Open Journal of Blood Diseases
Vol.3 No.3(2013), Article ID:36934,6 pages DOI:10.4236/ojbd.2013.33019
Cutaneous Manifestations of Sickle Cell Disease
![]()
1Division of Internal Medicine, Washington University in St. Louis, St. Louis, USA; 2Division of Hematology, Washington University in St. Louis, St. Louis, USA; 3Division of Dermatology, Washington University in St. Louis, St. Louis, USA.
Email: hjones@dom.wustl.edu
Copyright © 2013 Heather Jones et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received June 24th, 2013; revised July 24th, 2013; accepted August 1st, 2013
Keywords: Sickle Cell Disease; Leg Ulcers; Pseudoxanthoma Elasticum; Nitric Oxide Deficiency; Hydroxyurea
ABSTRACT
Sickle cell disease is an inherited erythrocyte disorder affecting multiple organ systems. As individuals are living well into middle age due to advances in diagnosis and treatment, further disease-related complications are being recognized. This article reviews the cutaneous manifestations of sickle cell disease including leg ulcers and acquired pseudoxanthoma elasticum.
1. Introduction
Sickle cell disease (SCD) is an inherited blood dyscrasia affecting 50,000 to 100,000 people in the United States [1]. Currently SCD is the genetic disease most commonly identified by the Newborn Screening Program in the US. It is a chronic and disabling condition disproportionately affecting those of African heritage.
A variety of sickle syndromes exist. The homozygous hemoglobin (Hb) SS genotype of SCD is the most clinically severe, accounting for 60% - 70% of SCD in the US [2]. Normal adult HbA contains two alpha and two beta-globin chains. The HbSS genotype of sickle cell anemia results from a point mutation in the beta-globin gene at position six, resulting in the substitution of valine for glutamic acid. The absence of a polar amino acid at the sixth position of the beta-globin gene promotes aggregation and distortion of the hemoglobin protein in conditions of low oxygen tension. Alterations in the shape of hemoglobin thus result in sickle-shaped erythrocytes with damaged cell membranes. The weakened cell membranes predispose cells to hemolysis, damage endothelial cells, and establish a favorable milieu for vascular obstruction.
2. Leg Ulceration
Recurrent sores and scars over the lower extremities of an African patient with SCD were first reported by Herrick in 1910 [3]. The causal link between SCD and leg ulcers was not established for years however, as most leg ulcers at that time were attributed to syphilis. It was not until thirty years later that Cummer et al. summarized existing case reports and determined that leg ulceration occurred in a definite proportion of sickle cell patients. The authors concluded that “within dermatologic practice, anemia should be considered as a possible explanation for chronic ulcers on the legs” [4].
2.1. Pathophysiology
Leg ulcers are considered a sub-phenotype of sickle cell disease. Leg ulcers in sickle cell patients have been consistently associated with increased levels of lactate dehydrogenase (LDH) and bilirubin, as well as increased reticulocyte counts, all of which are markers of increased hemolysis [5]. The pathophysiology of ulceration in sickle cell disease was formerly attributed to vaso-occlusive tissue ischemia and necrosis [6]. The lower extremity, an area of marginal vascularity [7] was thought to be more susceptible to trauma with resultant inflammation, decreased oxygen tension, and sickling of red cells leading to further vascular compromise and ultimately ulceration. However, recent literature has emerged suggesting a more prominent role of nitric oxide (NO) deficiency in the pathophysiology of ulceration.
