Journal of Biosciences and Medicines
Vol.2 No.5(2014), Article
ID:48215,14
pages
DOI:10.4236/jbm.2014.25003
Protein Kinase Cδ-Mediated Posttranslational Phosphorylation of Constitutive Nitric Oxide Synthase Regulates Gastric Mucosal Inflammatory Responses to Helicobacter pylori: Effect of Ghrelin
Bronislaw L. Slomiany, Amalia Slomiany
Research Center, Rutgers School of Dental Medicine, Rutgers, The State University of New Jersey, Newark, USA
Email: slomiabr@sdm.rutgers.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 20 May 2014; revised 25 June 2014; accepted 19 July 2014
ABSTRACT
Disturbances in constitutive nitric oxide synthase (cNOS) activation associated with H. pylori colonization of gastric mucosa are considered of major consequences in defining the extent of inflammatory involvement. As rapid changes in cNOS activation are linked to the enzyme phosphorylation at the specific Ser/Thr residues, we investigated the influence of H. pylori LPS and gastric hormone, ghrelin, on the processes of phosphorylation of these two critical sites in gastric mucosal cells. We show that the LPS-induced reduction in cNOS activity is reflected in the phosphorylation on Thr497, while the countering effect of ghrelin is associated with a rapid increase in cNOS phosphorylation on Ser1179. Further, we demonstrate that cNOS phosphorylation on Thr497 as well as Ser1179 displays dependence on PKCδ. However, while the LPS-induced suppression in cNOS activation shows reliance on the phosphorylation of PKCδ and PI3K on Ser, the effect of ghrelin is manifested by the increase in phosphorylation of PKCδ and PI3K on Tyr, as well as membrane translocation and phosphorylation of Akt on Ser473. Thus, our findings suggest that the LPS-induced suppression in cNOS activation is mediated by PKCδ-controlled phosphorylation of PI3K on Ser that interferes with the membrane recruitment of Akt and promotes cNOS phosphorylation on Thr497, and that ghrelin-elicited up-regulation in cNOS activation relies on the PKCδ-directed phosphorylation of PI3K on Tyr that stimulates the membrane localization of Akt and enhances cNOS phosphorylation on Ser1179.
Keywords:Gastric Mucosa, H. pylori, Ghrelin, PKCδ, PI3K, cNOS Phosphorylation

1. Introduction
Lipopolysaccharide (LPS), an outer membrane component of H. pylori, is recognized as a potent endotoxin capable of eliciting a course of mucosal inflammatory events that characterize gastritis and duodenal ulcers [1] -[4] . Indeed, gastric mucosal responses to H. pylori LPS or associated with gastritis caused by H. pylori infection are manifested by the increase in proinflammatory cytokine production, massive rise in epithelial cell apoptosis, and the disturbances in NO production owing to sustained activation of inducible nitric oxide synthase (iNOS) and the suppression of constitutive nitric oxide synthase (cNOS) systems [5] -[8] . Whilst the high output of NO generated by iNOS is of importance to host defense against bacterial invasion, its sustained activation associated with persistence of inflammatory stimulus is also known to have cytotoxic consequences reflected in transcriptional derangements and the induction of apoptosis [9] -[11] . On the other hand, the low level of NO generated by cNOS appears to access a pool of substrates associated with transient cell-signaling events that are of importance to the maintenance of normal physiological functions [6] [7] [12] . Therefore, the suppression in cNOS activation associated with H. pylori colonization may be of major consequence in defining the extent of gastric mucosal inflammatory involvement.
Whereas the induction of iNOS gene for the sustained increase in NO generation appears to be linked to NF-κB activation, the process of cNOS activation, underlying rapid changes in cNOS-derived NO production, is regulated by a complex set of preand post-translational factors that affect the dynamics of its subcellular targeting and the activity by exposing the enzyme to fatty acid modification through N-myristoylation and thiopalmitoylation, interaction with regulatory cofactors, S-nitrosylation, and the protein phosphorylation [9] [13] -[16] . The literature evidence indicates that the activity of cNOS may be influenced by phosphorylation of the enzyme protein at several residues of Ser, Thr and Tyr, but most extensively characterized are the functional consequences of cNOS phosphorylation on Ser1179and Thr497 [17] -[19] . These data clearly established that phosphorylation of cNOS at the critical Ser1179 with the involvement of PI3K-dependent protein kinase Akt is responsible for a rapid stimulus-promoted cNOS activation, whereas the phosphorylation of cNOS at Thr497 by PKC is associated with a decrease in the enzyme activity [18] -[20] . Thus, these two posttranslational modifications contribute directly to a rapid regulation of cNOS-derived NO release.
Other emerging pertinent data point increasingly convincing to the existence of cross talk among PKC, PI3K and Akt, and suggest that PKC is a major cellular target of LPS-induced Toll-like receptor 4 (TLR4) activation as well as G protein-coupled receptor stimulation [6] [21] -[24] . Indeed, stimulation of cells with LPS or PKC activator, phorbol esters that mimic diacylglycerol, leads to Ser phosphorylation of PI3K regulatory p85 subunit at the Tyr-binding site that impairs PI3K signaling to Akt, and thus accounts for the negative PI3K regulation through the PKC pathway [24] -[27] . Further, gastric hormone, ghrelin, recognized for its role in the NOS system regulation, relays on the GHS-R1a-mediated activation of G protein-dependent PKC pathway that relieves the p85 inhibition and facilitates the PI3K-directed Akt docking, and signaling to cNOS [16] [24] [28] -[30] .
Recently, we have reported on the role of PKCδ in modulation of gastric mucosal inflammatory responses to H. pylori LPS by ghrelin. Our results revealed that while the LPS-elicited PKCδ activation leads to PI3K phosphorylation on Ser that exerts the detrimental influence on the kinase activity, the effect of ghrelin is manifested by phosphorylation of PKCδ and PI3K on Tyr, and associated with up-regulation in the kinase activation [31] . In the present study, we investigated the involvement of PKCδ in controlling the posttranslational processes of cNOS activation through phosphorylation associated gastric mucosal inflammatory responses to H. pylori LPS and ghrelin.
2. Materials and Methods
2.1. Gastric Mucosal Cell Incubation
Gastric mucosal cells, collected by scraping the mucosa of freshly dissected rat stomachs with a blunt spatula, were suspended in five volumes of ice-cold Dulbecco’s modified (Gibco) Eagle’s minimal essential medium (DMEM), supplemented with fungizone (50 µg/ml), penicillin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf serum, and gently dispersed by trituration with a syringe, and settled by centrifugation [7] . Following rinsing, the cells were resuspended in the medium to a concentration of 2 × 107 cell/ml, transferred in 1 ml aliquots to DMEM in culture dishes and incubated under 95% O2 - 5% CO2 atmosphere at 37˚C for up to 2 h in the presence of 0 - 100 ng/ml H. pylori LPS [7] . H. pylori used for LPS preparation was cultured from clinical isolates obtained from ATCC No. 4350 [1] . In the experiments evaluating the effect of ghrelin (rat), PKC inhibitors, Gö6976 and GF109203X (Sigma), PI3K inhibitor, LY294002, and PLC inhibitor, U73122, (Calbiochem), the cells were first preincubated for 30 min with the indicated dose of the agent or vehicle before the addition of the LPS.
