calculated for the #1769 and #2889 data sets. Using the data sets of the mean RPKM values, the Global Normalization was performed between #10-10 and #1769. The fold-change (FC) values of #1769 against #10-10 were calculated from the normalized RPKM values in each gene. The FC values were converted to the base-2 logarithmic values. The fold-change values log2(FC) of #2889 against #10-10 were also calculated in the same way.
2.5. Construction of Fungal sp. No.11243-Specific CPM
The No.11243-specific CPM (11243CPM) was constructed (Figure 2(a)). In the KEGG database, the K number was assigned to each ortholog group [22] . Each node that was contained in the CPM corresponded to a K number. By using KAAS [23] , we assigned K numbers to the genes encoded in the No.11243 genome. The amino acid sequences of the deduced genes were retrieved from the DDBJ/EMBL/GenBank database (accession nos. DF938580~938599). Following the assignment of the K numbers, the whole CPM was constructed from KEGG (ver. 2014-04-07 release). In the CPM, the nodes and arcs corresponded to enzymes and compounds, respectively. In order to construct a 11243CPM, two simplification processes were performed as follows: 1st: Elimination of excess compounds. Some arcs indicating excess compounds were eliminated from the whole CPM [11] . Some specific compounds, for example H2O and oxygen, connected many pairs of enzymes in the CPM. We think that large fluctuations of these compounds rarely occur in a cell. Since these compounds are usually essential for growth, sufficient amounts are assumed to exist in a cell. Thus, the arcs corresponding to these compounds were eliminated from the CPM. 2nd: Selection of organism-specific enzymes. Some nodes were selected from the CPM. The selected nodes corresponded to K numbers that were assigned to the genes encoded in the genome of No.11243. In this way, 11243CPM was constructed. The 11243CPM was defined as graph D11243,
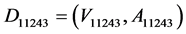 (1)
(1)
where V11243 and A11243 denote nodes and arcs, respectively. In D11243, the nodes correspond to the enzymatic genes encoded in the No.11243 genome.
2.6. Construction of Activated Pathway Model and Exploration of Pathways
The activated pathway model was constructed (Figure 2(b), Figure 2(c)). This model was defined as the graph Dact. In order to construct Dact, we used the FC values of gene expression. The FC values of a strain with high productivity against a strain with low productivity were used. The nodes corresponding to the gene with an FC value higher than the threshold were selected from D11243. The nodes that became alone by this process were eliminated. If some enzymatic genes were assigned to a node, then the gene that had the biggest fold-change value was adopted. The Dact is a subgraph of D11243.
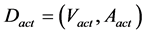 (2)
(2)
where Vact ⊆ V11243 and Aact ⊆ A11243 are satisfied. In this study, the gene expression changes were used from #10-10 to #1769 and from #10-10 to #2889. The constructed graphs were defined as 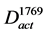 and
and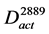 , respectively (Figure 2(b), Figure 2(c)).
, respectively (Figure 2(b), Figure 2(c)).

Figure 2. Flowchart for the construction of the fungal sp. No.11243-specific model and other activated pathway models. (a) Fungal sp. No.11243-specific model. From the whole CPM, this model was constructed by two simplification processes. (b) The activated pathway model of #1769 against #10-10. This model was constructed from D11243, by using gene expression profiles. (c) The activated pathway model of #2889 against #10-10. (d) The conserved activated pathway model between #1769 and #2889. The arcs that were included in both 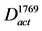 and
and 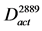 were kept.
were kept.
In order to explore the pathways that affected the productivity with high probability, the conserved activated pathways were constructed as a model (Figure 2(d)). Since random mutations occurred in the genome of the mutant strain, various genes were assumed to be induced. In order to select the genes that affect the productivity with high probability, the conserved activated pathway model was constructed. This model was defined as the graph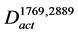 . The graph
. The graph 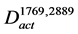 contains the common arcs between
contains the common arcs between 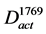 and
and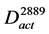 :
:
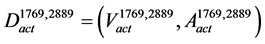 , where,
, where,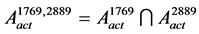 (3)
(3)
The 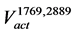 is the set of nodes that are connected by the arcs contained in the
is the set of nodes that are connected by the arcs contained in the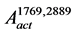 .
