International Journal of Clinical Medicine
Vol.06 No.11(2015), Article ID:61065,12 pages
10.4236/ijcm.2015.611107
Oligomerized Amyloid-β1-40 Peptide Favors Cholesterol, Oxysterol, and Fatty Acid Accumulation in Human Neuronal SK-N-BE Cells
Amira Zarrouk1,2*, Thomas Nury2, Mohamed Hammami1, Gérard Lizard2
1Laboratoire de Biochimie (LR12ES05)-Lab-NAFS “Nutrition, Aliments Fonctionnels et Santé Vasculaire”, Faculté de Médecine, Université de Monastir, Monastir, Tunisie
2Equipe “Biochimie du Peroxysome, Inflammation et Métabolisme Lipidique” (EA 7270), Université de Bourgogne-Franche Comté/INSERM, Dijon, France
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 21 August 2015; accepted 10 November 2015; published 13 November 2015

ABSTRACT
Amyloid peptide, the main component of senile plaques, is a major biological characteristic of Alzheimer’s disease (AD). The aim of the present study conducted on human neuronal SK-N-BE cells was to evaluate whether oligomerized Aβ1-40-induced cell damages was associated with lipid modifications. Under treatment with Aβ1-40 (10 - 100 µM; 24 - 48 h), cell viability was recorded with the MTT test and by measuring LDH activity. Mitochondrial transmembrane potential and ATP production were assessed using flow cytometry and a luciferase-based ATP bioluminescence assay, respectively. Annexin V-CF647 staining assay for cell apoptosis detection was performed using flow cytometry. Potentially intracellular cytotoxic lipids (oxysterols: 7α-hydroxycholesterol (7α-OHC), 7β-hydroxycholesterol (7β-OHC), and 7-ketocholesterol (7KC), 24(S)-hydroxycholes- terol; arachidonic acid (C20:4 n-6); VLCFAs (C22:0, C24:0, C24:6 and C26:0)) were measured using gas chromatography coupled with mass spectrometry. The cellular level of docosahexaenoic acid (C22:6 n-3), often altered in AD, was also quantified. In the presence of Aβ1-40, the percentage of MTT-positive cells decreased and was associated with an increase in LDH activity. In addition, treatment with oligomerized Aβ1-40 induced a decrease of mitochondrial transmembrane potential as well as an apoptotic cell death. Sterol analysis revealed a higher cholesterol level and a signifi- cant increase of cytotoxic oxysterols per cell (7KC + 7β-OHC), and of the [(7β-OHC + 7KC)/choles- terol] ratio, considered as a lipid peroxidation index, in Aβ1-40-treated cells. An enhancement of C20:4 n-6, C22:6 n-3 and saturated VLCFAs was also observed. Therefore, Aβ1-40-induced side ef- fects are associated with intracellular accumulation of lipids, especially cholesterol, oxysterols (7β-OHC, 7KC), C20:4 n-6, and saturated VLCFAs, which could in turn contribute to neurotoxicity.
Keywords:
SK-N-BE Cells, Oligomerized Aβ1-40, Cholesterol, Oxysterols, Very Long Chain Fatty Acids

1. Introduction
Alzheimer’s disease (AD) is the most predominant dementia in the elderly. Aggregated amyloid deposits are the main components of senile plaques, which are characteristics of the AD brain [1] . Amyloid beta peptide (Aβ), known to trigger numerous types of neuronal damages, is generated by sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretase [2] .
At the moment, AD has been associated with several risk factors, and among them lipid alterations have been suspected [3] [4] . The γ-secretase cleavage site, which is directly centered within the transmembrane domain, suggests that membrane composition, especially the lipid environment, may influence Aβ generation [5] . Furthermore, numerous studies support the notion that an alteration of cholesterol metabolism and cholesterol oxide production, particularly 24(S)-hydroxycholesterol (24S-OHC), 27-hydroxycholesterol (27-OHC), 7KC, and 7β-OHC, can play critical roles in degenerative diseases such as AD [3] [4] [6] . Currently, the relationship between hypercholesterolemia and dementia is not clearly understood [7] . In humans, it is however well established that the APOE polymorphism and in particular the presence of the ε4 isoform is associated with a greater risk of developing AD [8] . In contrast to cholesterol, several arguments suggest that oxysterols probably play critical roles in AD. Increased levels of 7KC and 7β-OHC, resulting from autoxidation of cholesterol during oxidative stress [9] , have been shown in brain lesions [10] as well as an increase in both 27-OHC and 24S-OHC in the frontal cortex of AD patients [11] . In addition, enhanced plasma levels of 24S-OHC, which could be a consequence of neuronal damages, have been reported during the first stages of dementia [12] . However, decreased plasma levels of 24S-OHC were reported in other stages and were associated with cerebral atrophy and severity of dementia [13] . It should be noted that 7KC, 7β-OHC, and 24S-OHC are potent inducers of cell death and also have pro-oxidant and pro-inflammatory activities on numerous cells, including those of the central nervous system [14] . Furthermore, it was reported that 24S-OHC downregulates APP trafficking resulting in suppression of Aβ production [15] . In contrast, 27-OHC enhances production of Aβ1-42 by up-regulating APP and β-secretase [16] .