2.2. cNOS Activity Assay
The activity of cNOS enzyme in the gastric mucosal cells was measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline using NOS-detect kit (Stratagene). The cells from the control and experimental treatments were homogenized in a sample buffer containing either 10 mM EDTA (to assess iNOS contribution) or 6 mM CaCl2 (for Ca2+-depenedent cNOS), and centrifuged. The aliquots of the resulting supernatant were incubated for 30 min at 25˚C in the presence of 50 µ Ci/ml of L-[3H] arginine, 10 mM NADPH, 5 µM tetrahydrobiopterin, and 50 mM Tis-HCl buffer, pH 7.4, in a final volume of 250 µl. Following addition of stop buffer and Dowex-50 W (Na+) resin, the mixtures were transferred to spin cups, centrifuged and the formed L-[3H] citrulline contained in the flow through was quantified by scintillation counting [16] .
2.3. Akt Activity Assay
The kinase activity of Akt in gastric mucosal cells was measured with the Akt Activity Kit (Calbiochem) by quantifying phosphorylation of a biotinylated peptide substrate (GRPRTSSFAEG). The cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1% deoxycholate, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM PAF, and 1 mM NaF), containing protease inhibitor cocktail (Sigma), at 4˚C for 30 min, centrifuged at 14,000 × g for 15 min, and immunoprecipitated with anti-Akt antibody for 1 h at 4˚C. Protein A/G agarose beads were then added for an additional 1 h, and the immune complex was recovered by centrifugation and thoroughly washed with lysis buffer [16] . The agarose beads were then suspended for 30 min at room temperature in the kinase assay buffer, centrifuged, and the supernatants used for the Akt activity assay by following the manufacturer’s instruction.
2.4. PI3K Activity Assay
The measurement of PI3K activity in gastric mucosal cells was conducted withPI3 Kinase Activity Assay kit, using GST tagged GRP1 protein containing pleckstrin homology (PH) domain that binds PIP3 (EMD Millipore). The mucosal cells from the control and various experimental conditions were lysed at 4˚C for 30 min in the lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM sodium orthovanadate, 1 mM PAF, and 1 mM NaF), containing protease inhibitor cocktail [24] . Following centrifugation at 12,000 × g for 10 min, the supernatant was subjected to protein determination using BCA protein assay kit (Pierce), and the samples containing equal amounts of total protein (500 µg) were immunoprecipitated with p110a antibody overnight at 4˚C. Protein A/G agarose beads were added for an additional 2 h, and the immune complex was recovered by centrifugation and washed two times with cold lysis buffer, one time with fresh cold buffer consisting of 20 mM Tris-HCl buffer, pH 7.4, 150 mM NaCl, 5 mM LiCl, and 0.1 mM sodium orthovanadate, and once with buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, and 0.1 mM sodium orthovanadate [32] [33] . The pellet was resuspended in the reaction buffer and incubated in the glutathione-coated 96 well plate for 1 h with the PI (4, 5) P2 substrate, and the production of generated PI (3, 4, 5) P3 product was determined using horseradish peroxidase/TMB spectrophotometric quantification at 450 nm.
2.5. PKC Activity Assay
Protein kinase C activity measurement in gastric mucosal cells was conducted with ELISA PKC Activity Assay Kit that utilizes microtiter wells pre-coated with a specific synthetic peptide substrate for PKC and phosphospecific substrate antibody (Stressgen). The cells were rinsed with 0.05 M phosphate buffer/saline, pH 7.4, settled by centrifugation, and suspended for 30 min at 4˚C in the lysis buffer consisting of 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 4 mM EGTA, 2 mM EDTA, 1% NP40, 1 mM PMSF, 1 mM sodium orthovanadate, and 10 µg/ml of leupeptin and aprotinin. Following a brief sonication (3 × 10 sec pulses), the samples were centrifuged at 12,000 × g for 10 min and the resulting supernatant was subjected to protein determination using BCA protein assay kit (Pierce). The samples from various experimental treatments were adjusted to 5 µg of crude protein/30 µl, and added to the wells for PKC activity measurement using peroxidase conjugated secondary antibody and TMB spectrophotometric quantification at 450 nm [31] .
2.6. Cell Membrane Preparation
To assess the requirements in terms of phosphorylation on membrane translocation and activation of PI3K and Akt, the gastric mucosal cells from the control and experimental treatments were subjected to cell membrane preparation. For this, the cell suspensions were homogenized for 10 s at 600 rpm in 3 volumes of 50 mM Tris-HCl buffer, pH 7.4, containing 0.25 M sucrose, 25 mM magnesium acetate, 1 mM EDTA, 1 mM dithiothreitol, 10 mM aprotinin, 10 mM leupeptin, 10 mM chymostatin, and 1 mM PMSF [34] . Following centrifugation at 5000 × g for 15 min, the supernatant was diluted with two volumes of cold homogenization buffer and centrifuged at 10,000 × g for 20 min. The resulting supernatant was the subjected to centrifugation at 100,000 × g for 1 h at 4˚C, and the obtained membrane pellet was suspended in the extraction buffer, containing 20 mM HEPES, pH 7.9, 25% glycerol, 0.4 M NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM PMSF. After 30 min of incubation at 4˚C, the suspension was centrifuged at 15,000 × g for 15 min, and the supernatant containing solubilized membrane fraction was collected and stored at −70˚C until use. Protein content of the prepared membrane fractions was analyzed using BCA protein assay kit (Pierce).
2.7. Immunoprecipitation and Immunoblotting
The gastric mucosal cells from the control and experimental treatments were collected by centrifugation and resuspended for 30 min in ice-cold lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 5 mM EDTA, 1 mM sodium orthovanadate, 4 mM sodium pyrophosphate, 1 mM PMSF, and 1 mM NaF), containing 1 µg/ml leupeptin and 1 µg/ml pepstatin [15] [24] . Following brief sonication, the lysates were centrifuged at 10,000 g for 10 min, and the supernatants were subjected to protein determination using BCA protein assay kit (Pierce). The lysates of whole cells as well as those of membrane preparations were then used either for immunoblots analysis, or proteins of interest were incubated with the respective primary antibodies for 2 h at 4˚C, followed by overnight incubation with protein G-Sepharose beads. The immune complexes were precipitated by centrifugation, washed with lysis buffer, boiled in SDS sample buffer for 5 min, and subjected to SDS-PAGE using 40 µg protein/lane. The separated proteins were transferred onto nitrocellulose membranes, blocked for 1 h with 5% skim milk in Tris-buffered Tween (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and probed with specific antibodies directed against cNOS, phospho-cNOS (Ser1179) and phospho-cNOS (Thr497), Akt and phospho-Akt (Ser473), PKCδ and phospho-PKCδ (Ser645), p85a and p110a, and phosphotyrosine (4G10) (EMD Millipore) and phosphoserine PKC substrate (Cell Signaling).
2.8. Data Analysis
All experiments were carried out using duplicate sampling, and the results are expressed as means ± SD. Analysis of variance (ANOVA) and nonparametric Kruskal-Wallis tests were used to determine significance. Any difference detected was evaluated by means of post hoc Bonferroni test, and the significance level was set at P < 0.05.