.
Following the construction of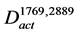 , we explored pathways that could be responsible for the substrate supply of FR901469 biosynthesis. FR901469 is considered to be biosynthesized from various types of amino acids [18] . Thus, the biosynthetic pathways of the substrate amino acids should be explored. The nodes corresponding to the enzymes that catalyzed the biosynthetic reactions of the substrate amino acids were defined as the beginning nodes of exploration. From the beginning nodes, the interactions between nodes were traced in the reverse direction. For the exploration of the pathways, we modified our developed Path Finding Algorithm [11] . Using this algorithm, the possible interactions between the nodes located within the constrained paths were explored comprehensively. Only the beginning node was fixed in this study, while both the beginning and end nodes were fixed in our previous study.
, we explored pathways that could be responsible for the substrate supply of FR901469 biosynthesis. FR901469 is considered to be biosynthesized from various types of amino acids [18] . Thus, the biosynthetic pathways of the substrate amino acids should be explored. The nodes corresponding to the enzymes that catalyzed the biosynthetic reactions of the substrate amino acids were defined as the beginning nodes of exploration. From the beginning nodes, the interactions between nodes were traced in the reverse direction. For the exploration of the pathways, we modified our developed Path Finding Algorithm [11] . Using this algorithm, the possible interactions between the nodes located within the constrained paths were explored comprehensively. Only the beginning node was fixed in this study, while both the beginning and end nodes were fixed in our previous study.
2.7. Overexpression of Target Gene
Overexpression plasmids were constructed by fusing four DNA fragments (ANO11243_065790 alcohol dehydrogenase promoter, ANO11243_080900 coding sequence and 300 bp downstream region (termination), Aureobasidin A resistance gene from pAUR316 (Takara), and the pUC19/BamH I-cleaved vector (Figure 3(a)), using the In-Fusion HD Cloning Kit (Clontech, CA, USA). PCR was performed using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan). The primer sets are listed in Table 1.
The preparation of protoplasts and the polyethylene glycol (PEG) transformation methods were described previously [8] . In the selection of transformants by drug resistance, Aureobasidin A (Clontech, CA, USA) was added to the regeneration agar medium, at a final concentration of 0.5 μg/mL. To confirm DNA fragment insertion, the genomic DNA of the transformant was isolated with a NucleoSpin Plant II kit (Macherey-Nagel, Düren, Germany), according to the manufacturer’s protocol. The isolated genomic DNA (20 ng) was amplified by PCR, using KOD-Plus DNA polymerase and the primer pair pUC19_pADH_F and AUR_080900_R (Table 1). The PCR reaction solution (1 μL) was electrophoresed in a 0.7% (wt/vol) agarose gel containing SYBR safe DNA gel stain (Invitrogen), at a constant voltage (100 V) for 30 min in Tris-acetate-EDTA buffer (pH 8.3). The 500 bp DNA ladder dye plus (TaKaRa) was used as a marker.

Figure 3. Construction of overexpression plasmid and PCR analysis. (a) Three DNA fragments, the endogenous alcohol dehydrogenase promoter (ANO11243_065790), the ANO11243_080900 coding/termination sequence, and the Aureobasidin resistance gene, were inserted into the pUC19 BamH I-cleaved vector by in-fusion cloning. (b) Gel electrophoresis analysis of PCR check for transformants. The primer and the amplified chain length are demonstrated in Figure 3(a). Lanes M: 500 bp DNA ladder marker (Takara), 1: #10-10, 2: #10-10+ANO11243_080900.
3. Results
3.1. Fungal sp. No.11243-Specific CPM and Activated Pathway Models
In order to explore the pathways related to FR901469 productivity, we constructed 11243CPM. In total, 1,999 K numbers were assigned to 2,172 genes by using KAAS. Using the information from the assigned K numbers, the graph D11243 was constructed. The D11243 was constructed from 549 nodes and 6,933 arcs (Table 2). The whole image of D11243 is shown in Figure 4(a). In this image, nodes and arcs represent enzymes and compounds, respectively. The multiplexing arcs between nodes are shown as single arcs.