Another finding relating lipid metabolism disorders to AD pathogenesis was the accumulation of saturated very long chain fatty acids (saturated VLCFAs: docosanoic acid (C22:0), tetracosanoic acid (C24:0) and hexacosanoic acid (C26:0)) in cortical regions of brains of AD patients with stages V-VI compared with those modestly affected (stages I-II) based on the neuropathological Braak classification [17] . In addition, in the plasma of demented patients, including AD patients, a marked accumulation of C26:0 was observed [18] . This fatty acid accumulation, in particular C24:0 and C26:0, was suspected of being the consequence of peroxisomal dysfunctions since these VLCFAs are metabolized in the peroxisome by β-oxidation [19] . In agreement with the possible alteration of peroxisomal metabolism suspected in AD, modifications of docosahexaenoic acid (DHA, C22:6 n-3) and plasmalogen levels were reported [19] . These various observations indicate substantial lipid alterations in AD, which may contribute to the initiation and/or progression of the disease.
With important roles attributed to Aβ in the development of AD due to its multiple neurotoxic activities [20] , and since major lipid modifications involving increased levels of cholesterol and neurotoxic lipids (oxysterols: 7KC, 7β-OHC, 24S-OHC; saturated VLCFAs: C22:0, C24:0, C26:0) can be observed in the brain, the cerebrospinal fluid, and/or the plasma of AD patients [4] [17] [18] , it was of interest to determine the ability of Aβ to induce lipid disorders on neuronal cells. So, the present study was realized on human neuroblastoma SK-N-BE cells to simultaneously evaluate the cytotoxic activity of the oligomerized Aβ1-40 and its ability to trigger lipid alterations. Cell viability was recorded with the MTT test, quantification of ATP level and LDH activity, and measurement of mitochondrial transmembrane potential with DiOC6(3). The induction of apoptosis was evaluated with Annexin V. The impact on cholesterol, oxysterols, and fatty acid levels was determined using gas chromatography coupled with mass spectrometry.
2. Material and Methods
2.1. Cells and Cell Treatments
As previously described, human neuronal cells (SK-N-BE) were seeded at 200,000 cells per well in 24-well microplates containing 1 mL of culture medium constituted by Dulbecco’s Modified Eagle Medium with L-glu- tamine (DMEM) (Lonza) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Pan Biotech) and 1% antibiotics (100 U/mL penicillin, 100 mg/mL streptomycin) (Pan Biotech) [21] . Aβ1-40 peptide (Sigma Aldrich) was solubilized in 1 mL of phosphate buffered saline exempt of calcium and incubated for 7 days at 37˚C.
2.2. Evaluation of Mitochondrial Activity with the Colorimetric MTT Assay
The MTT assay was carried as previously described [21] on SK-N-BE cells plated in 24-well flat-bottom culture plates with oligomeric Aβ1-40 (10 and/or 100 µM, 48 h). The MTT assay was used to evaluate the effects of oligomeric Aβ1-40 on mitochondrial activity and/or cell growth. Indeed, the tetrazolium salt (MTT) is reduced to formazan in the metabolic active cells by mitochondrial succinate dehydrogenase. A microplate reader was used to record mitochondrial activity and/or cell growth at a wavelength of 570 nm.
2.3. LDH Release Assay
Cytotoxicity induced by Aβ1-40 was assessed by lactate dehydrogenase (LDH) leakage into the culture medium. The LDH activity was determined using a commercially available kit (Cayman Chemical Company). The assay is based on the conversion of lactate to pyruvate in the presence of LDH with parallel reduction of NAD. NADH formed from the above reaction is used by diaphorase to catalyze the reduction of tetrazolium salt to formazan which is proportional to the quantity of LDH released in the medium. A microplate reader was used at a wavelength of 490 nm and LDH activity was determined from the calibration curve. This LDH activity was adjusted to the number of cells per well, and was expressed as µU/mg of protein.
2.4. Intracellular ATP Measurement
Intracellular ATP levels were measured using a luciferase-based ATP Bioluminescence Assay Kit CLS II (Roche Molecular Biochemicals). For ATP measurement, 100 µL of cell lysate was mixed with 50 µL of luciferase. Emitted bioluminescence was measured using a microplate reader. The protein of each treatment group was determined by the BCA Protein Assay Kit.
2.5. Flow Cytometric Measurement of Transmembrane Mitochondrial Potential with DiOC6 (3)
Variations of the transmembrane mitochondrial potential (ΔΨm) were measured with 3, 3’-dihexyloxacarbo- cyanine iodide (DiOC6 (3)) (Invitrogen), which allows the percentage of cells with low ΔΨm to be determined. With DiOC6 (3), mitochondrial depolarization is indicated by a decrease in green fluorescence collected through a 520/10-nm band pass filter. DiOC6 (3) was used at a 40 nM. Flow cytometric analyses were performed on a Galaxy flow cytometer (Partec). Ten thousand cells were acquired for each sample. Data were analyzed with Flomax software (Partec) or FlowJo software (Tree Star Inc.).
2.6. Cell Apoptosis Analysis
Apoptotic cell death was measured via Annexin V-CF647 (Millipore) staining followed by flow cytometry. Annexin V is a calcium-dependent phospholipid binding protein with high affinity for phosphatidylserine (PS), a membrane component normally localized to the internal face of the cell membrane and which is exposed on the cell surface upon induction of apoptosis. Annexin V, which is conjugated to CF647 (Abs/Em maxima: 650/665 nm), was excited by a red laser on a FACSalibur 4C flow cytometer (BD Biosciences) and the emission of fluorescence was collected with a 670 nm long pass filter. Five μL Annexin V-CF647 were added to the cells in the dark at 37˚C, in a humidified atmosphere containing 5% CO2. Around 15 min later, the stained cells were analyzed by flow cytometry. Ten thousand cells were acquired for each sample. Data were analyzed with Flomax software (Partec) or FlowJo software (Tree Star Inc.).