3. Results
Changes in cNOS activation through phosphorylation are considered of major consequences in defining the extent of gastric mucosal inflammatory involvement in response to H. pylori colonization [6] [7] . Hence, considering the central role of ghrelin in the regulation of NOS system activation [28] [35] , we employed rat gastric mucosal cells and examined the influence of H. pylori LPS and ghrelin on the activity of cNOS and the extent of its phosphorylation on Thr497 and Ser1179. As shown in Figure 1, incubation of gastric mucosal cells with the

Figure 1. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in gastric mucosal cell expression of cNOS activity. The cells, preincubated with 0 or 0.5 µg/ml Gh, were incubated for up to 2 h with the LPS at 100 ng/ml. Values represent the means ±SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS.
LPS caused a time-dependent reduction in cNOS activity reaching a 59% decrease after 2 h, while preincubation with ghrelin led to the reversal in the LPS-induced suppression of cNOS activity. Moreover, western blot analysis revealed that reduction in cNOS activity by the LPS was manifested in a time-dependent phosphorylation of the enzyme protein on Thr497 (Figure 2(a), Figure 2(b)), whereas the reversal by ghrelin in the in LPS-induced suppression in cNOS activity was reflected in a rapid decrease in Thr497 phosphorylation and a marked increase in the cNOS protein phosphorylation on Ser1179 (Figure 2(a), Figure 2(c)).
In further assessment of factors that influence the processes of cNOS activation in the presence of the LPS and ghrelin, we have employed specific pharmacological inhibitors of upstream effectors of cNOS processing, PLC, PKC and PI3K. For this, the gastric mucosal cells prior to incubation with the LPS or ghrelin, were pretreated with PLC inhibitor, U73122, PKC inhibitors, Gö6976 and GF109203X, or PI3K inhibitor, LY294002, and assayed for the activity of cNOS, Akt, PI3K, and PKC enzymes. As illustrated in Figure 3, the effect of the LPS was manifested by a significant increase in the activity of PKC, and the reduction in the activities of PI3K, Akt and cNOS, whereas ghrelin elicited further increase in the activity of PKC, and caused the reversal in the LPS-induced suppression in the activities of PI3K, Akt and cNOS. The activation of PKC by the LPS and ghrelin, moreover, was susceptible to suppression by PLC inhibitor, U73122, as well as the inhibitor of classical and novel PKC isoforms, GF109203X, but not the inhibitor of classical PKC isoforms, Gö6976, thus supporting the involvement of the novel PKC isozyme, identified earlier as PKCδ [31] . In addition, the PKC inhibitor, GF109203X, evoked a significant reduction in the ghrelin-induced activation of PI3K, Akt and cNOS. Furthermore, ghrelin-induced activation of PKCδ as well as that of PI3K, Akt and cNOS displayed susceptibility to LY294002, an inhibitor of PI3K. These results, thus underscore the reliance of the LPS as well as ghrelin on the common upstream PLC/PKCδ signaling pathway in cNOS processing through phosphorylation, and suggest that PI3K plays an essential role in ghrelin-induced gastric mucosal Akt and cNOS activation.
Consequently, to gain additional leads into the pathways utilized by the LPS and ghrelin to affect the cNOS activation, we examined the influence of PKC and PI3K inhibitors on cNOS phosphorylation on Thr497 and Ser1179. The results of western blot analysis revealed that cNOS protein phosphorylation on Thr497, induced by the LPS, was subject to suppression by ghrelin as well as PKC inhibitor, GF109203X, but not the inhibitor of PI3K, LY294002 (Figure 4(a), Figure 4(b)). We also observed that ghrelin-induced cNOS phosphorylation on Ser1179, associated with cNOS activation, was blocked by the inhibitor of PKC as well as that of PI3K. This suggests that cNOS phosphorylation on Thr497 as well as Ser1179 requires PKCδ participation, and that ghrelin-elicited up-regulation in cNOS activation shows dependence on PI3K activity.
Therefore, to address the character of the rapport between PKCδ and PI3K in mediation of cNOS phosphorylation, we assessed the changes in PKCδ and PI3K phosphorylation in response to the LPS and ghrelin. Probing gastric mucosal PKCδ immunoprecipitates with anti-pPKCδ (Ser) and anti-pTyr antibody, we revealed that
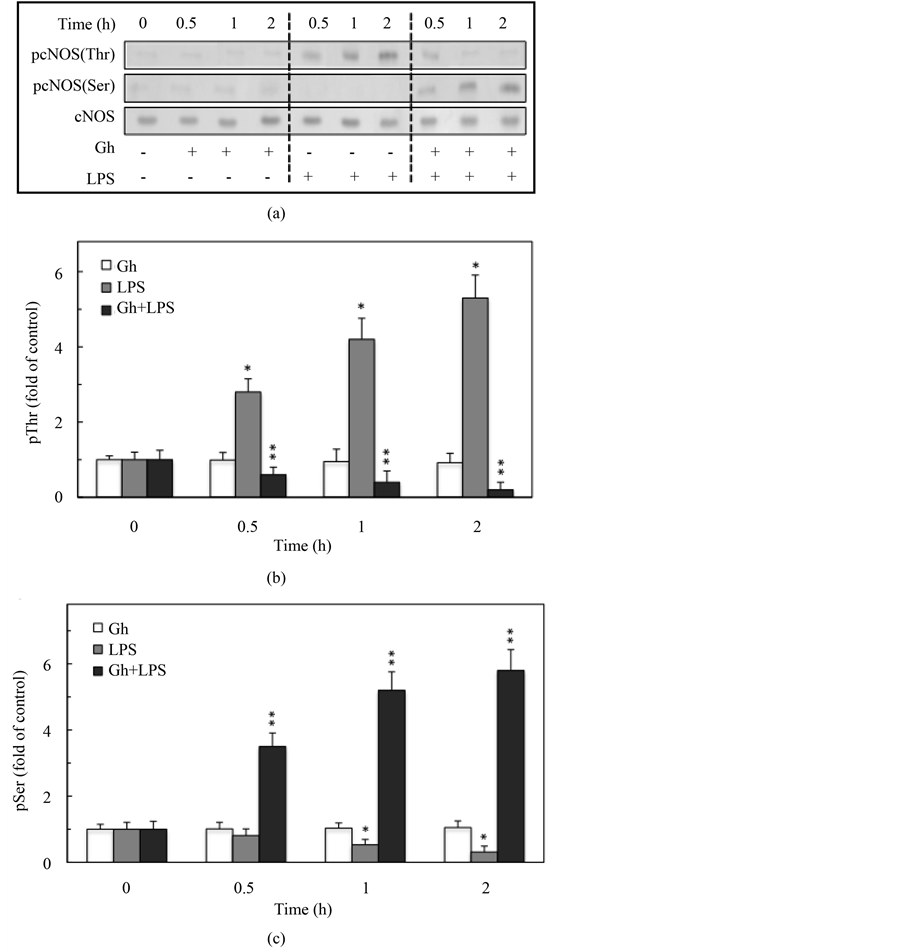
Figure 2. Effect of ghrelin on H. pylori LPS-induced changes in gastric mucosal cell cNOS threonine (Thr497) and serine (Ser1179) phosphorylation. The cells were treated with 0 or 0.5 µg/ml of ghrelin (Gh) and incubated for the indicated time periods with 0 or 100 ng/ml of the LPS. Cell lysates were analyzed by Western blotting with anti-cNOS for total cNOS as well as phosphorylation specific anti-pcNOS (Thr497) and anti-pcNOS (Ser1179) antibody (A), and the relative densities of phosphorylated proteins (B and C) are expressed as fold of control. The total cNOS was used as loading control. The data represent the mean ± SD of four separate experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS.
