Open Journal of Pathology
Vol.05 No.04(2015), Article ID:60329,7 pages
10.4236/ojpathology.2015.54016
Anaplastic Pleomorphic Xanthoastrocytoma: Morphological and Molecular Features of Three Cases
Nil Çomunoğlu1*, Pınar Karabağlı2, Şebnem Batur1, Büge Öz1
1Department of Pathology, Faculty of Medicine, Istanbul University, Istanbul, Turkey
2Department of Pathology, Faculty of Medicine, Selçuk University, Konya, Turkey
Email: *nilustundag@yahoo.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 1 September 2015; accepted 13 October 2015; published 16 October 2015
ABSTRACT
Pleomorphic xanthoastrocytoma (PXA) is usually a low grade astrocytic tumor. However, some cases show significant mitotic activity (5 or more mitosis per 10 High Power Field) and/or necrosis. These tumors are described as pleomorphic xanthoastrocytomas with anaplastic features and display increased risk of recurrence. We aimed to evaluate histopathological, immunohistochemical and molecular features of PXAs with anaplastic features, by reporting three primary cases displaying recurrence. Histopathologically we observed rhabdoid-like monomorphic atypical tumoral cells with increased mitotic activity, vascular endothelial proliferation and necrosis. Immunohistochemically, astrocytic and neuronal components displayed different specific staining properties. In 2 cases p53 was immunopositive. We detected BRAF V600E mutation in 2 cases and p16 mutation in one case, by fluorescence in situ hybridization (FISH) method. Anaplastic pleomorphic xanthoastrocytoma (APXA) is a rare tumor. We have presented 3 primary APXA cases displaying all characteristic histopathological features. Two of these cases were immunopositive for p53. Therefore, we think that this marker may not be so useful in differentiating APXA from glioblastoma (GBM). Two of our three cases display BRAF V600E mutation, which is compatible with the literature.
Keywords:
Anaplastic Pleomorphic Xanthoastrocytoma, Fluorescence in Situ Hybridization, Immunohistochemistry

1. Introduction
PXAs are astrocytic tumors occurring in children and young adults having a favorable prognosis. They arise at cerebral hemisheres, localized superficially, and at meninges. They are WHO grade II tumors containing a reticulin framework, consisting of pleomorphic and lipidized cells immunoreactive for glial fibrillary acidic protein (GFAP), and eosinophilic granular bodies (EGB) [1] [2] . Some PXA cases display anaplastic features such as high mitotic activity, necrosis. Giannini et al. described APXAs as PXAs containing five or more mitosis per 10 high power fields (HPF) and/or necrosis [3] . This group behaves like grade III gliomas biologically and shows frequent recurrences. Survival is shorter for these tumors. However, currently grading of these tumors according to WHO is not yet defined. Some of these tumors derive from PXA secondarily whereas some occur de novo. This latter group is described as primary APXA. Some of PXAs present as glioblastoma when recurs [2] [4] -[7] . BRAF V600E mutation is frequently seen in PXAs [8] [9] and APXAs [8] . In this report we will present clinical, morphological and molecular characteristics of three primary APXA cases.
2. Case Presentations
2.1. Case 1
A 32-year-old male patient presented with a generalized epileptic seizure. Neurological examination was normal. Cerebral computerized tomography (CT) and magnetic resonance imaging (MRI) revealed a small solid lesion localized at the frontal lobe, without clear dural attachment, inhomogeneously hyperdense/hyperintense, with irregular enhancement after contrast injection (Figure 1). The patient underwent operation with a preoperative diagnosis of high grade glioma. Histopathologically a dense hypercellular glial tumor was observed. Histopathological and immunohistochemical features (Figure 2 and Figure 3) are summarized in Table 1. A diagnosis of APXA was given. Six months later control MRI revealed a recurrent mass lesion. The morphologic features of the recurrent tumor were similar with that of the primary tumor except for that necrosis was present in the recurrent tumor. Histopathological and immunohistochemical features (Figure 4) are summarized in Table 2. Recurrent tumor was also diagnosed as APXA. In both tumors BRAF V600E mutation was detected by FISH method. p16 mutation was not observed.
2.2. Case 2
A 32-year-old female patient presented with headache. MRI revealed well circumscribed small, solid, contrast
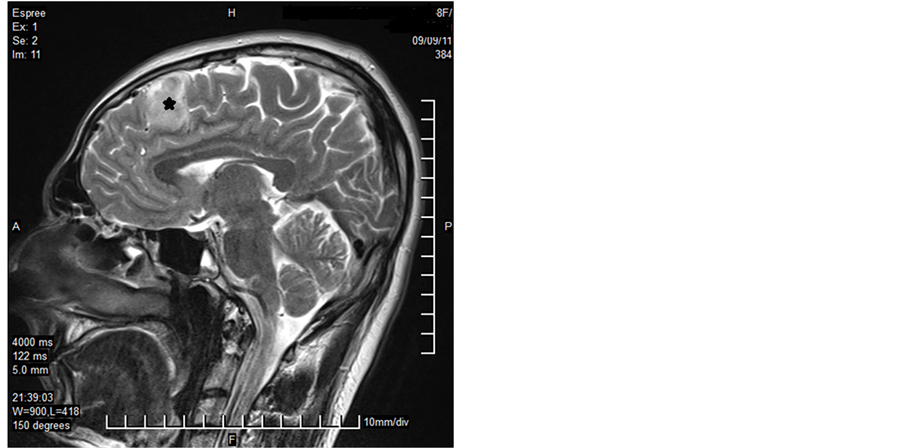
Figure 1. MRI revealed a small solid lesion at the frontal lobe, without clear dural attachment (Case 1―primary tumor; asterix).