In order to represent the dynamics of the metabolism, we constructed the activated pathway models  and
and . The gene expression profiles were measured by RNA-Seq. The expression changes were calculated from #10-10 to #1769, and from #10-10 to #2889. The nodes with a base-2 logarithmic fold- change value of 1.0 and over were kept in the
. The gene expression profiles were measured by RNA-Seq. The expression changes were calculated from #10-10 to #1769, and from #10-10 to #2889. The nodes with a base-2 logarithmic fold- change value of 1.0 and over were kept in the  and
and . In this way,
. In this way,  and
and  were constructed. The
were constructed. The and
and  contained 44 and 80 nodes, respectively (Table 2). The whole images of
contained 44 and 80 nodes, respectively (Table 2). The whole images of  and
and  are shown in Figure 4(b) and Figure 4(c), respectively.
are shown in Figure 4(b) and Figure 4(c), respectively.
We constructed the conserved activated pathway model  (Figure 4(d)). The #1769 and #2889 were developed independently from #10-10 (Figure 1(a)). Even though #1769 and #2889 independently contained the factor for improving the productivity, a common subgraph was found between
(Figure 4(d)). The #1769 and #2889 were developed independently from #10-10 (Figure 1(a)). Even though #1769 and #2889 independently contained the factor for improving the productivity, a common subgraph was found between  and
and . Thus, it is considered that the activated pathways in
. Thus, it is considered that the activated pathways in  probably affect the FR901469 productivity. The graph
probably affect the FR901469 productivity. The graph  comprised 30 nodes and 82 arcs, which were conserved between
comprised 30 nodes and 82 arcs, which were conserved between  and
and  (Table 2).
(Table 2).

Table 1. Primers used in this study.
Table 2. Overview of each model.
aNumber of nodes in each graph; bNumber of arcs in each graph; cKinds of compounds in each graph.

Figure 4. Whole Image of 11243CPM. (a) The whole image of D11243 is shown. In this image, the green nodes are common nodes between  and
and . The yellow nodes are unique nodes in the
. The yellow nodes are unique nodes in the . The blue nodes are unique nodes in the
. The blue nodes are unique nodes in the . (b) The whole image of
. (b) The whole image of . (c) The whole image of
. (c) The whole image of . (d) The whole image of
. (d) The whole image of .
.
3.2. Exploration of Substrate Supply Pathways for FR901469 Biosynthesis
In order to predict the important substrate supply pathways for FR901469 biosynthesis, we explored the activated pathways from , while assuming that the FR901469 productivity is affected by the amounts of substrates available for biosynthesis. Thus, we explored the pathways related to the biosynthesis and decomposition of the amino acids that are substrates of FR901469. In the CPM, it is assumed that all enzymatic reactions have two-directions. Consequently, the pathways were explored without any distinction between biosynthesis and decomposition. The beginning nodes of exploration were determined in
, while assuming that the FR901469 productivity is affected by the amounts of substrates available for biosynthesis. Thus, we explored the pathways related to the biosynthesis and decomposition of the amino acids that are substrates of FR901469. In the CPM, it is assumed that all enzymatic reactions have two-directions. Consequently, the pathways were explored without any distinction between biosynthesis and decomposition. The beginning nodes of exploration were determined in . The cyclic peptide, FR901469, is thought to be synthesized from nine kinds of amino acids, L-Alanine, L-Glutamine, Glycine, L-Ornithine, L-Proline, L-Threo- nine, L-allo-Threonine, L-Tyrosine, and L-Valine [18] . We searched for the enzymes catalyzing the reaction with formulae containing the substrate amino acids. From
. The cyclic peptide, FR901469, is thought to be synthesized from nine kinds of amino acids, L-Alanine, L-Glutamine, Glycine, L-Ornithine, L-Proline, L-Threo- nine, L-allo-Threonine, L-Tyrosine, and L-Valine [18] . We searched for the enzymes catalyzing the reaction with formulae containing the substrate amino acids. From , 3 ortholog groups were found. The nodes corresponding to these 3 ortholog groups were defined as beginning nodes. From these beginning nodes, the interactions between the nodes were traced in the reverse direction. In this way, the activated pathways that reached these nodes were explored comprehensively. As a result, the maximum length of the found pathways was 3 steps. The pathways that reached the biosynthesis of L-Alanine and L-Tyrosine are shown in Figure 5. The same structures are shown in Figure 5(a) and Figure 5(b), for the common activated pathways in #1769 and #2889, respectively. In Figure 5, the activated pathways reached the nodes corresponding to K00814 and K00838. For visualization, the L-Alanine and the L-Tyrosine pathways were merged. The K number information and the Gene ID corresponding to each node are described in Table 3.
