Open Journal of Ophthalmology
Vol.3 No.2(2013), Article ID:31268,4 pages DOI:10.4236/ojoph.2013.32009
Spinocerebellar Ataxia with Oculomotor Apraxia and Severe Corneal Astigmatism*
![]()
1IRCCS Eugenio Medea, Polo di Conegliano and Pieve di Soligo (TV), Italy; 2Institute for Maternal and Child Health—IRCCS Burlo Garofolo, Trieste, Italy.
Email: #pensiero@burlo.trieste.it
Copyright © 2013 Paola Michieletto et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 2nd, 2013; revised April 1st, 2013; accepted May 1st, 2013
Keywords: Spinocerebellar Ataxia; Oculomotor Apraxia; Corneal Astigmatism
ABSTRACT
Purpose: To disclose the association between spinocerebellar ataxia with oculomotor apraxia and high grade (7 diopters) congenital astigmatism. Methods: Single observational case report. A 39-year-old patient affected by spinocerebellar ataxia from the age of 20 was submitted to genetic and ophthalmic investigations to reach a diagnosis. Results: Genetic testing did not lead to a sure diagnosis, while clinical and instrumental ophthalmic examinations pointed out an oculomotor apraxia and a congenital severe astigmatism. Conclusion: To conclude the eye movement recording permitted to identify an oculomotor apraxia in this case of spinocerebellar ataxia. This is the first report of severe astigmatism in cases of ataxia with oculomotor apraxia.
1. Introduction
The term ataxia denotes a clinical syndrome of incoordination caused by lesions of the cerebellum and its afferent and/or efferent connections. A complete classification of cases, divided in hereditary ataxias, non hereditary degenerative ataxias and acquired ataxias, is shown in Klockgether & Paulson [1].
Ataxia with oculomotor apraxia (AOA) is a subgroup of autosomal recessive cerebellar ataxias (ARCAs) characterized in most of cases by ocular motor apraxia (OMA) [2]. Rather than a true apraxia (that is the inability to generate voluntary horizontal saccades with compensatory headthrusting and synkinetic blinking), OMA is better described as intermittent saccade failure (saccadic initiation failure, SIF) [3].
2. Case Report
We report on an adult male, 39 years old, who presented progressive cerebellar ataxia from the age of 20 years with consistent cerebellar atrophy, affecting predominantly the vermis, and a left temporal arachnoidal cyst on resonance magnetic imaging (RMI). No other case of cerebellar ataxia was present in his family. Moreover he presented signs of axonal sensorimotor neuropathy at the lower arms from the age of 28 years.
Genetic examination of genes ATXN1 (on chromosome 6p23), ATXN2 (12q23-q24.1) and ATXN3 (14q21), responsible of types 1, 2 and 3 of autosomal dominant spinocerebellar ataxias (SCA) did not show any alteration.
A progressive decline of cognitive functions was also observed. At the Wechsler Adult Intelligence Scale, revised (WAIS-R), which measures two different aspects of intelligence relating to verbal skills (VIQ) and performance skills (PIQ), at the last examination (39 years) he presented a VIQ of 49 and a PIQ of 50 (total IQ 45).
At this clinical control, normal alpha-fetoprotein, normal albumine, high cholesterol and normal immunoglobulin (IgG, IgA, IgE) levels were the more important serum evidences.
The ophthalmic examination showed convergent strabismus (variable angle esotropia) with bilateral limited abduction, and suspected an OMA, owing to some episodes of head rotation, before eyes, during voluntary lateral movements. Therefore an eye movement recording was performed (using an infrared limbus-tracking technique). During visually-guided saccade testing some tracts of SIF were evident, saccadic latencies were longer (mean 250 ms), saccadic peak velocities were normal, gazeevoked nystagmus to the right (of cerebellar origin, as shown by slow phases with linear velocity profile) was observed (Figure 1). So, it was a case of AOA.
OMA can be present only in some autosomal recessive ataxias: atassia-telangectasia (AT), ataxia telangectasialike disorder (ATLD), ataxia with oculomotor apraxia type 1 (AOA1) and type 2 (AOA2) [2]. AT was excluded for the age of onset (normally 2 - 3 years of life) and the lack of oculocutaneous telangectasias in our patient. ATLD is very similar to AT but has later onset with slower progression; patients lack telangiectasias and do not have raised concentrations of serum alpha-fetoprotein, but present low immunoglobulin levels. No cognitive impairment is reported. A cognitive impairment is present only in AOA1 and AOA2, high cholesterol in both and low albumin only in AOA1. Strabismus was reported only in AOA2 [4].