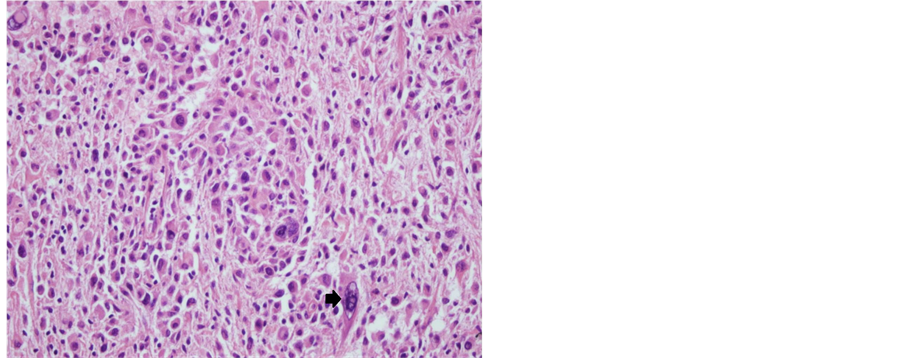
Figure 2. Tumoral cells were prominently pleomorphic cells with foamy, xanthomatous cytoplasm, nuclear inclusions and multinuclear giant cells (arrow) (Case 1―primary tumor; H&E, 200×).

Figure 3. GFAP immunopositivity in tumoral cells (Case 1― primary tumor; GFAP, 100×).
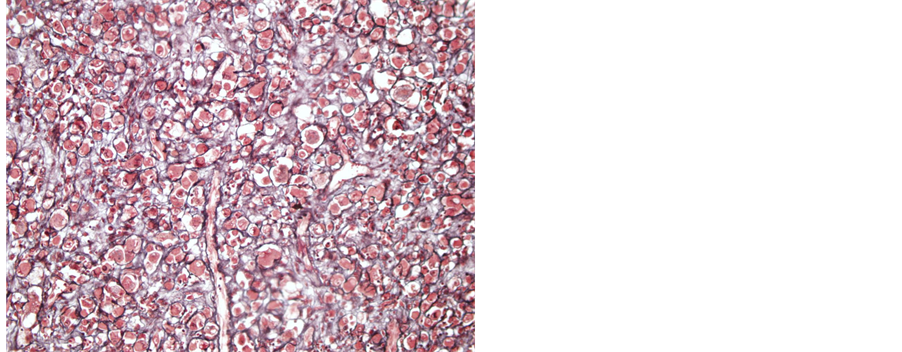
Figure 4. Reticulin surrounded individual tumoral cells (Case 1―recurrent tumor; reticulin, 400×).

Table 1. Histopathological features of APXA cases.
enhancing mass at the left temporal lobe. Histopathological evaluation showed an infiltrative, cellular glial tumor. Histopathological and immunohistochemical features are summarized in Table 1. A diagnosis of APXA was given. Control MRI 1 year later revealed isointense, dense, homogenously contrast enhancing 3 mass lesions located at extraaxial part of left frontal lobe, superior vertex (subarachnoidal dissemination) and left inferotemporal lobe. Re-operation was performed. Histopathologically prominent reticulogenesis was detected in addition to the features seen in the primary tumor. Histopathological and immunohistochemical features are summarized in Table 2. Recurrent tumor was also given a diagnosis of APXA. BRAF V600E mutation was not detected.
2.3. Case 3
A 23-year-old female patient presented with epileptic seizure. MRI revealed intraparenchymal mass lesion in the right temporal lobe, measuring approximately 5 × 5 cm. Lesion showed indentation into cavernous sinus from the medial aspect and Meckel’s cave. Lesion had irregular borders, lobulated contours and showed heterogenous contrast enhancement. Histopathologically cellular glial tumor was observed. Histopathological and immunohistochemical features are summarized in Table 1. A diagnosis of APXA was given. Six months later control MRI revealed a recurrent lesion. Re-operation was performed. Histopathological and immunohistochemical features are summarized in Table 2. Recurrent tumor was also diagnosed as APXA. BRAF V600E and p16 mutations were detected.
2.4. Molecular Analysis
2.4.1. Fluorescence in Situ Hybridization
Fluorescence in situ hybridization (FISH) was performed on four micron tissue sections according to the methods previously described [9] .
Table 2. Histopathological features of recurrent APXA cases.