PKCδ in response to the LPS showed a marked increase in serine phosphorylation, whereas the effect of ghrelin was manifested by PKCδ phosphorylation at tyrosine (Figure 5(a), Figure 5(b)). Further, we found that the LPS elicited phosphorylation of PI3K regulatory p85 subunit on serine, while the effect of ghrelin was reflected in the increase in phosphorylation of PI3K on tyrosine. The effect of ghrelin, moreover, was associated with the
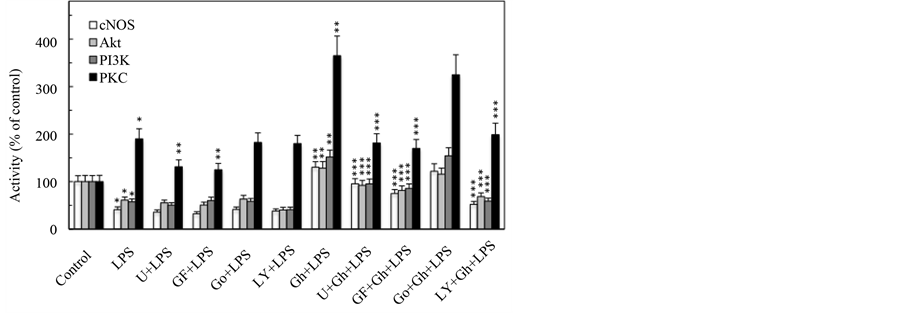
Figure 3. Effect of PLC, PKC and PI3K inhibitors on ghrelin (Gh)-induced changes in the expression of cNOS, Akt, PI3K and PKC activities in gastric mucosal cells exposed to H. pylori LPS. The cells, preincubated with 15 µM of PLC inhibitor, U73122 (U), 5 µM of wide spectrum PKC inhibitor, GF109203X (GF), 10 µM of classical PKC inhibitor, Gö6976 (Go), or 25 µM of PI3K inhibitor, LY294002 (LY), were treated with 0.5 µg/ml Gh, and incubated for 2 h in the presence of 100 ng/ml LPS. The data represent the means ±SD of five separate experiments. *P < 0.05 compared with that of control. **P < 0.5 compared with that of LPS. ***P < 0.05 compared with that of Gh + LPS.
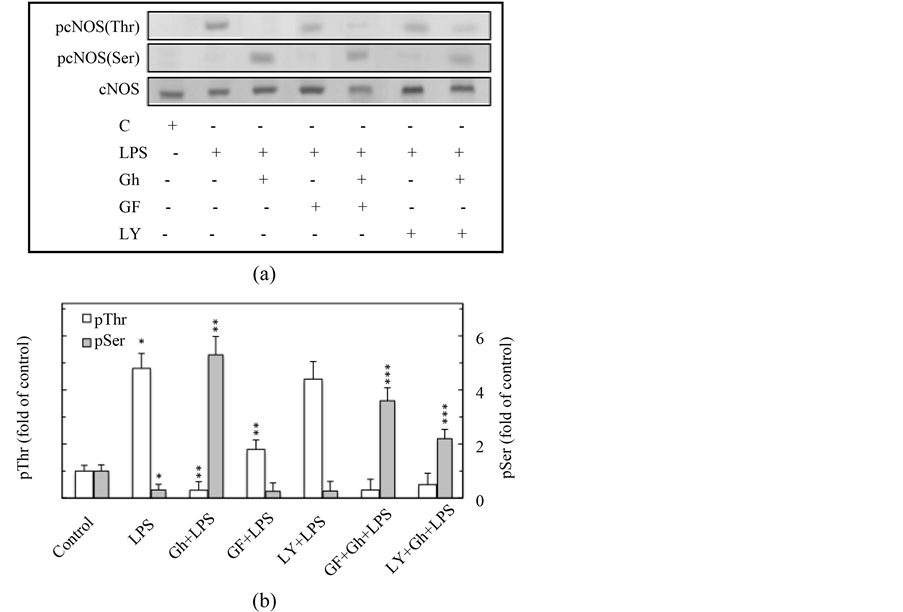
Figure 4. Effect of PKC inhibitor, GF109203X (GF), and PI3K inhibitor, LY2944002 (LY), on ghrelin (Gh)-induced changes in the expression of pcNOS (Thr497) and pcNOS (Ser1179) in gastric mucosal cells exposed to H. pylori LPS. The cells, preincubated with 5 µM GF or 25 µM LY, were treated with Gh at 5 µg/ml and incubated for 2 h in the presence of 100 ng/ml LPS. Cell lysates were analyzed by Western blotting (A) for total cNOS, pcNOS (Thr497) and pcNOS (Ser1179) proteins, and the relative densities of phosphorylated proteins are expressed as fold of control (B). The total cNOS was used as loading control. The data represent the means ± SD of four separate experiments. *P < 0.05 compared with that of control. **P < 0.5 compared with that of LPS. ***P < 0.05 compared with that of Gh + LPS.