2.7. Quantification of Cholesterol, Cholesterol Oxide Derivatives and Fatty Acids by Gas Chromatography Coupled with Mass Spectrometry
Cholesterol oxide derivatives (also called oxysterols), including 7α-OHC (mainly formed via CYP7A1 [22] but which can also arise from the decomposition of 7α-hydroperoxycholesterol produced by free radical oxidation of cholesterol [23] ), those oxidized at C7 resulting from cholesterol autoxidation (7KC and 7β-OHC) [9] , as well as 24S-OHC, and cholesterol were quantified as follow. After trypsinization, cells were suspended in ethanol containing butylated hydroxytoluene (Sigma; 50 μg/mL) and EDTA (Sigma; 50 μg/mL). 7β-OHC (d7) (Avanti Polar lipids/Coger), 24S-OHC (d6) (Avanti Polar lipids/Coger), and Epicoprostanol (Sigma) were added as internal standards. Samples were then subjected to alkaline hydrolysis with 0.35 M KOH for 2 h at room temperature. The reaction mixture was adjusted to pH 7 with phosphoric acid, and lipids were extracted with hexane. After solvent evaporation, 100 μL of a mixture of N, O-bis (trimethylsilyl) trifluoroacetamide, and trimethylchlorosilane (4/1, v/v) (Acros Organics, Fisher Scientific) were added, and samples were incubated at 80˚C for 60 min to form trimethylsilyl ethers. After evaporation, the residue was dissolved in 100 μL hexane for gas chromatography coupled with mass spectrometry (GC-MS) analysis. GC-MS was performed using an Agilent Technology 6890 GC equipped with an HP7683 injector and a 5973 mass selective detector (Agilent Technologies). Chromatography was performed using a HP-5MS-fused silica capillary column (length: 25 m; inner diameter: 0.25 mm; film thickness: 0.25 μm; Agilent Technologies). GC-MS conditions were as follows: carrier gas, helium at a flow-rate of 1.1 mL/min; injector temperature, 250˚C; oven temperature, 180˚C increased at 10˚C/min to 260˚C, then at 1˚C/min to 280˚C and held for 5 min. The mass spectrometer was operated in the electron impact mode with an electron energy of 70 eV. The ion source temperature and the quadrupole temperature were 230˚C and 150˚C, respectively. The ions used for analysis were 24S-OHC 145 m/z, 24S-OHC (d6) 151 m/z, (25-OHC) 131 m/z, cholesterol 368 m/z, epicoprostanol 370 m/z, 7α-OHC 456 m/z, 7β-OHC 456 m/z, 7β-OHC (d7) 463 m/z, and 7KC 472 m/z. Calibration curves were obtained using authentic standards extracted with the method used for cell samples.
C22:0, C24:0, C26:0, C20:4, C22:6, and C24:6 were quantified using a HP7890A gas chromatograph equipped with an HP7683 injector and a HP5975C mass selective detector (Agilent Technologies). Chromatography was performed using an HP-5MS-fused silica capillary column (length: 30 m; inner diameter: 0.25 mm; film thickness: 0.25 mm; Agilent Technologies). The GC-MS conditions were as follows: carrier gas, helium at a flow rate of 1.1 mL/min; injector temperature, 250˚C, split mode; oven temperature, 140˚C increased at 5˚C/min to 300˚C and held for 10 min. The mass spectrometer was operated under negative chemical ionization mode with methane as the reactant gas. The ion source temperature and the quadrupole temperature were 150˚C and 106˚C, respectively. A SIM program was used for mass spectrometry with [M-181] (−) ions as quantification.
3. Results
3.1. Effect of Aβ1-40 on Cell Viability
The ability of Aβ1-40 to induce neurotoxicity was estimated using i) the MTT test, which reflects mitochondrial activity and/or cell growth, and ii) by LDH activity. SK-NB-E cells were cultured without or with Aβ1-40 (10 - 100 µM, 24 - 48 h). A significant decrease in the percentage of MTT-positive cells was observed after 24 and 48 h of treatment with the two concentrations of Aβ1-40 used (Figure 1(a)). With Aβ1-40 (10-100 µM), as cytotoxic effects in the same range of order were observed with the MTT test, LDH activity was only measured on SK-N- BE cells treated with Aβ1-40 (10 µM). An increase in LDH activity was observed after 24 - 48 h of treatment. However, significant differences between control (untreated cells) and Aβ1-40-treated cells were only found at 48 h (Figure 1(b)).
3.2. Effect of Aβ1-40 on ATP Production and Transmembrane Mitochondrial Potential
Data obtained with the MTT test support that mitochondrial activity and/or cell growth is affected under treatment

Figure 1. Effects of oligomerized Aβ1-40 on cell viability. SK-N-BE cells were incubated with or without Aβ1-40 (10 - 100 µM) for 24 and/or 48 h. Cell proliferation and/or mitochondrial metabolism was evaluated using the MTT test (a) and cell death by LDH activity (b). Data shown are mean ± SD from two to three separate experiments conducted in triplicate. Significance of the difference is indicated by * (Mann-Whitney test; *P < 0.05).
with Aβ1-40. To determine the impact of Aβ1-40 at the mitochondrial level, ATP production and mitochondrial transmembrane potential (ΔΨm) were measured. A significant increase of the percentage of DiOC6 (3) negative cells (with low ΔΨm) was observed with 100 µM at 24 h, and with 10 and 100 µM at 48 h of treatment with oligomerized Aβ1-40 (Figure 2(a)).