Nitric oxide is an important regulator of vascular tone, cell adhesion, and blood flow. Its production in SCD patients is dramatically reduced, as is its bioavailability, through the breakdown of NO substrates, increases in free radical production, and uncoupling of endothelial nitric oxide synthase (eNOS). Cell free hemoglobin released during hemolysis affects the bioavailability of NO as deoxy-HbS and oxy-HbS react with NO forming a stable Fe2+ Hb-NO complex, methemoglobin, and nitrate [8]. Also released from damaged erythrocytes during hemolysis is the enzyme Arginase. Once soluble, Arginase competes with endothelial nitric oxide synthase (eNOS) to metabolize arginine, the precursor to NO formation, into ornithine and urea. As L-arginine is the rate limiting substance in the NOS pathway, its absence disrupts electron transport through eNOS and diminishes the amount of NO produced. This consumption of NO and its precursor causes such downstream sequelae as impaired vascular smooth muscle relaxation, increased adhesion molecule and endothelin-1 expression, and platelet activation and aggregation [9]. The resulting vasculopathy skews the normal balance between vasoconstriction and vasodilatation toward vasoconstriction, endothelial activation, and proliferation [7]. Ultimately, the resulting deficiency of NO and aberrant vascular activity is thought to lead to tissue ischemia and ulcer formation (Figure 1).
Chronic oxidative stress caused by repeated episodes of HbS de-oxygenation and re-oxygenation has also been implicated in the pathogenesis of leg ulceration [8]. Re-oxygenation of hemoglobin is a major source of free radical formation generated by an electron transfer be-
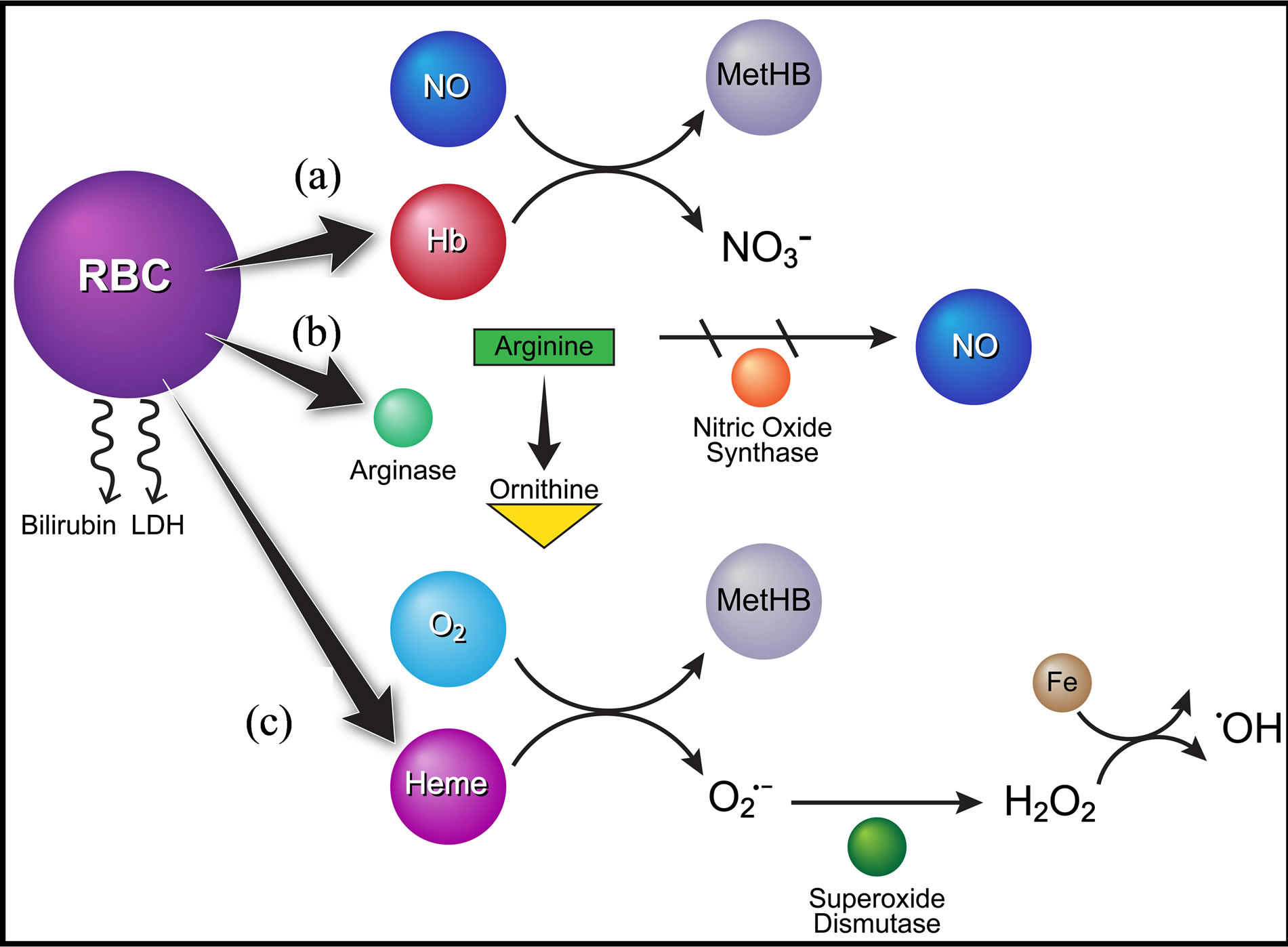
Figure 1. Diagram of intravascular hemolysis pathways. Intravascular hemolysis releases hemoglobin, bilirubin, LDH and Arginase. (a) Nitric oxide (NO) reacts with freed oxyhemoglobin to form methemoglobin and intert nitrate. (b) Arginine, the substrate for NO synthetase (NOS), is degraded to ornithine in the presence of Arginase released by erythrocytes, thereby decreasing the amount of NO in plasma. (c) In the presence of oxygen, heme auto-oxides to produce methemoglobin and superoxide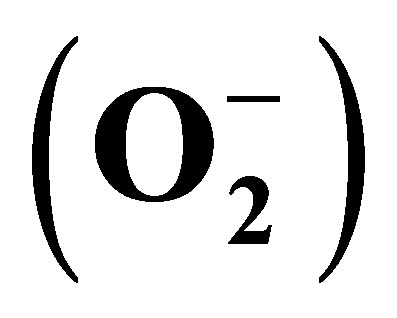 , which is then acted upon by superoxide dismutase (SOD) to form hydrogen peroxide (H2O2), which in turn degrades methemoglobin, releasing free iron (Fe). The freed iron can then react with H2O2 to produce hydroxyl radicals (OH), the most harmful of all reactive oxygen species.