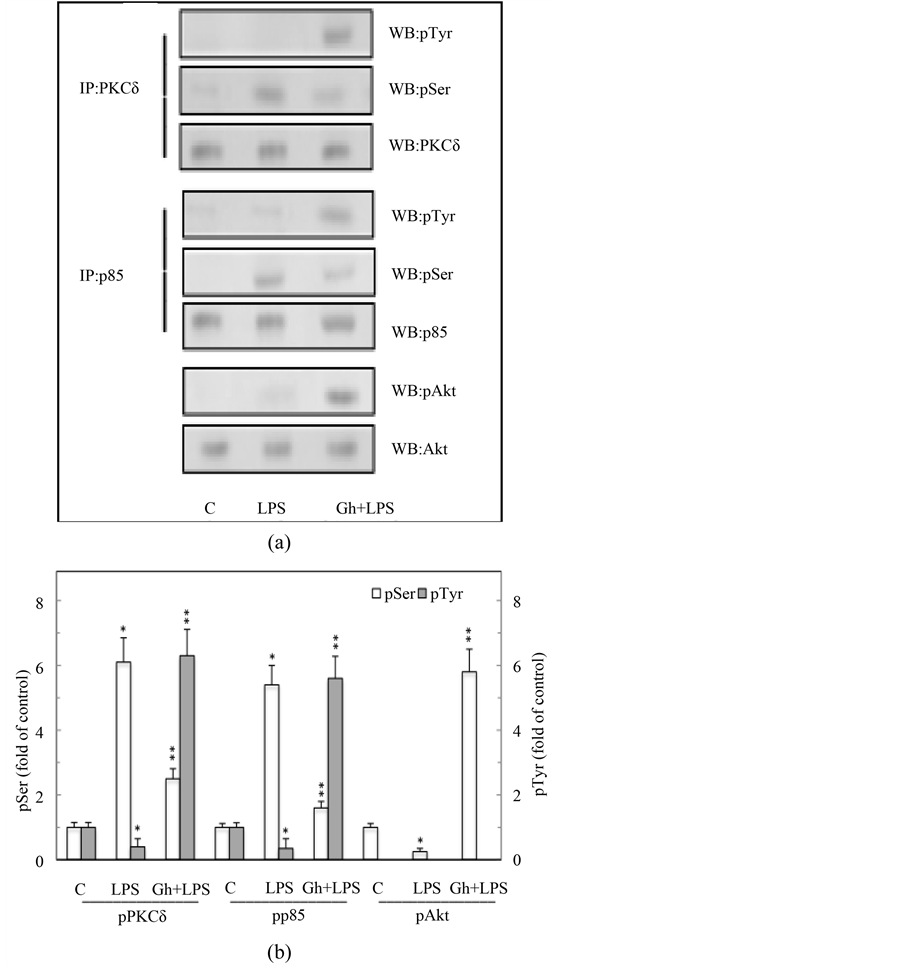
Figure 5. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in gastric mucosal cell PKCδ, PI3K, and Akt phosphorylation. The cells were treated with the LPS at 100 ng/ml or Gh at 0.5 ng/ml + LPS and incubated for 2 h. A, cell lysates were immunoprecipitated (IP) with anti-p85 (PI3K) or anti PKCδ antibody and immunoblotted (WB) with anti-pSer-PKC substrate and anti-pTyr (G410) antibody for phosphorylated p85, while the PKCd immunoprecipitates were immunoblotted with anti-pPKCδ (Ser) and anti-pTyr (4G10). Cell lysates were also blotted with anti-pAkt (Ser) and anti-Akt antibodies. B, the relative densities of phosphorylated proteins are expressed as fold of control. Total PKCδ, p85, and Akt were used as loading control. The data represent the mean ± SD of four separate experiments. *P < 0.05 compared with that of control. **P < 0.5 compared with that of LPS.
phosphorylation of Akt kinase on Ser473 (Figure 5(a), Figure 5(b)). Thus, the LPS-induced suppression in PI3K activation through the regulatory p85 subunit phosphorylation on Ser is of direct relevance to cNOS phosphorylation on Thr497.
Moreover, as the activation of Akt through phosphorylation occurs downstream of PI3K and requires membrane localization [16] [29] [36] , we evaluated the influence of H. pylori LPS and ghrelin on membrane translocation and phosphorylation of PI3K and Akt. For this, the lysates of whole gastric mucosal cells as well as the membrane fraction were immunoprecipitated with anti-p85 (PI3K) or anti-Akt antibody, and subjected to western blot analysis using anti-pSer-PKC substrate and anti-pTyr antibody for phosphorylated p85, and with anti-pAkt (Ser473) for pAkt. The results of analyses revealed that in the absence of ghrelin, the LPS caused the elevation in membrane translocation of Ser-phosphorylated PI3K (p85 subunit), while the effect ghrelin was reflected in a marked drop in the membrane content of Ser-phosphorylated PI3K (p85), rise in phosphorylation of the membrane associated PI3K on Tyr, and the increase in the membrane content of pAkt (Figure 6(a), Figure 6(b)). We have also noticed that the LPS-induced membrane translocation of Ser-phosphorylated PI3K was blocked by the pretreatment of the cells with PKC inhibitor, GF109203X. Further, we observed that PKC inhibitor, GF109203X, exerted the inhibitory effect on ghrelin-induced membrane translocation of Tyr-phosphorylated PI3K as well as pAkt. Therefore, the LPS-induced and PKCδ-mediated PI3K phosphorylation on Ser interferes with the membrane recruitment and activation of Akt.

Figure 6. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in phosphorylation and membrane translocation of PI3K and Akt in gastric mucosal cells. The cells were treated with the LPS at 100 ng/ml, or Gh at 0.5 µg/ml+ LPS, or GF109203X (GF) at 5 µM + LPS, or GF + Gh + LPS, and incubated for 2 h. The lysates of whole cells (T) as well as the membrane (M) fraction were immunoprecipitated (IP) with anti-p85(PI3K) or anti-Akt antibody, and immunoblotted (WB) with anti-pSer-PKC substrate and anti-pTyr (G410) antibody for phosphorylated p85, and with anti-pAkt (Ser1179) for pAkt (A). The relative densities of phosphorylated proteins are expressed as fold of control (B). Total p85 and Akt were used as loading control. The data represent the mean ± SD of four separate experiments. *P < 0.05 compared with that of control. **P < 0.5 compared with that of LPS. ***P < 0.05 compared with that of Gh + LPS.
4. Discussion
The activity of cNOS and the consistently low and measured quantities of NO generated by this isoform in conjunction with local signaling events is recognized of a key importance to the maintenance of gastric mucosal integrity and its normal physiological function. Hence, to maintain the local transient NO demands, the activity of cNOS is tightly regulated by a diverse array of preand post-translational factors that affect its subcellular targeting, interaction with regulatory cofactors, S-nitrosylation, and phosphorylation [9] [13] -[16] [19] . Indeed, the cNOS enzyme protein phosphorylation at the critical Ser1179 with the involvement of PI3K/Akt is now recognized as an important post-translational mechanism for rapid stimulus-promoted cNOS activation, whereas phosphorylation of cNOS at theThr497 with PKC participation is associated with a decrease in the enzyme activity [18] -[20] . As disturbances in cNOS activation are considered of major consequences in defining the extent of gastric mucosal inflammatory involvement in response to H. pylori colonization and the PKC is also regarded of pivotal importance in the PI3K/Akt signaling cascade [6] [24] [31] , we investigated the influence of H. pylori LPS and gastric hormone, ghrelin, on the processes of cNOS activation through phosphorylation.
Our results revealed that incubation of gastric mucosal cells with the LPS led to a time-dependent reduction in cNOS activity that was reflected in a gradual phosphorylation of the enzyme protein on Thr497. Furthermore, we found that preincubation with ghrelin, recognized for its prominent role in modulation of gastric mucosal inflammatory responses to H. pylori infection [7] [15] [37] -[39] , exerted counteractive effect on the LPS-induced suppression in cNOS activity as well as the extent of its phosphorylation on Thr497, and was manifested by the rapid increase in the activity and phosphorylation of cNOS on Ser1179. These findings are in keeping with the literature data demonstrating that post-translational phosphorylation of cNOS protein at the critical Thr/Ser plays an essential role in modulation of cNOS-dependent NO production [17] -[19] , and underscore the importance of a coordinated dephosphorylation of Thr497 and phosphorylation of Ser1179 in a rapid up-regulation in cNOS activation by ghrelin in gastric mucosal response to H. pylori colonization. Further, we observed that the LPS-elicited drop in cNOS activity was accompanied by a marked increase in the activity of PKC, and the suppression in the activities of PI3K and Akt, while ghrelin evoked further rise in PKC activity as well as exerted counteracting effect on the LPS-induced suppression in the activities of PI3K, Akt and cNOS. The activation of PKC by the LPS and ghrelin, moreover, was susceptible to suppression by PLC inhibitor, U73122. Hence, we concluded that up-regulation in PLC/PKC activity plays a pivotal role in modulation of cNOS activation by the LPS as well as ghrelin. This interpretation of our results is in accord with the preponderant evidence indicating that PKC is a major cellular target of LPS-induced TLR4 activation as well as G protein-coupled GHS-R1a receptor stimulation by ghrelin [6] [22] -[24] [30] [31] [39] .