The ATP level was measured on SK-N-BE cells treated with oligomerized Aβ1-40 (10 µM, 48 h). A significant increase in intracellular ATP supporting mitochondrial dysfunctions was revealed in treated cells compared to the control (Figure 2(b)).
3.3. Effect of Aβ1-40 on Apoptotic Cell Death Induction
Annexin V-CF647 staining assay for cell apoptosis detection was performed using flow cytometry. A significant increase of the percentage of Annexin V positive cells was observed in cells treated with oligomerized Aβ1-40 (100 µM, 24 h) and with 10 and 100 µM at 48 h (P < 0.05) (Figure 3).
3.4. Effect of Aβ1-40 on Lipid Profile
The effects of Aβ1-40 (10 µM, 48 h) on the intracellular levels of cholesterol, oxysterols (7α-OHC, 7β-OHC, 7KC, 24S-OHC) (Table 1), and fatty acids (C20:4 n-6 (AA), C22:0, C22:6 n-3 (DHA), C24:0, C24:6) (Table 2) was investigated using GC-MS on SK-N-BE cells. Substantial modifications of the intracellular levels of cholesterol and oxysterols were observed (Table 1).
Cholesterol analysis revealed a significant increase (Mann-Whitney test; P < 0.05) in Aβ1-40-treated cells. In addition, oxysterol analysis in SK-N-BE-treated cells revealed a significant increase (Mann-Whitney test; P < 0.05) in 7β-OHC and in the sum (7β-OHC + 7KC), reflecting cholesterol autoxidation. Furthermore, the [(7β- OHC + 7KC)/cholesterol] ratio, considered as a lipid peroxidation index, was significantly enhanced (Mann- Whitney test; P < 0.05) in Aβ1-40-treated cells. However, no significant difference in the sum of cytotoxic oxysterols (7α-OHC + 7β-OHC + 7KC + 24S-OHC) was observed between untreated cells (control) and Aβ1-40- treated cells, whereas it was highest under treatment with Aβ1-40.
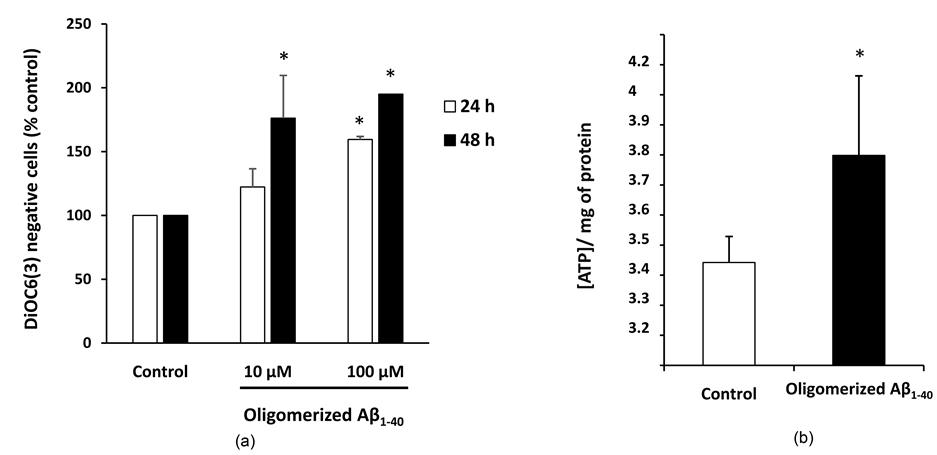
Figure 2. Effects of oligomerized Aβ1-40 on transmembrane mitochondrial potential and ATP production. SK-N-BE cells were incubated without (control) or with Aβ1-40 (10 µM, 48 h). (a): effect on transmembrane mitochondrial potential measured by flow cytometry with DiOC6 (3); (b): effect of oligomerized Aβ1-40 (10 µM, 48 h) on ATP level measured using a luciferase-based ATP bioluminescence assay. Data shown are mean ± SD from two or three separate experiments conducted in triplicate. Significance of the difference is indicated by * (Mann-Whitney test; *P < 0.05).