, which is then acted upon by superoxide dismutase (SOD) to form hydrogen peroxide (H2O2), which in turn degrades methemoglobin, releasing free iron (Fe). The freed iron can then react with H2O2 to produce hydroxyl radicals (OH), the most harmful of all reactive oxygen species.
tween heme iron and oxygen [8]. Although both HbA and HbS auto-oxidize in the presence of oxygen to form methemoglobin and superoxide, HbS has been demonstrated to auto-oxidize 1.7 times faster than HbA [8]. The increased formation of superoxide in turn causes increased production of hydrogen peroxide (H2O2), which then reacts with methemoglobin to produce hydroxyl radicals (OH−). These reactive oxygen species then induce oxidative damage to erythrocytes resulting in lipid peroxidation and cell damage [8].
2.2. Prevalence
Estimates of the incidence and prevalence of leg ulcerations attributable to SCD vary. The results of the United States Cooperative Study of Sickle Cell Disease (CSSCD) found that about 25% of patients with SCD over the age of ten had a history of leg ulcers, with active ulcers at entry or development of ulcers during the eight year study period [10]. Other worldwide estimates differ. In a recently published study of 457 SCD patients followed for ten years in a French referral center, 5.5% of patients had leg ulcerations [11]. In contrast, Serjeant reported 75% of Jamaican SCD patients older than 30 years had either active or healed ulcers [12]. This wide variability may be partially explained by discrepancies in socioeconomic status between the groups of individuals studied (Table 1).
2.3. Risk Factors
Leg ulcers in sickle cell patients are more common with increasing age [8,10]. Genotype also modifies risk with ulceration more prevalent in the homozygous SS patients with SCD [8,10]. The intensity of hemolysis may play a role as well. Table 1 lists other proposed risk factors and biomarkers for leg ulcer development.