, 3 ortholog groups were found. The nodes corresponding to these 3 ortholog groups were defined as beginning nodes. From these beginning nodes, the interactions between the nodes were traced in the reverse direction. In this way, the activated pathways that reached these nodes were explored comprehensively. As a result, the maximum length of the found pathways was 3 steps. The pathways that reached the biosynthesis of L-Alanine and L-Tyrosine are shown in Figure 5. The same structures are shown in Figure 5(a) and Figure 5(b), for the common activated pathways in #1769 and #2889, respectively. In Figure 5, the activated pathways reached the nodes corresponding to K00814 and K00838. For visualization, the L-Alanine and the L-Tyrosine pathways were merged. The K number information and the Gene ID corresponding to each node are described in Table 3.

Figure 5. Conserved activated pathways. All pathways explored from  are shown. The labels of the nodes correspond to the K numbers in the KEGG database. These pathways reach the nodes corresponding to K00814 and K00838. (a) The fold- change values log2(#1769/#10-10) are reflected to the explored pathways. The degrees of induction are represented by the colors of the nodes. Bright green represents large induction. White is used for genes with unchanged expression. Red represents repression. (b) The fold-change values log2(#2889/#10-10) are reflected to the explored pathways. The color code is the same as in Figure 5(a).
are shown. The labels of the nodes correspond to the K numbers in the KEGG database. These pathways reach the nodes corresponding to K00814 and K00838. (a) The fold- change values log2(#1769/#10-10) are reflected to the explored pathways. The degrees of induction are represented by the colors of the nodes. Bright green represents large induction. White is used for genes with unchanged expression. Red represents repression. (b) The fold-change values log2(#2889/#10-10) are reflected to the explored pathways. The color code is the same as in Figure 5(a).
Table 3. Enzymatic genes in the common activated pathways.
aLogarithmic induction ratio of 1769 to 10-10; bLogarithmic induction ratio of 2889 to 10-10; cLogarithmic induction ratio of 2889 to 10-10+ANO11243_080900.
3.3. Verification of the Effect of a Selected Gene from the Explored Pathways
In order to find the important key gene for improving productivity, we selected the gene, which was assumed to affect the FR901469 productivity, from the explored pathways. In order to select the target gene, we reflected the fold-change value of #1769 against #10-10, and #2889 against #10-10 to the explored pathways (Figure 5(a), Figure 5(b)). Each node was colored according to the fold- change value. The fold-change values corresponding to each node are described in Table 3. The 10 enzymatic genes that were included in these conserved activated pathways were assumed to affect the FR901469 productivity. Then, we tried to identify the key gene that affects the productivity the most among these 10 genes, by considering the following points. First, the expression of the genes was induced largely in the mutant strains. It is assumed that the inductions of the expression of these genes were important for improving the FR901469 productivity. Second, the nodes were connected with many nodes. The enzymatic gene corresponding to such a node is assumed to affect many other enzymatic reactions. Among the 10 genes, the ANO11243_080890 and ANO11243_ 080900 genes were induced ten-fold in both #1769 and #2889 (Table 3). Furthermore, the node corresponding to ANO11243_080900 had more connections than the node corresponding to ANO11243_080890. Consequently, we considered that ANO11243_080900 was the most likely candidate for improving the FR901469 productivity, among the 10 genes.