Moreover, the assays of PKC activity in the in the presence of Gö6976, a classical PKC inhibitor, as well as the classical and novel PKC inhibitor, GF109203X, revealed that only GF109203X attenuated the activation of PKC by the LPS and ghrelin. In addition, GF109203X also elicited a distinct reduction in the ghrelin-induced activation of PI3K, Akt and cNOS, thus supporting the involvement of the novel isoform of PKC, identified earlier as PKCδ [31] , in cNOS processing through phosphorylation. These findings are consistent with several previous studies where PKCδ has been implicated in TLR-mediated regulation of NO production in response to LPS [23] [40] [41] . Therefore, to gain further leads into the pathways utilized by H. pylori LPS and ghrelin to affect the cNOS activation we examined the influence of PKC and PI3K inhibitors on cNOS phosphorylation at Thr497 and Ser1179. We found that the LPS-induced phosphorylation of cNOS on Thr497 was not only the subject to suppression by ghrelin but also displayed susceptibility to PKC inhibitor, GF109203X, whereas the ghrelin-elicited cNOS phosphorylation on Ser1179 was blocked by the inhibitor of PKC, GF109203X, as well as the inhibitor of PI3K, LY294002. Accordingly, we asserted that while cNOS phosphorylation on Thr497 as well as Ser1179 requires PKCδ participation, the up-regulation in cNOS activation by ghrelin also shows dependence on PI3K. Indeed, the role of PI3K in mediation of Akt directed cNOS activation through phosphorylation on Ser1179 is well recognized [24] [28] -[33] .
As PI3K is linked to majority of TLR-initiated downstream signaling events, and the evidence points to the existence of cross talk among PKC and PI3K [24] [29] [31] [33] [42] , we next addressed the character of the rapport between PKCδ and PI3K in mediation of cNOS phosphorylation in response to the LPS and ghrelin. By examining the gastric mucosal PKCδ and PI3K regulatory p85 subunit phosphorylation patterns, we established that in response to stimulation by the LPS both PKCδ and PI3K showed a marked increase in their Ser phosphorylation, whereas the effect of ghrelin was associated with the phosphorylation of PKCδ and PI3K on Tyr, and that of Akt on Ser473. Interestingly, the LPS-induced phosphorylation of PI3K on Ser was reflected not only in the suppression of PI3K activity but also that of Akt and cNOS. Furthermore, studies indicate that stimulation of cells with LPS or PKC activator, phorbol esters leads to Ser phosphorylation of the SH2 (Src homology-2) domain of the p85 regulatory subunit at the Tyr-binding site, and results in the impairment of PI3K activation and the inhibition of Akt activation [26] [27] . Indeed, the activation of Akt is known to occur downstream of PI3K and involves the PI3K-catalyzed generation of the lipid second messenger, PIP3, which serves as a recognition site for N-terminal PH (pleckstrin homology) domain of Akt [15] [16] [30] [36] [43] . Hence, it is apparent that in the absence of PI3K platform for Akt-mediated cNOS activation, the LPS-induced up-regulation in PKCδ activation leads to the increase in cNOS phosphorylation on Thr497. This interpretation is supported by our findings showing that phosphorylation of PI3K on Ser by the LPS as well as that on Tyr induced by ghrelin takes place downstream of PKCδ as both events exhibited susceptibility to the PKC inhibitor, GF109203X, and the reports indicating that PKC through the interaction with PH domain-containing partners can influence the process of PI3K activation [31] [33] [39] .
The evidence from a variety of cellular systems indicates that subcellular localization plays an important regulatory role determining the activation and substrate specificity of Akt PI3K and PKC [22] [24] [27] [29] [44] Indeed, we have shown recently that H. pylori LPS caused the translocation of Ser-phosphorylated PKCδ from the cytosol to the cell membrane fraction, while the effect of ghrelin was reflected in phosphorylation of the membrane-associated PKCδ on Ser as well as Tyr [31] . The results reported herein revealed that the LPS-elicited increase in gastric mucosal cell membrane translocation of Ser-phosphorylated PI3K (p85) was associated with cNOS phosphorylation on Thr497, whereas the effect of ghrelin, manifested by the rise in phosphorylation of the membrane associated PI3K on Tyr and that of pAkt on Ser473, was coupled to cNOS phosphorylation on Ser1179. Equally significant is to note that in our study the membrane translocation of PI3K phosphorylated on Ser as well as Tyr and that of Akt phosphorylated on Ser473 exhibited susceptibility to PKC inhibitor GF109203X, thus indicating that PKCδ is a primary linchpin in controlling the processes of cNOS activation through phosphorylation at the posttranslational level.
5. Conclusion
In summary, our study demonstrates that PKCδ plays a critical role in the modulation of gastric mucosal in flammatory responses to H. pylori LPS by ghrelin (Figure 7). While the LPS-elicited PKCδ activation leads to
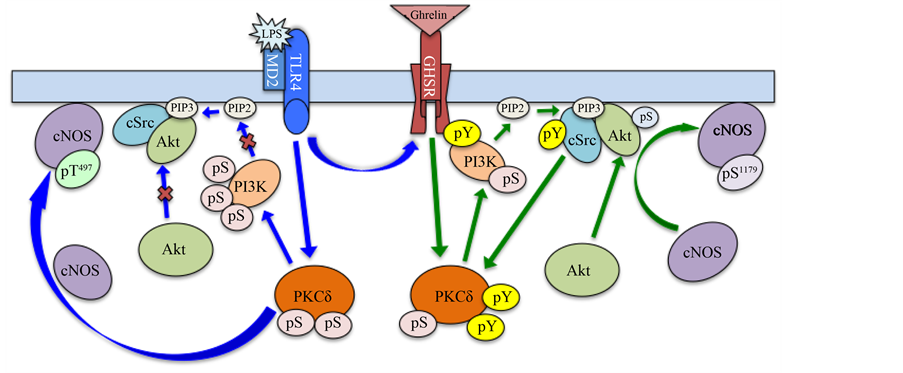
Figure 7. Schematic representation of the signaling pathways involved in the regulation of gastric mucosal posttranslational cNOS activation through phosphorylation in response to H. pylori LPS and ghrelin. Stimulation of Toll-like receptor 4 (TLR4)/MD2 by the LPS triggers the activation of PKCδ that leads to the increase in PI3K phosphorylation on Ser [31] , which interferes with the membrane recruitment of Akt and promotes the PKCδ-induced cNOS phosphorylation on Thr497 that results in a decrease in cNOS activity. Ligation of the growth hormone secretagogue receptor (GHS-R1a) by ghrelin leads to PKCδ-directed activation of PI3K through phosphorylation on Tyr that stimulates the membrane localization of Akt and results in the up-regulation in cNOS activation through phosphorylation on Ser1179. PIP2 phosphatidylinositol 4, 5-bisphosphate, PIP3 phosphatidylinositol 3, 4, 5-tirisphosphate, pS phosphoserine, pT phosphothreonine, pY phosphotyrosine.
the increase in PI3K phosphorylation on Ser that interferes with the membrane recruitment of Akt and promotes cNOS phosphorylation on Thr497, the effect of ghrelin is manifested by phosphorylation of PKCδ and PI3K on Tyr, and that of Akt on Ser473, and associated with up-regulation in cNOS activation through phosphorylation on Ser1179.