2.4. Clinical Features and Assessment
Ulcers occurring in patients with SCD typically occur in areas with less subcutaneous fat, thin skin, and decreased blood flow [6]. Most ulcers occur around the medial and lateral malleoli. Other locations include the anterior tibia, dorsum of the foot, and the region overlying the Achilles

Table 1. Other proposed risk factors and biomarkers for sickle cell leg ulcers.
tendon [13-15]. They are round and punched-out in appearance with raised margins, deep bases, and necrotic slough [5,13]. Ulceration size varies from a few millimeters in diameter to large circumferential ulcers that involve the entire distal lower extremity [13]
Leg ulceration is typically precluded by minor trauma which leads to a small lesion that progressively enlarges [14]. Significant pain is the rule. Bacterial colonization of ulcers is frequent, usually aerobic and polymicrobial with Staphylococcus aureus, Pseudomonas aeruginosa, and group A Streptococci most commonly isolated [16]. One recent study found that 85% of patients had wound infections as a complication of ulceration, ranging from localized super-infection to recurrent cellulitis of the affected limb [16]. Healing of ulcers is typically slow, resulting in hypopigmentation and chronic scarring. Patients and healthcare workers alike should adjust their expectations appropriately.
The initial health assessment includes work-up for contributing co-morbidities with an emphasis on hydration status. Physical examination is performed to identify lower extremity edema and signs of super-infection. Pretreatment evaluation often includes a non-invasive arterial flow study with ankle-brachial index measurement as well as a venous duplex ultrasound.
2.5. Treatment
Lower extremity ulcers in sickle cell patients are difficult to treat and often recur with discontinuation of therapy. Healing of these ulcers occurs 20 times more slowly than healing of similar types of refractory vascular lower extremity wounds [17]. Common preventative measures include reducing trauma with properly fitted shoes, minimizing venous stasis with compression stockings, and patient education. Emollients to prevent drying and cracking and proper hygiene are also recommended.
Wound care is often initiated in a wound care or dermatology clinic. Treatment begins with moist supportive dressings. If ulcer drainage is an issue, absorbent dressings can be used. Super-infection is treated with topical antibiotic ointments of neomycin, polymyxin B, or bacitracin [16]. Debridement is typically reserved for necrotic ulcerations. Additionally, oral zinc sulphate as an adjunct to local wound care has been shown to accelerate healing of ulcerations [18].
Hydroxyurea (HU) is a cytotoxic, cytostatic drug used for preventative treatment of SCD. It is currently the only FDA approved treatment for adults with SCD in the United States [19,20]. Hydroxyurea has been shown to reduce the frequency of vaso-occlusive crises, pulmonary events, and blood transfusions. Its primary mechanism of action is inducing production of HbF, which lowers the relative percentage of HbS and prevents polymerization of sickled hemoglobin. Hydroxyurea also contributes to NO donation, eNOS stabilization, and myeloperoxidase reduction [21]. It has been shown to significantly increase plasma concentrations of erythrocyte iron-nitrosyl-hemoglobin (NO-heme), NO metabolites, and cGMP within two hours of administration [21]. Erythrocyte iron-nitrosyl-hemoglobin serves as a carrier of NO and facilitates delivery to peripheral tissues, which is thought to improve microvascular perfusion [21]. Nevertheless, the role of NO remains poorly defined in part because of the possibility of newly developed ulcers in patients treated with HU.
Additional therapy aimed at increasing NO production includes L-arginine and tetrahydrobiopterin supplementation. The administration of L-arginine alone has yielded mixed results, thought to be due to its consumption by erythrocyte Arginase in plasma [9]. Co-administration of L-arginine with an inhibitor of Arginase may protect the substrate for use by eNOS [9]. Further supplementation with tetrahydrobiopterin, a cofactor for eNOS, may restore NO synthesis and thus vascular homeostasis.