We empirically evaluated the effect of this selected gene by overexpression. First, ANO11243_080900 was overexpressed in the #10-10 strain (Figure 3(b)). The strain in which ANO11243_080900 was overexpressed was defined as #10- 10+ANO11243_080900. Second, the FR901469 productivities of #10-10 and #10-10+ANO11243_080900 were evaluated in the same way as the other strains. The results confirmed that #10-10+ANO11243_080900 produced about 1.5-fold more FR901469 than #10-10 (Figure 6(a)). Finally, we performed RNA-Seq for #10-10 and #10-10+ANO11243_080900, under the same conditions used for #1769 and #2889. We determined the RPKM values and calculated the fold- change values of #10-10+ANO11243_080900 against #10-10. The fold-change values log2(FC) were reflected to the explored pathways (Figure 6(b)). The ANO11243_080900 expression was induced by 1,022-fold. In Figure 6(b), the node corresponding to ANO11243_080900 is represented as a square node. Therefore, it was confirmed that the ANO11243_080900 gene affected the FR901469 productivity. In this study, only ANO11243_080900 was overexpressed. However, some other genes, for example ANO11243_080890 and ANO11243_005270, were also induced.
4. Discussion
In the explored pathways, we compared the degrees of gene expression induction between #1769 and #2889, and found that the degrees of induction were similar. The pathways for substrate supply were explored from . The
. The

Figure 6. Relative FR901469 productivity and activated pathways in the #10-10+ANO11243_080900 transformant. (a) The relative productivities of #10-10 and 10-10+ANO11243_080900 after a 6 day culture. The bar graph represents mean values of three replicate experiments. The error bars are Mean value ± Standard error of three replicate experiments. *P ? 0.043. (b) The fold- change values log2(#10-10+ANO11243_080900/#10-10) are reflected to the explored pathways. The color code is the same as in Figure 5. The node corresponding to ANO11243_080900 is represented as a square node. The nodes framed in blue are BAL metabolism genes.
explored pathways were constructed from 10 genes (Figure 5, Table 3). The fold-change values corresponding to each node are described in Table 3. Between #1769 and #2889, the Pearson's correlation coefficient, which was calculated from these 10 data points, was approximately 0.92. Even though the parent of #1769 and #2889 branched at #10-10, the induction patterns of the configuration genes were similar. Thus, the alteration of these induction patterns is important for improving the FR901469 productivity.
We considered that the beta-alanine (BAL) metabolic genes affected the FR901469 productivity. In this study, we assigned K13524 to ANO11243_ 080900. Since the gamma-aminobutyric acid aminotransferase (GABA-AT) and BAL aminotransferase (BAL-AT) reactions are catalyzed by the same enzyme in human [24] , K13524 corresponded to GABA-AT and BAL-AT in the KEGG database. Thus, K13524 has connections with both the GABA and BAL metabolic genes in our CPM. GABA-AT is known as a transaminase catabolizing GABA to succinate semialdehyde (SSA) in the GABA-shunt, an alternative route for the conversion of alpha-ketoglutarate to succinate [25] . The amino-acid sequence of ANO11243_080900 shows strong overall similarity to the sequences of the GABA-AT enzymes from Aspergillus nidulans GatA and Saccharomyces cerevisiae Uga1p, with identities of 67% and 53%, respectively. GatA is conserved in Aspergillus species. When grown with GABA as the sole nitrogen source, the expression of GatA in A. oryzae was strongly induced [26] . Upon the overexpression of ANO11243_080900 in this study, the GABA-shunt related genes, ANO11243_054840 (K01580) and ANO11243_010070 (K00135), which catabolize L-glutamate to GABA and SSA to succinate, respectively, remained uninduced (log2(FC) = −0.37 and −1.19, respectively). In contrast, the beta-alanine metabolic genes, ANO11243_080890 (K00129) and ANO11243_080880 (K00276), were co-upregulated by the ANO11243_080900 overexpression (log2(FC) = 8.64 and 1.16, respectively) (Table 3). Quite interestingly, these three genes including ANO11243_080900 are clustered on the genome, which strongly suggests that ANO11243_080900 is a member of the beta-alanine metabolism genes. Considering the co-upregulation and the co-localization of these three genes, the ANO11243_080900 product may be involved in the BAL metabolism (Figure 7). The overexpression of ANO11243_080900 could have co-upregulated the expression of the two genes encoding K00129 and K00276, which might stimulate the conversion between 1,3-diamino-propane and malonic semialdehyde through BAL metabolism. Considering the finding that the three genes are commonly induced in the two mutants, #1769 and #2889, activating the BAL transamination reaction could be at least one of the effective means to enhance the production of this compound, possibly by increasing particular or overall amino acid biosynthesis.