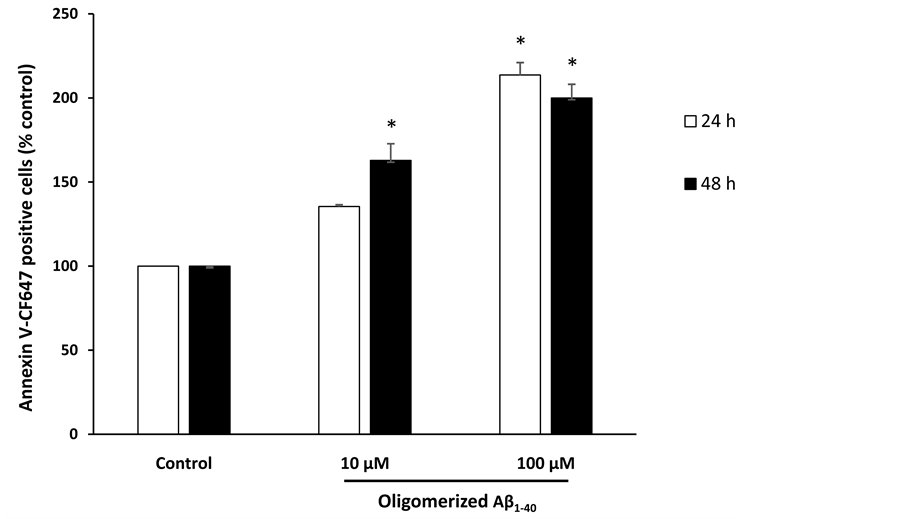
Figure 3. Effects of oligomerized Aβ1-40 on apoptosis induction. SK-N-BE cells were incubated without (control) or with Aβ1-40 (10 and 100 µM, 24 and 48 h). Cells were stained with Annexin V-CF647 and analyzed by flow cytometry. Data shown are mean ± SD from two or three separate experiments conducted in triplicate. Significance of the difference is indicated by * (Mann-Whitney test; *P < 0.05).
Considerable modifications in the intracellular levels of fatty acids were also revealed (Table 2). Under treatment with Aβ1-40 significant accumulations (Mann-Whitney test; P < 0.05) of AA (C20:4 n-6), C22:0, DHA (C22:6 n-3), and the sum of saturated VLCFAs (C22:0 + C24:0 + C26:0) was observed.

Table 1.Evaluation of the effects of oligomerized Aβ1-40 on oxysterols profile.

Table 2.Evaluation of the effects of oligomerized Aβ1-40 on fatty acids profile.
4. Discussion
Amyloid peptide (Aβ), the main component of senile plaques, was shown to be neurotoxic in several studies but no data are available to evaluate the relation between this molecule and lipid metabolism disorders associated with AD pathogenesis [7] [14] [18] . To attain a better understanding of the neurotoxicity of Aβ1-40, its effects on the cellular lipid profile were considered. On human neuronal SK-N-BE cells, our data show that Aβ1-40 favors the accumulation of cholesterol, oxysterols, and fatty acids, which are known to play critical roles in the development of AD [14] .
On SK-N-BE cells, the neurotoxicity of oligomerized Aβ1-40 evaluated with the MTT test and LDH activity showed both a significant increase in the percentage of MTT-positive cells and an increase in LDH activity, which supports the ability of Aβ1-40 to induce cell death [24] . This decrease of mitochondrial succinate dehydrogenase activity associated to the loss of transmembrane mitochondrial potential observed after staining with DiOC6 (3) suggests that Aβ1-40 can induce mitochondrial alterations that are assumed to contribute to the pathogenesis of AD [25] . The oligomerized Aβ1-40, as its isomer the Aβ1-42, was able to induce apoptotic cell death in SK-N-BE cells evaluated by PS externalization revealed with Annexin V. Aβ1-42 also induces apoptosis in cultured FVB mouse hippocampal neurons [26] .
The significant increase in intracellular ATP observed in SK-N-BE cells treated with Aβ1-40 supports the hypothesis that stressed cells may require more energy to counteract various side effects resulting from stress conditions and to preserve their vital functions [27] . This adaptive response of neural cells to an environmental stress could also explain (at least in part) the ability of Aβ1-40 to disturb lipid homeostasia.
The increased intracellular level of cholesterol detected in Aβ1-40-treated cells supports the notion that the cellular stress triggered by Aβ1-40 can promote cholesterol synthesis and/or accumulation [28] . There is also a great deal of evidence on cultured neurons [29] and in transgenic mouse models [30] suggesting that cholesterol accumulation is linked to Aβ. Recent evidence using mouse models of cholesterol loading demonstrates that cholesterol sensitizes neurons to Aβ-induced oxidant cell death [31] . As it is known that Aβ1-40 is a pro-oxidant molecule [32] , we determined the impact of Aβ1-40 on lipid peroxidation via the generation of cholesterol oxide derivatives resulting from cholesterol autoxidation (7β-OHC and 7KC, mainly). Interestingly, a significant accumulation of 7β-OHC and (7β-OHC + 7KC) was detected. Moreover, the [(7β-OHC + 7KC)/cholesterol] ratio (considered as a lipid peroxidation index) was also significantly enhanced. These oxysterols produced in the cells through the autoxidation of cholesterol not only argue in favor of the occurrence of an oxidative stress induced by oligomerized Aβ1-40, but this also provides information on the potential cytotoxic pathways adopted by oligomerized Aβ1-40. Indeed, some oxysterols, mainly those oxidized at C7 (7α-OHC, 7β-OHC, 7KC), are cytotoxic and able to induce cell death associated with oxidative processes [14] [33] . Therefore, the oxysterols could in turn contribute to the cytotoxic effects of Aβ1-40.
Although not significant, the enhancement of the intracellular level of 24S-OHC (a potent liver X receptor (LXR) agonist produced by enzymatic oxidation of cholesterol via CY46A1) [4] [14] may have negative consequences. It could contribute to disturb cholesterol level via LXR, and/or participate in the cytotoxic effects of Aβ1-40. Indeed, it is well established that 24S-OHC has a wide range of activities that depend on its concentration.