Surgical interventions are typically reserved for ulcers that are recalcitrant to conservative therapies. Such interventions include split-thickness skin grafting and myocutaneous flaps [22-24]. By bringing the native blood supply to the area of ulceration, myocutaneous flaps have been found to increase healing [22]. However, success with these surgical methods has been inconsistent [7,12].
3. Pseudoxanthoma Elasticum
Classic Pseudoxanthoma elasticum (PXE) is a heritable, multisystem disease which is characterized by ectopic mineralization of connective tissue. It is inherited in an autosomal recessive manner and is caused by mutation in the Abcc6 gene which encodes an efflux transporter primarily expressed in the liver, kidneys, and intestines [25].
Skin lesions similar to PXE have been reported after contact with saltpeter, ingestion of L-tryptophan, and in patients with blood dyscrasias [26-28]. PXE-like lesions described in patients with beta-thalassemia [29]. were thought to be caused by genetically distinct dermal alterations than those found in classic PXE as the genes responsible for both conditions are located on separate chromosomes, 11 and 16 respectively. In an effort to compare the structure of PXE skin lesion, biopsy samples were taken from patients with classic PXE and those with PXE-like lesions and beta-thalassemia [28]. Interestingly, in both disorders, elastic fiber polymorphism, fragmentation, and mineralization were structurally identical, as were abnormalities of collagen fibrils and filament aggregates [28]
3.1. Pathophysiology
The pathophysiology underlying both classic and acquired PXE has not been fully elucidated. The majority of cases of PXE are caused by mutation in the Abcc6 gene. Two general mechanisms have been proposed to explain the consequences of Abcc6 mutations in PXE. The prevailing hypothesis argues that absence of functional ABCC6 activity results in deficiency of circulating factors required for prevention of ectopic mineralization of tissue. The competing argument postulates that the lack of ABCC6 expression in the resident cells alters the biologic profile of the cell [30]. To test these competing hypotheses, Jiang et al. performed an elegant experiment using Abcc6−/−knockout (KO) mice [30]. Skin grafts from genetically engineered KO mice were transplanted on the backs of wild type (WT) mice and subsequent tissue mineralization was measured. For comparison, skin grafts from WT mice were also transplanted to KO mice, and for control, WT skin grafts were transplanted on to WT mice. The survival of the skin grafts was dependent on establishing adequate blood circulation from the recipient animal. The results of the experiment showed Abcc6−/−mice skin grafts did not develop mineralization when grafted on to Abcc6+/+mice, however skin from WT mice would mineralize after grafting on to Abcc6−/−mice [30]. The findings of this experiment supported the metabolic hypothesis and the authors concluded that circulating factors in the recipients blood were likely responsible in determining the degree of mineralization of the graft, regardless of the graft genotype. Thus far, the specific circulating factors influencing tissue mineralization have yet to be identified.
3.2. Prevalence
The precise prevalence of acquired PXE in patients with SCD remains unknown. Angiod streaks, a characteristic ocular manifestation of PXE, have been reported in patients with SCD since the 1950s [31]. In a review of 100 patients with thalassemia major and thalassemia intermedia, Aessopos found that 16% had PXE cutaneous lesions confirmed by skin histopathology and 20% had angiod streaks [31]. This finding was reproduced by Aessopossix years later while focusing on patients with thalassemia and arterial calcifications at which time 20% of patients manifested PXE-like cutaneous lesions [31].
3.3. Clinical Features
In the initial report of acquired PXE in patients with SCD, angiod streaks and PXE-like skin changes of the neck were described [26,27]. Dermatologic manifestations of both classic PXE and acquired PXE are caused by accumulation of calcium phosphate complexes in the skin and typically do not manifest until the second or third decade of life. Clinical symptoms vary and are generally milder in acquired PXE when compared to classic PXE [32,28]. Primary skin lesions are small, yellowish papules that may coalesce into plaques, occurring most commonly in the neck and flexural areas [33,34].