Furthermore, fungal sp. No.11243 has a duplicated gene encoding K13524, which is ANO11243_016490. The expression of this gene remained uninduced in #1769, #2889, and ANO11243_080900 overexpression (log2(FC) = 0.80, 0.49, and −0.13, respectively). In fungi, some species reportedly have gene duplications of K13524, but the substrates for only a few enzymes have been verified in detail. In Saccharomyces kluyveri, the functions reportedly differ between the duplicated genes. One of them catalyzed the reaction as GABA-AT and the other catalyzed the reaction as BAL-AT [27] . Therefore, the two duplicated genes assigned to K13524 in this study may also have different functions.
Besides the above-mentioned BAL-related genes, the FR901469 productivity might be improved by the overexpression of other target genes. The fold-change

Figure 7. Location of BAL metabolite genes on the fungal sp. No.11243 genome and the BAL metabolite reaction. ANO11243_ 080880, ANO11243_080890, and ANO11243_080900 form a cluster on the genome. The encoded proteins catalyze the BAL metabolic pathway centered on Beta-alanine. ANO11243_080880 and ANO11243_080890 were co-upregulated with ANO11243_ 080900 overexpression.
values of #10-10+ANO11243_080900 against #10-10 were reflected to the explored pathways (Figure 6(b)). The fold-change values corresponding to each node are described in Table 3. Upon the overexpression of ANO11243_080900, the ANO11243_005270, ANO11243_077720, and ANO11243_034720 genes were also induced by more than two-fold. The expressions of the ANO11243_005270 and ANO11243_077720 genes were induced equally to those in the other two mutant strains. In contrast, the expression of other genes, such as ANO11243_ 007360 and ANO11243_092160, were scarcely induced. It is assumed that the productivity in the #10-10+ANO11243_080900 strain increases to the productivity levels in the two mutant strains by the overexpression of these genes.
In this study, we applied the CPM to the genome of fungal sp. No.11243, which produces FR901469. Some mutant strains of No.11243 have been developed independently. The conserved activated pathway model  was constructed by the combination of the gene expression profiles measured in these mutant strains. Consequently, the important pathways that affect the FR901469 productivity were predicted, by using the characteristics of the independent development. We can improve the productivity of secondary metabolites effectively from many candidate pathways, which may affect the productivity with high probability. Thus, the development of many independent mutant strains is one of the important methods for exploring the key genes in bioproduction. The various target genes for improving the secondary metabolite productivity will be contained in the explored pathways. Using a genetic technology approach to identify these genes will facilitate the development of highly productive strains.
was constructed by the combination of the gene expression profiles measured in these mutant strains. Consequently, the important pathways that affect the FR901469 productivity were predicted, by using the characteristics of the independent development. We can improve the productivity of secondary metabolites effectively from many candidate pathways, which may affect the productivity with high probability. Thus, the development of many independent mutant strains is one of the important methods for exploring the key genes in bioproduction. The various target genes for improving the secondary metabolite productivity will be contained in the explored pathways. Using a genetic technology approach to identify these genes will facilitate the development of highly productive strains.
Acknowledgements
This work was supported by a grant from the commission for the Development of Artificial Gene Synthesis Technology for Creating Innovative Biomaterial, from the Ministry of Economy, Trade and Industry (METI), Japan.