The analysis of the aprataxin gene (APTX) on chromosome 9p13 did not show APTX mutations in our patient, excluding AOA1. As regards AOA2, the spinocerebellar ataxia, with onset between 12 and 22 years, is the most frequent presenting sign, as in our patient. It is characterized by cerebellar atrophy, peripheral neuropathy (100% of cases), OMA (50% of cases), and frequently elevated serum alpha-fetoprotein, while our patient presented normal values.
Genetic examination on chromosome 9q34 showed SETX mutation: three nucleotidic variations in heterozygous state were present: c.2975A > G(K992R), c.4365T > C(S1459P) and c.6341A > G(E2114G). From the data available in the literature, these variations alone are not able, in heterozygous state, to cause the disease.
Patient’s father died many years ago for a heart attack; his mother presented only one of the three SETX variations: E2114G.
Therefore it was not possible to obtain a genetic diagnosis of the illness.
Apart from convergent strabismus and OMA, the ophthalmic examination showed a prominent congenital alteration.
Cycloplegic refraction (obtained using cyclopentolate drops) was characterized by a mixed astigmatism of high grade in both eyes:
Right eye (RE) +3.00 sph −6.75 cyl (175); left eye (LE) +2.75 sph −4.75 cyl (175).
The best corrected visual acuity was of 20/32 in RE and 20/30 in LE. Glasses for the astigmatism were worn till age of six years.
In Figure 2 the corneal topography shows a bilateral severe corneal astigmatism of congenital type: keratometry of RE is 46.04 D on the steeper axis (89˚) and 40.85 D on the flatten axis (179˚), with a resultant cylinder of 5.19 D; LE 45.45 D at 94˚ and 41.48 D at 4˚, with a cylinder of 3.97 D.
No other case of high astigmatism was present in the family.
3. Discussion
It was not possible to classify our case among the various types of ataxias. Eye movements recording permitted a sure evidence of OMA in this case, but his membership to a type of AOA was not proved even using the genetic examination.

Figure 1. A short tract of saccadic initiation failure (SIF) and gaze-evoked nystagmus. In abscissa the time (in seconds) from the beginning of the recording. In ordinate the degrees of ocular deviation to the right from the primary position (0). Black line: position of the stimulus. Red line: position of the right eye. Blue line: position of the left eye.
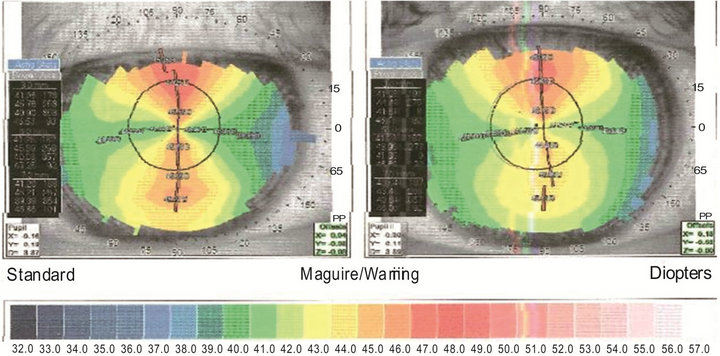
Figure 2. Corneal topography of the right (left image) and left (right image) eye. In both eyes a clepsydra-like image, typical of congenital astigmatism, is clearly observed. The difference (that is the astigmatism) between the steeper (vertical) and flatten (horizontal) axis is 5.19 diopters (D) in the right eye and 3.97 D in the left eye.
In any case, severe congenital corneal astigmatism was never reported in cases of AOA.
Refraction is determined by means of the coordinated contributions of ocular biometric components, such as axial length, anterior chamber depth, corneal curvature, and lens thickness. Now there are evidences of the existence of genetic causes of refractive errors [5], in particular of an autosomal dominant inheritance of astigmatism [6]. In some cases of irregular astigmatism (cornea plana, keratoconus) a genetic cause was clearly identified [7,8].