3.4. Treatment
Treatment for both classic and acquired PXE can be directed at either the organ systems involved or at the underlying aberrant mineralization of tissues. Regarding ocular manifestations of PXE, patients with angiod streaks are generally asymptomatic unless the lesions extend toward the fovea or develop macular chorodial neovascularization [35]. Recent clinical trials organized by the National Eye Institute and the National Institutes of Health have compared different anti-angiogenic compounds to counteract neovascularization of the retina in PXE patients [25]. In contrast to the organ system approach, studies have emerged which look more closely at ways to reduce mineralization of all tissues. These studies include those in which the mineral content of diet and the severity of disease are explored. Interestingly, studies using Abcc6−/−knockout mice have shown that magnesium, when added to the mouse diet in significant amounts, can prevent the aberrant mineralization associated with PXE [25].
4. Conclusion
Lower extremity ulcers are a source of significant morbidity in sickle cell patients (Figure 2). The etiology ofulcer formation remains poorly understood and treatment woefully inadequate. Acquired PXE in patients

Figure 2. Sickle cell ulcer of the lower extremity.
with hemoglobinopathies also presents a unique challenge with manifestations ranging from benign cutaneous lesions to decreased visual acuity. While recent studies have brought to light the importance of nitric oxide in the formation and treatment of ulcers in SCD, further work will be required to establish improved treatment regimens for patients. Similarly, elucidation of the substrate of the Abcc6 export protein will likely prove important for continued translational research on acquired PXE.
REFERENCES
- O. Brawley, L. Cornelius, L. Edwards, V. Gamble, B. Green, C. Inturrisi, et al., “NIH Conference Annals of Internal Medicine National Institutes of Health Consensus Development Conference Statement: Hydroxyurea Treatment for Sickle Cell Disease,” Annals of Internal Medicine, Vol. 148, No. 12, 2008, pp. 932-938. doi:10.7326/0003-4819-148-12-200806170-00220
- M. Bender and W. Hobbs, “Gene Reveiws,” University of Washington, Seattle, 1993.
- J. Herrick, “Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia. 1910,” Yale Journal of Biology and Medicine, Vol. 74, No. 3, 2001, pp. 179-184.
- C. Cummer and C. LaRocco, “Ulcers of the Legs in Sickle Cell Anemia,” Archives of Dermatology, Vol. 42, No. 6, 1940, pp. 1015-1039. doi:10.1001/archderm.1940.01490180024002
- V. Nolan, A. Adewoye, C. Baldwin, L. Wang, D. Wyszzynski, J. Farrell, et al., “Sickle Cell Leg Ulcers: Associations with Haemolysis and SNPs in Klotho, TEK and Genes of the TGF-B/BMP Pathway,” British Journal of Haematology, Vol. 133, No. 5, 2006, pp. 570-578. doi:10.1111/j.1365-2141.2006.06074.x
- J. Trent and R. Kirsner, “Leg Ulcers in Sickle Cell Disease,” Advances in Skin & Wound Care, Vol. 17, No. 8, pp. 410-416. doi:10.1097/00129334-200410000-00010
- F. Wolfort and T. Krizek, “Skin Ulceration in Sickle Cell Anemia,” Plastic and Reconstructive Surgery, Vol. 43, No. 1, 1969, pp. 71-77. doi:10.1097/00006534-196901000-00011
- E. Chirico and V. Pialoux, “Role of Oxidative Stress in the Pathogenesis of Sickle Cell Disease,” IUBMB Life, Vol. 64, No. 1, 2012, pp. 72-80. doi:10.1002/iub.584
- K. Wood, L. Hsu and M. Gladwin, “Sickle Cell Disease Vasculopathy: A State of Nitric Oxide Resistance,” Free Radical Biology & Medicine, Vol. 44, No. 8, 2008, pp. 1506-1528. doi:10.1016/j.freeradbiomed.2008.01.008
- M. Koshy, R. Entsuah, A. Koranda, A. Kraus, R. Johnson, R. Bellvue, et al., “Leg Ulcers in Patients with Sickle Cell Disease,” Blood, Vol. 74, No. 4, 1989, pp. 1403-1408.