Cite this paper
Takeda, I., Itoh, H., Matsui, M., Shibata, T., Machida, M. and Aburatani, S. (2017) Exploration of Activated Pathways for Improving Anti- fungal Agent FR901469 Productivity in Fungal Species No.11243 Using Compre- hensive Pathway Model. Journal of Bio- sciences and Medicines, 5, 16-31. https://doi.org/10.4236/jbm.2017.57003
References
- 1. Bentley, R. (2006) From Miso, Sake and Shoyu to Cosmetics: A Century of Science for Kojic Acid. Natural Product Reports, 23, 1046-1062.
https://doi.org/10.1039/b603758p - 2. Hwang, K.S., Kim, H.U., Charusanti, P., Palsson, B.O. and Lee, S.Y. (2014) Systems Biology and Biotechnology of Streptomyces Species for the Production of Secondary Metabolites. Biotechnology Advances, 32, 255-268.
https://doi.org/10.1016/j.biotechadv.2013.10.008 - 3. Wiemann, P. and Keller, N.P. (2014) Strategies for Mining Fungal Natural Products. Journal of Industrial Microbiology & Biotechnology, 41, 301-313.
https://doi.org/10.1007/s10295-013-1366-3 - 4. Berdy, J. (2005) Bioactive Microbial Metabolites. The Journal of Antibiotics (Tokyo), 58, 1-26.
https://doi.org/10.1038/ja.2005.1 - 5. Kanda, M., Tsuboi, M., Sakamoto, K., Shimizu, S., Yamashita, M. and Honda, H. (2009) Improvement of FR901379 Production by Mutant Selection and Medium Optimization. Journal of Bioscience and Bioengineering, 107, 530-534.
https://doi.org/10.1016/j.jbiosc.2009.01.002 - 6. Matsui, M., Yokoyama, T., Kumagai, T., Nemoto, K., Terai, G., Machida, M., Shibata, T. and Aburatani, S. (2016) Genomic Analysis of Fungal Species No.11243 Mutant Strains Provides Insights into the Relationship between Mutations and High Productivity. British Biotechnology Journal, 10, 1-13.
https://doi.org/10.9734/BBJ/2016/19400 - 7. Schroeckh, V., et al. (2009) Intimate Bacterial-Fungal Interaction Triggers Biosynthesis of Archetypal Polyketides in Aspergillus nidulans. Proceedings of the National Academy of Sciences of the United States of America, 106, 14558-14563.
https://doi.org/10.1073/pnas.0901870106 - 8. Matsui, M., Yokoyama, T., Nemoto, K., Kumagai, T., Terai, G., Tamano, K., Machida, M. and Shibata, T. (2017) Identification of a Putative FR901469 Biosynthesis Gene Cluster in Fungal sp. No. 11243 and Enhancement of the Productivity by Overexpressing the Transcription Factor gene frbF. Journal of Bioscience and Bioengineering, 123, 147-153.
https://doi.org/10.1016/j.jbiosc.2016.08.007 - 9. Bok, J.W. and Keller, N.P. (2004) LaeA, a Regulator of Secondary Metabolism in Aspergillus spp. Eukaryot Cell, 3, 527-535.
https://doi.org/10.1128/EC.3.2.527-535.2004 - 10. Kittila, T., Mollo, A., Charkoudian, L.K. and Cryle, M.J. (2016) New Structural Data Reveal the Motion of Carrier Proteins in Nonribosomal Peptide Synthesis. Angewandte Chemie International Edition, 55, 9834-9840.
https://doi.org/10.1002/anie.201602614 - 11. Takeda, I., Machida, M. and Aburatani, S. (2015) Development of an Algorithm for Reconstructing a Comprehensive Pathway Model: Application to Saccharomyces cerevisiae. Journal of Biomedical Science and Engineering, 8, 500-510.
https://doi.org/10.4236/jbise.2015.88047 - 12. Bro, C., Knudsen, S., Regenberg, B., Olsson, L. and Nielsen, J. (2005) Improvement of Galactose Uptake in Saccharomyces cerevisiae through Overexpression of Phosphoglucomutase: Example of Transcript Analysis as a Tool in Inverse Metabolic Engineering. Applied and Environmental Microbiology, 71, 6465-6472.
https://doi.org/10.1128/AEM.71.11.6465-6472.2005 - 13. Conrad, M., Schothorst, J., Kankipati, H.N., Van Zeebroeck, G., Rubio-Texeira, M. and Thevelein, J.M. (2014) Nutrient Sensing and Signaling in the Yeast Saccharomyces cerevisiae. FEMS Microbiology Reviews, 38, 254-299.
https://doi.org/10.1111/1574-6976.12065 - 14. Johnston, M. (1987) A Model Fungal Gene Regulatory Mechanism: The GAL Genes of Saccharomyces cerevisiae. Microbiological Reviews, 51, 458-476.