References
- Piotrowski, J., Piotrowski, E., Skrodzka, D., Slomiany, A. and Slomiany, B.L. (1997) Induction of Acute Gastritis and Epithelial Apoptosis by Helicobacter pylori Lipopolysaccharide. Scandinavian Journal of Gastroenterology, 32, 203-211. http://dx.doi.org/10.3109/00365529709000195
- De Boer, W.A. (2000) Helicobacter pylori Infection: Focus on a “Search-and-Treat” Strategy for Ulcer Disease. Scandinavian Journal of Gastroenterology, 35, 4-9.
- Reider, G., Hoffman, J.A., Hatz, R.A., Stolte, M. and Enders, G.A. (2003) Up-Regulation of Inducible Nitric Oxide Synthase in Helicobacter pylori-Associated Gastritis May Represent an Increased Risk Factor to Develop Gastric Carcinoma of the Intestinal Type. International Journal of Medical Microbiology, 293, 403-412. http://dx.doi.org/10.1078/1438-4221-00280
- Slomiany, B.L. and Slomiany, A. (2000) Blockade of p38 Mitogen-Activated Kinase Pathway Inhibits Inducible Nitric Oxide Synthase and Gastric Mucosal Inflammatory Reaction to Helicobacter pylori Lipopolysaccharide. Inflammo-pharmacology, 8, 371-382. http://dx.doi.org/10.1163/156856000750264438
- Fu, S., Ramanujam, K.S., Wong A., et al. (1999) Increased Expression and Cellular Localization of Inducible Nitric Oxide Synthase and Cyclooxygenase-2 in Helicobacter pylori Gastritis. Gastroenterology, 116, 1319-1329. http://dx.doi.org/10.1016/S0016-5085(99)70496-8
- Backert, S. and Neumann, M. (2010) What a Disorder: Proinflammatory Signaling Pathways Induced by Helicobacter pylori. Trends in Microbiology, 18, 479-486. http://dx.doi.org/10.1016/j.tim.2010.08.003
- Slomiany, B.L. and Slomiany, A. (2010) Ghrelin Protection against Lipopolysaccharide-Induced Gastric Mucosal Cell Apoptosis Involves Constitutive Nitric Oxide Synthase-Mediated Caspase-3 S-Nitrosylation. Mediators of Inflammation, 2010, Article ID: 280464.
- Slomiany, B.L. and Slomiany, A. (2011) Role of Constitutive Nitric Oxide Synthase in Regulation of Helicobacter pylori-Induced Gastric Mucosal Cyclooxygenase-2 Activation through S-Nitrosylation: Mechanism of Ghrelin Action. Open Journal of Gastroenterology, 1, 13-22. http://dx.doi.org/10.4236/ojgas.2011.12003
- Korhonen, R., Lahti, A., Kankaanrata, H. and Moilanen, E. (2005) Nitric Oxide Production and Signaling in Inflammation. Current Drug Targets: Inflammation & Allergy, 4, 471-479. http://dx.doi.org/10.2174/1568010054526359
- Cuzzocrea, S. and Salvemini, D. (2007) Molecular Mechanisms Involved in the Reciprocal Regulation of Cyclooxygenase and Nitric Oxide Synthase Enzymes. Kidney International, 71, 290-297. http://dx.doi.org/10.1038/sj.ki.5002058
- Maa, M.C., Chang, M.Y., Chen, Y.J., et al. (2008) Requirement of Inducible Nitric-Oxide Synthase in Lipopolysaccharide-Mediated Src Induction and Macrophage Migration. Journal of Biological Chemistry, 283, 31408-31416. http://dx.doi.org/10.1074/jbc.M801158200
- Chanvorachote, P., Nimmannit, U., Wang, L., et al. (2005) Nitric Oxide Negatively Regulates Fas CD95-Induced Apoptosis through Inhibition of Ubiquitin-Proteasome-Mediated Degradation of FLICE Inhibitory Protein. Journal of Biological Chemistry, 280, 42044-42050. http://dx.doi.org/10.1074/jbc.M510080200
- Fulton, D., Gratton, J.P. and Sessa, W.C. (2001) Post-Translational Control of Endothelial NO Synthase: Why Isn’t Calcium/Calmodulin Enough? Journal of Pharmacology and Experimental Therapeutics, 299, 818-824.
- Erwin, P.A., Mitchell, D.A., Sartoretto, J., et al. (2006) Subcellular Targeting and Differential S-Nitrosylation of Endothelial Nitric-Oxide Synthase. Journal of Biological Chemistry, 281, 151-157. http://dx.doi.org/10.1074/jbc.M510421200
- Slomiany, B.L. and Slomiany, A. (2010) Role of Constitutive Nitric Oxide Synthase S-Nitrosylation in Helicobacter pylori-Induced Gastric Mucosal Cell Apoptosis: Effect of Ghrelin. Inflammopharmacology, 18, 233-240. http://dx.doi.org/10.1007/s10787-010-0051-7
- Slomiany, B.L. and Slomiany, A. (2010) Helicobacter pylori Induces Disturbances in Gastric Mucosal Akt Activation through Inducible Nitric Oxide Synthase-Dependent S-Nitrosylation: Effect of Ghrelin. ISRN Gastroenterology, 2011, Article ID: 308727.
- Sessa, W.C. (2004) eNOS at a Glance. Journal of Cell Science, 117, 2427-2429.
- Chen, C.A., Druhan, L.J., Varadharaj, S., Chen, Y.R. and Zweier, J.L. (2008) Phosphorylation of Endothelial Nitric-Oxide Synthase Regulates Superoxide Generation from the Enzyme. Journal of Biological Chemistry, 283, 27038-27047.
- Fleming, I. (2010) Molecular Mechanisms Underlying the Activation of eNOS. Pflugers Archives: European Journal of Physiology, 459, 793-806.
- Oubaha, M. and Gratton, J.P. (2009) Phosphorylation of Endothelial Nitric Oxide Synthase by Atypical PKCz Contributes to Angiopoietin-1-Dependent Inhibition of VEGF-Induced Endothelial Permeability. Blood, 114, 3343-3351.
- Lutrell, D.K. and Lutrell, L.M. (2004) Not So Strange Bedfellows: G-Protein-Coupled Receptors and Src Family Kinases. On-cogene, 23, 7969-7978. http://dx.doi.org/10.1038/sj.onc.1208162
- Kazi, J.U. (2011) The Mechanism of Protein Kinase C Regulation. Frontiers in Biology, 6, 328-336.