On the other hand, analysis of intracellular fatty acids conducted on SK-N-BE cells treated with Aβ1-40 revealed a significant increase of AA (C20:4 n-3), C22:0 and DHA (C22:6 n-3) and the sum of VLCFAs ((C22:0 + C24:0 + C26:0). These results underline that Aβ1-40 could disrupt the metabolism of fatty acids and especially affect the peroxisomal β-oxidation of VLCFAs given that the β-oxidation or the synthesis of some of these lipids (C22:6 n-3, C24:0, and C26:0) occurs, at least in part, in the peroxisome [19] . As a cortical accumulation of C22:0, C24:0, and C26:0 has been found in patients with stages V and VI pathology compared with those modestly affected (stages I and II) based on the neuropathological Braak staging for AD patients [17] , our data obtained on SK-N-BE cells support the hypothesis that Aβ1-40 could favor peroxisomal dysfunctions leading to reduced peroxisomal β-oxidation, which could thus contribute to the development of AD, as previously suggested [17] . However, as a simultaneous increase in DHA produced by β-oxidation was simultaneously observed, it cannot be excluded that abnormal elongase activities could be also activated, and contribute to the accumulation of C22:0, C24:0, and C26:0 [19] . Nevertheless, the ability of Aβ1-40 to favor the accumulation of VLCFAs, which are strong inducers of mitochondrial dysfunctions and trigger oxidative stress on various neuronal cells [21] , reinforces the hypothesis that these fatty acids could constitute potential risk factors contributing to the development of brain lesions in AD [19] .
The important accumulation of arachidonic acid (AA), the precursor of leukotrienes and prostaglandins, on SK-N-BE cells treated with Aβ1-40 also contributes new insights into the biological activities of this molecule. This finding is in agreement with data reporting that eicosanoids might participate in Aβ1-40 toxicity in neurons and that noncytokinic inflammation contributes to the development of AD [34] . It is also known that AA can participate to neurotoxicity via its ability to decrease neuroprotectins [35] . However, recent findings also suggest that prostaglandin derived from AA might also have neuroprotective effects [36] . Therefore, the increase in AA could be an adaptive response that could either contribute to Aβ1-40 cytotoxic effects or to counteract its side effects.
In addition to its ability to trigger cell death, our data establish that Aβ1-40 favors a substantial cellular accumulation of lipids: cholesterol, oxysterols (especially those resulting from cholesterol autoxidation), and fatty acids. Since a marked accumulation of VLCFAs and DHA was observed, modifications of lipid metabolism, including peroxisomal dysfunctions, are suspected. It is suggested that the accumulation of cholesterol, oxysterols, and fatty acids could in turn contribute to the cytotoxic effects of Aβ1-40. Consequently, the identification of molecules capable of counteracting the different side effects of these lipids may be advantageous in preventing the neurotoxicity induced by Aβ1-40.
Acknowledgements
This work was funded by grants from INSERM, University of Bourgogne (Dijon, France), and ABASIM (Dijon, France).
Cite this paper
AmiraZarrouk,ThomasNury,MohamedHammami,GérardLizard, (2015) Oligomerized Amyloid-β1-40 Peptide Favors Cholesterol, Oxysterol, and Fatty Acid Accumulation in Human Neuronal SK-N-BE Cells. International Journal of Clinical Medicine,06,813-824. doi: 10.4236/ijcm.2015.611107
References
- 1. Selkoe, D. (1994) Alzheimer’s Disease: A Central Role for Amyloid. Journal of Neuropathology & Experimental Neurology, 53, 438-447.
http://dx.doi.org/10.1097/00005072-199409000-00003 - 2. Haass, C., Schlossmacher, M.G., Hung, A.Y., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B.L., Lieberburg, I., Koo, E.H., Schenk, D., Teplow, D.B. and Selkoe, D.J. (1992) Amyloid Beta-Peptide Is Produced by Cultured Cells during Normal Metabolism. Nature, 359, 322-325.
http://dx.doi.org/10.1038/359322a0 - 3. Bjõrkhem, I. (2006) Crossing the Barrier: Oxysterols as Cholesterol Transporters and Metabolic Modulators in the Brain. Journal of Internal Medicine, 260, 493-508.
http://dx.doi.org/10.1111/j.1365-2796.2006.01725.x - 4. Grimm, M.O., Zimmer, V.C., Lehmann, J., Grimm, H.S. and Hartmann, T. (2013) The Impact of Cholesterol, DHA, and Sphingolipids on Alzheimer’s Disease. BioMed Research International, 814390.
- 5. Grziwa, B., Grimm, M.O., Masters, C.L., Beyreuther, K., Hartmann, T. and Lichtenthaler, S.F. (2003) The Transmembrane Domain of the Amyloid Precursor Protein in Microsomal Membranes Is on Both Sides Shorter than Predicted. The Journal of Biological Chemistry, 278, 6803-6808.
http://dx.doi.org/10.1074/jbc.M210047200 - 6. Leoni, V. and Caccia, C. (2013) 24S-Hydroxycholesterol in Plasma: A Marker of Cholesterol Turnover in Neurodegenerative Diseases. Biochimie, 95, 595-612.
http://dx.doi.org/10.1016/j.biochi.2012.09.025 - 7. Wood, W.G., Li, L., Müller, W.E. and Eckert, G.P. (2014) Cholesterol as a Causative Factor in Alzheimer’s Disease: A Debatable Hypothesis. Journal of Neurochemistry, 129, 559-572.
http://dx.doi.org/10.1111/jnc.12637 - 8. Seripa, D., D’Onofrio, G., Panza, F., Cascavilla, L., Masullo, C. and Pilotto, A. (2001) The Genetics of the Human APOE Polymorphism. Rejuvenation Research, 14, 491-500.