- M. Halabi-Tawil, F. Lionnet, R. Girot, C. Bachmeyer, P. Lévy and S. Aractingi, “Sickle Cell Leg Ulcers: A Frequently Disabling Complication and a Marker of Severity,” British Journal of Dermatology, Vol. 158, No. 2, 2008, pp. 339-344. doi:10.1111/j.1365-2133.2007.08323.x
- G. Serjeant, “Leg Ulceration in Sickle Cell Anemia,” Archives of Internal Medicine, Vol. 133, No. 4, 1974, pp. 690-694. doi:10.1001/archinte.1974.00320160184017
- J. Eckman, “Leg Ulcers in Sickle Cell Disease,” Hematology/Oncology Clinics of North America, Vol. 10, No. 6, 1996, pp. 1333-1344. doi:10.1016/S0889-8588(05)70404-4
- G. Serjeant, B. Serjeant, J. Mohan and A. Clare, “Leg Ulceration in Sickle Cell Disease: Medieval Medicine in a Modern World,” Hematology/Oncology Clinics of North America, Vol. 19, No. 5, 2005, pp. 943-956. doi:10.1016/j.hoc.2005.08.005
- S. Robinson and S. Tasker, “Chronic Leg Ulcers of Sickle Cell Anemia; Report of Case; with Reference to Recognition,” California Western Journal of Medicine, Vol. 64, No. 4, 1946, pp. 250-2.
- S. Sehgal and B. Arunkumar, “Microbial Flora and Its Significance in Pathology of Sickle Cell Disease Leg Ulcers,” Infection, Vol. 20, No. 2, 1992, pp. 86-88. doi:10.1007/BF01711070
- L. McMahon, H. Tamary, M. Askin, P. Adams-Graves, R. Eberhardt, M. Sutton, et al., “A Randomized Phase II Trial of Arginine Butyrate with Standard Local Therapy in Refractory Sickle Cell Leg Ulcers,” British Journal of Haematology, Vol. 15, No. 5, 2010, pp. 516-524. doi:10.1111/j.1365-2141.2010.08395.x
- G. Serjeant, R. Galloway and M. Gueri, “Oral Zinc Sulphate in Sickle-Cell Ulcers,” Lancet, Vol. 31, No. 2, 1970, pp. 891-892. doi:10.1016/S0140-6736(70)92067-2
- R. Ware and B. Aygun. Advances in the Use of Hydroxyurea,” Hematology ASH Education Program, Vol. 2009, No. 1, 2009, pp. 62-69. doi:10.1182/asheducation-2009.1.62
- O. Platt, “Hydroxyurea for the Treatment of Sickle Cell Anemia,” The New England Journal of Medicine, Vol. 358, No. 13, 2008, pp. 1362-1369. doi:10.1056/NEJMct0708272
- M. Gladwin, J. Shelhamer, F. Ognibene, M. Pease-Fye, J. Nichols, B. Link, et al., “Nitric Oxide Donor Properties of Hydroxyurea in Patients with Sickle Cell Disease,” British Journal of Haematology, Vol. 116, No. 2, 2002, pp. 436-444. doi:10.1046/j.1365-2141.2002.03274.x
- R. Khouri and J. Upton, “Bilateral Lower Limb Salvage with Free Flaps in a Patient with Sickle Cell Ulcers,” Annals of Plastic Surgery, Vol. 27, No. 6, 1991, pp. 574- 576. doi:10.1097/00000637-199112000-00011
- R. Richards, C. Bowen and M. Glynn. Microsurgical Free Flap Transfer in Sickle Cell Disease,” Annals of Plastic Surgery, Vol. 29, No. 3, 1992, pp. 278-281. doi:10.1097/00000637-199112000-00011
- M. Gueri and G. Serjeant, “Leg Ulcers in Sickle-Cell Anaemia,” Tropical and Geographical Medicine, Vol. 22, No. 2, 1970, pp. 155-160.