- Loegering, D.J. and Lennartz, M.R. (2011) Protein Kinase C and Toll-Like Receptor Signaling. Enzyme Research, 2011, Article ID: 537821.
- Slomiany, B.L. and Slomiany, A. (2014) Role of Ghrelin-Induced Phosphatidylinositol 3-Kinase Activation in Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori. Inflammopharmacology, 22, 169-177.http://dx.doi.org/10.1007/s10787-013-0190-8
- Guha, M. and Mackman, N. (2002) The Phosphatidylinositol 3-Kinase-Akt Pathway Limits Lipopolysaccharide Activation of Signaling Pathways and Expression of Inflammatory Mediators in Human Monocytic Cells. Journal of Biological Chemistry, 277, 32124-32132.
- Fukao, T. and Koyasu, S. (2003) PI3K and Negative Regulation of TLR Signaling. Trends in Immunology, 24, 358-363. http://dx.doi.org/10.1016/S1471-4906(03)00139-X
- Lee, J.Y., Chiu, Y.H., Asara, J. and Cantley, L.C. (2011) Inhibition of PI3K Binding to Activators by Serine Phosphorylation of PI3K Regulatory Subunit p85α Src Homology-2 Domains. Proceedings of the National Academy of Sciences of the United States of America, 108, 14157-14162. http://dx.doi.org/10.1073/pnas.1107747108
- Xu, X., Bong, S.J., Chang, H.H. and Jin, Z.G. (2008) Molecular Mechanism of Ghrelin-Mediated Endothelial Nitric Oxide Synthase Activation. Endocrinology, 149, 4183-4192. http://dx.doi.org/10.1210/en.2008-0255
- Cahill, C.M., Rogers, J.T. and Walker, W.A. (2012) The Role of Phosphoinositide 3-Kinase Signaling in Intestinal Inflammation. Journal of Signal Transduction, 2012, Article ID: 358476. http://dx.doi.org/10.1155/2012/358476
- Lodeiro, M., Alén, B.O., Mosteiro, C.S., Beiroa, D., Nogueiras, R., Theodoropoulou, M., et al. (2011) The SHP-1 Protein Tyrosine Phosphatase Negatively Modulates Akt Signaling in the Ghrelin/GHSR1a System. Molecular Biology of the Cell, 22, 4182-4191. http://dx.doi.org/10.1091/mbc.E11-04-0373
- Slomiany, B.L. and Slomiany, A. (2014) Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori via Ghrelin-Induced Protein Kinase Cδ Tyrosine Phosphorylation. Inflammopharmacology, 22, 251-262.
- Slomiany, A. and Slomiany, B.L. (2012) Phosphatidylglycerol-Containing ER-Transport Vesicles Built and Restore Outer Mitochondrial Membrane and Deliver Nuclear DNA Translation Products to Generate Cardiolipin in the Inner Mitochondrial Membrane. Advances in Biological Chemistry, 2, 132-145. http://dx.doi.org/10.4236/abc.2012.22016
- Slomiany, B.L. and Slomiany, A. (2011) Ghrelin Suppression of Helicobacter pylori-Induced Gastric Mucosal Expression of iNOS Is Mediated through the Inhibition of IKK-β Activation by cNOS-Dependent S-Nitrosylation. Open Journal of Cell Biology, 1, 1-10.
- Wiles, T.J., Dhakal, B.K., Eto, D.S. and Mulvey, M.A. (2008) Inactivation of Host Akt/Protein Kinase B Signaling by bacterial Pore-Forming Toxins. Molecullar Biology of the Cell, 19, 1427-1438. http://dx.doi.org/10.1091/mbc.E07-07-0638
- Cao, X., Kambe, F., Moeller, L.C., Refetoff, S. and Seo, H. (2005) Thyroid Hormone Induces Rapid Activation of Akt/ Protein Kinase B-Mammalian Target of Rapamycin p70S6K Cascade through Phosphatidylinositol 3-Kinase in Human Fibroblasts. Molecular Endocrinology, 19, 102-112. http://dx.doi.org/10.1210/me.2004-0093
- Walker, V.G., Ammer, A., Cao, Z., Clump, A.C., Jiang, B.H., Kelley, L.C., et al. (2007) PI3K Activation Is Required for PMA-Directed Activation of cSrc by AFAP-110. American Journal of Physiology, Cell Physiology, 293, C119-C132.
- Osawa, H., Nakazato, M., Date, Y., Kita, H., Ohnishi, H., Ueno, H., et al. (2005) Impaired Production of Gastric Ghrelin in Chronic Gastritis Associated with Helicobacter pylori. Journal of Clinical Endocrinology and Metabolism, 90, 10-16. http://dx.doi.org/10.1210/jc.2004-1330
- Waseem, T.M., Duxbury, M., Ito, H., Ashley, S.W. and Robinson, M.K. (2008) Exogenous Ghrelin Modulates Release of Pro-Inflammatory and Anti-Inflammatory Cytokines in LPS-Stimulated Macrophages through Distinct Signaling Pathways. Surgery, 143, 334-342. http://dx.doi.org/10.1016/j.surg.2007.09.039
- Slomiany, B.L. and Slomiany, A. (2013) Induction in Gastric Mucosal Prostaglandin and Nitric Oxide by Helicobacter pylori Is Dependent on MAPK/ERK-Mediated Activation of IKK-β and cPLA2: Modulatory Effect of Ghrelin. Inflammopharmacology, 21, 241-251. http://dx.doi.org/10.1007/s10787-013-0169-5
- Kubo-Murai, M., Hazeki, K., Sukenobu, N., Yoshikawa, K., Nigorikawa, K., Inoue, K., et al. (2007) Protein Kinase Cδ Binds TIRAP/Mal to Participate in TLR Signaling. Molecular Immunology, 44, 2257-2264. http://dx.doi.org/10.1016/j.molimm.2006.11.005
- Wen, J., Ribeiro, R. and Zhang, Y. (2011) Specific PKC Isoforms Regulate LPS-Stimulated iNOS Induction in Murine Microglial Cells. Journal of Neuroinflammation, 8, 38.
- Carpenter, S. and O’Neill, L.A.J. (2009) Recent Insights into the Structure of Toll-Like Receptors and Post-Translational Modifications of Their Associated Signaling Proteins. Biochemical Journal, 422, 1-10.http://dx.doi.org/10.1042/BJ20090616
- Handa, M., Feng, J. and Hemmings, B.A. (2004) Structure, Regulation and Function of PKB/AKT—A Major Therapeutic Target. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1697, 3-16.http://dx.doi.org/10.1016/j.bbapap.2003.11.009
- Matsuda, N., Hayashi, Y., Takahashi, Y. and Hattori, Y. (2006) Phosphorylation of Endothelial Nitric-Oxide Synthase Is Diminished in Mesenteric Arteries from Septic Rabbits Depending on the Altered Phosphatidylinositol 3-Kinase/Akt Pathway: Reversal Effect of Fluvastatin Therapy. Journal of Pharmacology and Experimental Therapeutics (JPET), 319, 1348-1354.