http://dx.doi.org/10.1089/rej.2011.1169 - 9. Iuliano, L. (2011) Pathways of Cholesterol Oxidation via Non-Enzymatic Mechanisms. Chemistry and Physics of Lipids, 164, 457-468.
http://dx.doi.org/10.1016/j.chemphyslip.2011.06.006 - 10. Vaya, J. and Schipper, H.M. (2007) Oxysterols, Cholesterol Homeostasis, and Alzheimer Disease. Journal of Neurochemistry, 102, 1727-1737.
http://dx.doi.org/10.1111/j.1471-4159.2007.04689.x - 11. Gamba, P., Guglielmotto, M., Testa, G., Monteleone, D., Zerbinati, C., Gargiulo, S., Biasi, F., Iuliano, L., Giaccone, G., Mauro, A., Poli, G., Tamagno, E. and Leonarduzzi, G. (2014) Up-Regulation of β-Amyloidogenesis in Neuron-Like Human Cells by Both 24- and 27-Hydroxycholesterol: Protective Effect of N-Acetyl-Cysteine. Aging Cell, 13, 561-572.
http://dx.doi.org/10.1111/acel.12206 - 12. Lütjohann, D., Papassotiropoulos, A., Bjõrkhem, I., Locatelli, S., Bagli, M., Oehring, R.D., Schlegel, U., Jessen, F., Rao, M.L., von Bergmann, K. and Heun, R. (2000) Plasma 24S-Hydroxycholesterol (Cerebrosterol) Is Increased in Alzheimer and Vascular Demented Patients. Journal of Lipid Research, 41, 195-198.
- 13. Solomon, A., Leoni, V., Kivipelto, M., Besga, A., Oksengard, A., Julin, P., Svensson, L., Wahlund, L., Andreasen, N., Winblad, B., Soininen, H. and Bjorkhem, I. (2009) Plasma Levels of 24S-Hydroxycholesterol Reflect Brain Volumes in Patients without Objective Cognitive Impairments but Not with Alzheimer’s Disease. Neuroscience Letters, 462, 89-93.
http://dx.doi.org/10.1016/j.neulet.2009.06.073 - 14. Zarrouk, A., Vejux, A., Mackrill, J., O’Callaghan, Y., Hammami, M., O’Brien, N. and Lizard, G. (2014) Involvement of Oxysterols in Age-Related Diseases and Ageing Processes. Ageing Research Reviews, 18, 148-162.
http://dx.doi.org/10.1016/j.arr.2014.09.006 - 15. Urano, Y., Ochiai, S. and Noguchi, N. (2013) Suppression of Amyloid-β Production by 24S-Hydroxycholesterol via Inhibition of Intracellular Amyloid Precursor Protein Trafficking. The FASEB Journal, 27, 4305-4315.
http://dx.doi.org/10.1096/fj.13-231456 - 16. Prasanthi, J.R., Huls, A., Thomasson, S., Thompson, A., Schommer, E. and Ghribi, O. (2009) Differential Effects of 24-Hydroxycholesterol and 27-Hydroxycholesterol on Beta-Amyloid Precursor Protein Levels and Processing in Human Neuroblastoma SH-SY5Y Cells. Molecular Neurodegeneration, 4, 1.
http://dx.doi.org/10.1186/1750-1326-4-1 - 17. Kou, J., Kovacs, G.G., Hoftberger, R., Kulik, W., Brodde, A., Forss-Petter, S., Hõnigschnabl, S., Gleiss, A., Brügger, B., Wanders, R., Just, W., Budka, H., Jungwirth, S., Fischer, P. and Berger, J. (2011) Peroxisomal Alterations in Alzheimer’s Disease. Acta Neuropathologica, 122, 271-283.
http://dx.doi.org/10.1007/s00401-011-0836-9 - 18. Zarrouk, A., Riedinger, J.M., Ahmed, S.H., Hammami, S., Chaabane, W., Debbabi, M., Ben Ammou, S., Rouaud, O., Frih, M., Lizard, G. and Hammami, M. (2015) Fatty Acid Profiles in Demented Patients: Identification of Hexacosanoic Acid (C26:0) as a Blood Lipid Biomarker of Dementia. Journal of Alzheimer’s Disease, 44, 1349-1359.
- 19. Lizard, G., Rouaud, O., Demarquoy, J., Cherkaoui-Malki, M. and Iuliano, L. (2012) Potential Roles of Peroxisomes in Alzheimer’s Disease and in Dementia of the Alzheimer’s Type. Journal of Alzheimer’s Disease, 29, 241-254.