- J. Uitto, L. Bervcovitch, S. Terry and P. Terry, “Pseudoxanthoma Elasticum: Progress in Diagnostics and Research towards Treatment: Summary of the 2010 PXE International Research Meeting,” American Journal of Medical Genetics Part A, Vol. 155A, No. 7, 2011, pp. 1517- 1526. doi:10.1002/ajmg.a.34067
- W. Geeraets and D. Guerry, “Angioid Streaks and Sickle Cell Disease,” American Journal of Ophthalmology, Vol. 49, 1960, pp. 450-470.
- K. Suerig and F. Siefert, “Pseudoxanthoma Elasticum and Sickle Cell Anemia,” Archives of Internal Medicine, Vol. 113, No. 1, 1964, pp. 135-141. doi:10.1001/archinte.1964.00280070137022
- E. Fabbri, G. Forni, G. Guerrini and C. Borgna-Pignatti, “Pseudoxanthoma-Elasticum-Like Syndrome and Thalassemia: An Update,” Dermatology Online Journal, Vol. 15, No. 7, 2009, p. 7.
- A. Aessopos, P. Savvides, G. Stamatelos, I. Rombos, T. Tassiopoulos, M. Karagiorga, et al., “Pseudoxanthoma Elasticum-Like Skin Lesions and Angioid Streaks in Beta Thalassemia,” American Journal of Hematology, Vol. 41, No. 3, 1992, pp. 159-164. doi:10.1002/ajh.2830410304
- Q. Jiang, M. Endo, F. Dibra, K. Wang and J. Uitto, “Pseudoxanthoma Elasticum Is a Metabolic Disease,” Journal of Investigative Dermatology, Vol. 129, No. 2, 2009, pp. 348-354. doi:10.1038/jid.2008.212
- A. Aessopos, D. Farmakis and D. Loukopoulos, “Elastic Tissue Abnormalities Resembling Pseudoxanthoma Elasticum in Beta Thalassemia and the Sickling Syndromes,” Blood, Vol. 99, No. 1, 2002, pp. 30-35. doi:10.1182/blood.V99.1.30
- T. van Meurs, J. van Hagen, M. van de Scheur, H. Vermaat, M. Ruijs, H. van den Hoogenband, et al., “Classic Pseudoxanthoma Elasticum in a Patient with Sickle Cell Disease,” Journal of the American Academy of Dermatology, Vol. 56, No. 1, 2007, pp. 170-171. doi:10.1016/j.jaad.2006.10.023
- A. Adam and M. Maritim, “Pseudoxanthoma Elasticum in a Patient with Sickle Cell Disease: Case Report,” East African Medical Journal, Vol. 85, No. 2, 2008, pp. 98- 101. doi:10.4314/eamj.v85i2.9613
- Q. Li, Q. Jiang, E. Pfendner, A. Váradi and J. Uitto, “Pseudoxanthoma Elasticum: Clinical Phenotypes, Molecular Genetics and Putative Pathomechanisms,” Experimental Dermatology, Vol. 18, No. 1, 2009, pp. 1-11. doi:10.1111/j.1600-0625.2008.00795.x
- I. Georgalas, I. Tservakis, D. Papaconstaninou, M. Kardara, C. Koutsandrea and I. Ladas, “Pseudoxanthoma Elasticum, Ocular Manifestations, Complications and Treatment,” Clinical and Experimental Optometry, Vol. 94, No. 2, 2011, pp. 169-80. doi:10.1111/j.1444-0938.2010.00559.x
- E. Cacciola, R. Musso, R. Giustolisi, E. Cacciola and M. Alessi, “Blood Hypercoagulability as a Risk Factor for Leg Ulcers in Sickle Cell Disease,” Blood, Vol. 75, No. 12, 1990, pp. 2467-2478.