The extracellular matrix of many connective tissues contains a class of proteoglycans known as small leucine rich repeat proteoglycans (SLRPs). These proteins are involved in the regulation of collagen fibril diameter, interfibrillar spacing, and the adhesive properties of different types of collagens. The SLRPs have also been recognized as molecules that exhibit direct and indirect cell signaling properties, thereby influencing cellular growth, differentiation and migration. At a genomic level, the various members of the SLRP gene family map to relatively few chromosomes: Opticin, PRELP, and fibromodulin map to human chromosome 1q3; asporina (ASPN), osteoadherin/ osteomodulin (OMD), and OGN/mimecan map to human chromosome 9q2; epiphycan, keratocan, lumican, and decorin to human chromosome 12q2; chondroadherin (Chad) to human chromosome 17q21 [9].
All these evidences show that some genetic factors able to interfere in corneal structure, also located in chromosome 9, could be involved in astigmatism formation and so a genetic cause of both AOA and astigmatism must be sought. Otherwise a random combination must be taken into account.
4. Conclusion
We think it is important to point out this first case of AOA with severe astigmatism to add more information about the cases of ataxia not yet classifiable.
5. Acknowledgements
Work partially supported by IRCCS Eugenio Medea.
REFERENCES
- T. Klockgether and H. Paulson, “Milestones in Ataxia,” Movement Disorders, Vol. 26, No. 6, 2011, pp. 1134-1141. doi:10.1002/mds.23559
- B. L. Fogel and S. Perlman, “Clinical Features and Molecular Genetics of Autosomal Recessive Cerebellar Ataxias,” The Lancet Neurology, Vol. 6, No. 3, 2007, pp. 245- 257. doi:10.1016/S1474-4422(07)70054-6
- C. M. Harris, F. Shawkat, I. Russell-Eggitt, J. Wison and D. Taylor, “Intermittent Horizontal Saccade Failure (Ocular Motor Apraxia) in Children,” British Journal of Ophthalmology, Vol. 80, No. 2, 1996, pp. 151-158. doi:10.1136/bjo.80.2.151
- M. Anheim, B. Monga, M. Fleury, P. Charles, C. Barbot, M. Salih, J. P. Delaunoy, M. Fritsch, L. Arning, M. Synofzik, L. Schöls, J. Sequeiros, C. Goizet, C. Marelli, I. Le Ber, J. Koht, J. Gazulla, J. De Bleecker, M. Mukhtar, N. Drouot, L. Ali-Pacha, T. Benhassine, M. Chbicheb, A. M’Zahem, A. Hamri, B. Chabrol, J. Pouget, R. Murphy, M. Watanabe, P. Coutinho, M. Tazir, A. Durr, A. Brice, C. Tranchant and M. Koenig, “Ataxia with Oculomotor Apraxia Type 2: Clinical, Biological and Genotype/Phenotype Correlation Study of a Cohort of 90 Patients,” Brain, Vol. 132, No. 10, 2009, pp. 2688-2698. doi:10.1093/brain/awp211
- T. L. Young, R. Metlapally and A. E. Shay, “Complex Trait Genetics of Refractive Error,” Archives of Ophthalmology, Vol. 125, No. 1, 2007, pp. 38-48. doi:10.1001/archopht.125.1.38
- M. Clementi, M. Angi, P. Forabosco, E. Di Gianantonio and R. Tenconi, “Inheritance of Astigmatism: Evidence for a Major Autosomal Dominant Locus,” The American Journal of Human Genetics, Vol. 63, 1998, pp. 825-830. doi:10.1086/302014
- E. Tahvanainen, A. S. Villanueva, H. Forsius, P. Salo and A. de la Chapelle, “Dominantly and Recessively Inherited Cornea Plana Congenita Map to the Same Small Region of Chromosome 12,” Genome Research, Vol. 6, No. 4, 1996, pp. 249-254. doi:10.1101/gr.6.4.249
- N. S. Pellegata, J. L. Dieguez-Lucena, T. Joensuu, S. Lau, K. T. Montgomery, R. Krahe, T. Kivelä, R. Kucherlapati, H. Forsius and A. de la Chapelle, “Mutations in KERA, Encoding Keratocan, Cause Cornea Plana,” Nature Genetics, Vol. 25, No. 1, 2000, pp. 91-95. doi:10.1038/75664
- E. S. Tasheva, A. Ke and G. W. Conrad, “Analysis of the Expression of Chondroadherin in Mouse Ocular and NonOcular Tissues,” Molecular Vision, Vol. 10, 2004, pp. 544- 554.
NOTES
*Conflict of Interest and Source of Funding: No conflicts of interest are declared.
#Corresponding author.