- 20. Wang, X., Wang, W., Li, L., Perry, G., Lee, H.G. and Zhu, X. (2014) Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochimica et Biophysica Acta, 1842, 1240-1247.
http://dx.doi.org/10.1016/j.bbadis.2013.10.015 - 21. Zarrouk, A., Vejux, A., Nury, T., El Hajj, H.I., Haddad, M., Cherkaoui-Malki, M., Riedinger, J.M., Hammami, M. and Lizard, G. (2012) Induction of Mitochondrial Changes Associated with Oxidative Stress on Very Long Chain Fatty Acids (C22:0, C24:0, or C26:0)-Treated Human Neuronal Cells (SK-NB-E). Oxidative Medicine and Cellular Longevity, 2012, Article ID: 623257.
http://dx.doi.org/10.1155/2012/623257 - 22. Pikuleva, I.A. (2006) Cholesterol-Metabolizing Cytochromes P450. Drug Metabolism and Disposition, 34, 513-520.
http://dx.doi.org/10.1124/dmd.105.008789 - 23. Smith, L.L. (1996) Review of Progress in Sterol Oxidations 1987-1995. Lipids, 3, 453-487.
http://dx.doi.org/10.1007/BF02522641 - 24. Ferrera, P., Mercado-Gómez, O., Silva-Aguilar, M., Valverde, M. and Arias, C. (2008) Cholesterol Potentiates Beta-Amyloid-Induced Toxicity in Human Neuroblastoma Cells: Involvement of Oxidative Stress. Neurochemical Research, 33, 1509-1517.
http://dx.doi.org/10.1007/s11064-008-9623-y - 25. García-Escudero, V., Martín-Maestro, P., Perry, G. and Avila, J. (2013) Deconstructing Mitochondrial Dysfunction in Alzheimer Disease. Oxidative Medicine and Cellular Longevity, 2013, Article ID: 162152.
http://dx.doi.org/10.1155/2013/162152 - 26. Jiang, F., Mao, Y., Liu, H., Xu, P., Zhang, L., Qian, X. and Sun, X. (2015) Magnesium Lithospermate B Protects Neurons against Amyloid β (1-42)-Induced Neurotoxicity through the NF-κB Pathway. Neurochemical Research, 40, 1954-1965.
http://dx.doi.org/10.1007/s11064-015-1691-1 - 27. Volonté, C., Amadio, S., Cavaliere, F., D’Ambrosi, N., Vacca, F. and Bernardi, G. (2003) Extracellular ATP and Neurodegeneration. Current Drug Targets—CNS and Neurological Disorders, 2, 403-412.
http://dx.doi.org/10.2174/1568007033482643 - 28. Barbero-Camps, E., Fernández, A., Baulies, A., Martinez, L., Fernández-Checa, J.C. and Colell, A. (2014) Endoplasmic Reticulum Stress Mediates Amyloid β Neurotoxicity via Mitochondrial Cholesterol Trafficking. American Journal of Pathology, 184, 2066-2081.
http://dx.doi.org/10.1016/j.ajpath.2014.03.014 - 29. Galbete, J.L., Martin, T.R., Peressini, E., Modena, P., Bianchi, R. and Forloni, G. (2000) Cholesterol Decreases Secretion of the Secreted form of Amyloid Precursor Protein by Interfering with Glycosylation in the Protein Secretory Pathway. Biochemical Journal, 348, 307-313.
http://dx.doi.org/10.1042/bj3480307 - 30. Refolo, L.M., Malester, B., La Francois, J., Bryant-Thomas, T., Wang, R., Tint, G.S., Sambamurti, K., Duff, K. and Pappolla, M.A. (2000) Hypercholesterolemia Accelerates the Alzheimer’s Amyloid Pathology in a Transgenic Mouse Model. Neurobiology of Disease, 7, 321-331.
http://dx.doi.org/10.1006/nbdi.2000.0304 - 31. Colell, A., Fernández, A. and Fernández-Checa, J.C. (2009) Mitochondria, Cholesterol and Amyloid Beta Peptide: A Dangerous Trio in Alzheimer Disease. Journal of Bioenergetics and Biomembranes, 41, 417-423.
http://dx.doi.org/10.1007/s10863-009-9242-6 - 32. Cutler, R.G., Kelly, J., Storie, K., Pedersen, W.A., Tammara, A., Hatanpaa, K., Troncoso, J.C. and Mattson, M.P. (2004) Involvement of Oxidative Stress-Induced Abnormalities in Ceramide and Cholesterol Metabolism in Brain Aging and Alzheimer’s Disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 2070-2075.
http://dx.doi.org/10.1073/pnas.0305799101 - 33. Zarrouk, A., Nury, T., Samadi, M., O’Callaghan, Y., Hammami, M., O’Brien, N.M., Lizard, G. and Mackrill, J.J. (2015) Effects of Cholesterol Oxides on Cell Death Induction and Calcium Increase in Human Neuronal Cells (SK-N-BE) and Evaluation of the Protective Effects of Docosahexaenoic Acid (DHA; C22:6 n-3). Steroids, 99, 238-247.
http://dx.doi.org/10.1016/j.steroids.2015.01.018 - 34. Prasad, K.N., Hovland, A.R., La Rosa, F.G. and Hovland, P.G. (1998) Prostaglandins as Putative Neurotoxins in Alzheimer’s Disease. Proceedings of the Society for Experimental Biology and Medicine, 219, 120-125.
http://dx.doi.org/10.3181/00379727-219-44323 - 35. Bazan, N.G. (2009) Cellular and Molecular Events Mediated by Docosahexaenoic Acid-Derived Neuroprotectin D1 Signaling in Photoreceptor Cell Survival and Brain Protection. Prostaglandins, Leukotrienes and Essential Fatty Acids, 81, 205-211.
http://dx.doi.org/10.1016/j.plefa.2009.05.024 - 36. Fattahi, M.J. and Mirshafiey, A. (2014) Positive and Negative Effects of Prostaglandins in Alzheimer’s Disease. Psychiatry and Clinical Neurosciences, 68, 50-60.
http://dx.doi.org/10.1111/pcn.12092
NOTES

*Corresponding author.