- 15. Matsui, M., Yokoyama, T., Nemoto, K., Kumagai, T., Terai, G., Arita, M., Machida, M. and Shibata, T. (2015) Genome Sequence of Fungal Species No. 11243, Which Produces the Antifungal Antibiotic FR901469. Genome Announcements, 3, e00118-e00115.
https://doi.org/10.1128/genomeA.00118-15 - 16. Fujie, A., et al. (2000) FR901469, a Novel Antifungal Antibiotic from an Unidentified Fungus No. 11243. I. Taxonomy, Fermentation, Isolation, Physico-Chemical Properties and Biological Properties. The Journal of Antibiotics (Tokyo), 53, 912- 919.
https://doi.org/10.7164/antibiotics.53.912 - 17. Fujie, A., et al. (2000) FR901469, a Novel Antifungal Antibiotic from an Unidentified Fungus No. 11243. II. In Vitro and in Vivo Activities. The Journal of Antibiotics (Tokyo), 53, 920-927.
https://doi.org/10.7164/antibiotics.53.920 - 18. Fujie, A., Muramatsu, H., Yoshimura, S., Hashimoto, M., Shigematsu, N. and Takase, S. (2001) FR901469, a Novel Antifungal Antibiotic from an Unidentified Fungus No. 11243. III. Structure Determination. The Journal of Antibiotics (Tokyo), 54, 588-594.
https://doi.org/10.7164/antibiotics.54.588 - 19. Matsui, M., Yokoyama, T., Nemoto, K., Kumagai, T., Tamano, K., Machida, M. and Shibata, T. (2017) Further Enhancement of FR901469 Productivity by Co-Overex- pression of cpcA, a Cross-Pathway Control Gene, and frbF in Fungal sp. No. 11243. Journal of Bioscience and Bioengineering, 124, 8-14.
- 20. Langmead, B., Trapnell, C., Pop, M. and Salzberg, S.L. (2009) Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biology, 10, R25.
https://doi.org/10.1186/gb-2009-10-3-r25 - 21. Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R. and Salzberg, S.L. (2013) TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biology, 14, R36.
https://doi.org/10.1186/gb-2013-14-4-r36 - 22. Kanehisa, M. (2013) Chemical and Genomic Evolution of Enzyme-Catalyzed Reaction Networks. FEBS Letters, 587, 2731-2737.
https://doi.org/10.1016/j.febslet.2013.06.026 - 23. Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A.C. and Kanehisa, M. (2007) KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Research, 35, W182-W185.
https://doi.org/10.1093/nar/gkm321 - 24. Schor, D.S., Struys, E.A., Hogema, B.M., Gibson, K.M. and Jakobs, C. (2001) Development of a Stable-Isotope Dilution Assay for Gamma-Aminobutyric Acid (GABA) Transaminase in Isolated Leukocytes and Evidence That GABA and Beta-Alanine Transaminases Are Identical. Clinical Chemistry, 47, 525-531.
- 25. Bach, B., Meudec, E., Lepoutre, J.P., Rossignol, T., Blondin, B., Dequin, S. and Camarasa, C. (2009) New Insights into γ-Aminobutyric Acid Catabolism: Evidence for γ-Hydroxybutyric Acid and Polyhydroxybutyrate Synthesis in Saccharomyces cerevisiae. Applied and Environmental Microbiology, 75, 4231-4239.
https://doi.org/10.1128/AEM.00051-09 - 26. Sano, M., Dohmoto, M. and Ohashi, S. (2016) Characterization of the gatA Gene from Aspergillus oryzae. Journal of Biological Macromolecules, 16, 9-15.
- 27. Andersen, G., Andersen, B., Dobritzsch, D., Schnackerz, K.D. and Piskur, J. (2007) A Gene Duplication Led to Specialized γ-Aminobutyrate and β-Alanine Aminotransferase in Yeast. The FEBS Journal, 274, 1804-1817.
https://doi.org/10.1111/j.1742-4658.2007.05729.x
NOTES

*These authors contributed equally to this work.