2.4.2. Mutational Analysis
A DNA sample from the patients’ brain tumour tissue was extracted using a commercial extraction kit (Qiagen, Valencia, CA, USA) for mutational analysis of p16 gene. Polymerase chain reaction (PCR)-single strand conformational polymorphism (PCR-SSCP) was performed according to the methods previously described [10] .
3. Discussion
PXA is firstly described by Kepes et al. in 1979 [10] . In pre-immunohistochemistry era it has been considered as a mesenchymal tumor showing morphologically malignant features, originating from meninges and generally occurring in children and young adults [2] . At the end of 70’s Kepes and Rubinstein applied immunohistochemistry for these morphologically and clinically distinct tumors and observed that tumor cells expressed GFAP supporting their astrocytic origin [11] . To date, in several reports it has been supported that molecular and subcellular features of PXA’s had indicated their astrocytic and neurocytic genotype and phenotype. Tumoral cells show immunopositivity for glial markers such as GFAP and S-100. Eight to seventy three percent positivity has also been reported for neuronal markers, such as class III beta tubulin, synaptophysin, NFP, SMI-31 and MAP-2 [2] [12] [13] . Ultrastructural investigations further showed astrocytic (intermediate filament, lipid droplets, lysosomes) and neuronal (microtubules, dense core granules) features of tumoral cells [14] . In 1989, Weldon- Linne et al. described PXA with anaplastic features [15] . In 1999, Giannini et al. suggested the usage of the terminology of “PXA with anaplastic features” for the tumors that contains 5 mitosis per 10 HPF and/or necrosis [3] .
Classical PXA shows superficial localization, leptomeningeal involvement and invasion through duramater. Resulting from superficial localization, most of the patients present with epileptic seizures. Radiologically, PXA is seen mostly as a solid mass having a cystic component. It may have an infiltrative cortical component, complicating differential diagnosis with diffuse astrocytoma [1] [2] . Multicentricity may occur. In contrast, it has been reported that anaplastic PXAs could show extracranial involvement in paranasal sinuses, nasal cavity, orbita and skull bone [16] . Vu et al. reviewed the literature and observed that 91% of the cases had had supratentorial localization [17] .
Several investigators examined morphological features of primary and secondary APXA. Hirose et al. reported 6 APXA cases in their series. Histopathological features of these APXA cases included intranuclear inclusions in 3 cases, xanthomatous cells in 2 cases, desmoplasia in 4 cases, EGB in 3 cases, calcification in 2 cases, lymphocytes in 3 cases, ganglioside cells in 1 case, monotonous cell population in all 6 cases and necrosis in 4 cases. Mitotic activity was 1 - 15 per 10 HPF and Ki-67 proliferation index was 11.8% - 33.2% [5] . Hirose et al. claimed that the criteria proposed by Giannini et al. [3] were difficult to apply [5] . In report of Hirose et al., 2 cases showed mitosis below 5 per 10 HPF. They have given the diagnosis with the aid of observation of necrosis, microvascular proliferation, prominent cellular anaplasia and high Ki-67 score [5] . In their review of APXA cases, Vu et al. [17] observed necrosis in 12 cases, VEP in 13 cases and found that mitotic index had varied between 1 and 30 per 10 HPF.
In all three patients of our case series, we observed xanthomatous cells, EGB, intranuclear inclusions and perivascular lymphocytes. Calcification, necrosis, vascular endothelial proliferation (VEP) were present in 2 cases. In all cases increase in reticulin fibers was seen. Mitotic index was between 5 and 11 per 10 HPF. Ki-67 labeling index varied between 20% and 80%. We observed monotonous rhabdoid-like cells.
In PXAs it has been demonstrated that astrocytic and neurocytic immunohistochemical markers are positive. However, in English literature, immunostaining properties of APXA cases have been reported to be variable. In APXA, GFAP may be focally or weakly positive. Immunopositivity of neuronal markers such as synaptophysin, Class III beta tubulin, neurofilament protein had been reported [11] [12] . As an astrocytic marker, p53 is negative or focally positive [18] . Vu et al. observed GFAP positivity in 22 cases, S-100 positivity in 11 cases, Neurofilament positivity in 7 of 13 tested cases, synaptophysin in 5 of 12 tested cases [17] . Hirose et al. detected GFAP positivity in 5 of 5 tested cases, S-100 positivity in 3 of 3 tested cases, olig-2 positivity in 5 of 5 tested cases, Neurofilament positivity in 3 of 5 tested cases, synaptophysin positivity in 2 of 5 tested cases, chromogranin positivity in 1 of 3 tested cases in their study. p53 labeling index was between 2% and 59.2% [5] . In 2 of our 3 cases p53 was positive. Perry et al. claimed that PXAs with neuronal differentiation showed anaplastic features more frequently [19] although Hirose et al. did not observe this feature [5] . Interpretation and determination of the discriminating role of immunohistochemical stains is difficult regarding the existence of cases in the literature reported as immunonegative for neuronal and astrocytic markers. Giannini et al. specifically suggested that p53 was strongly positive in GBM’s and therefore this feature could be useful in differential diagnosis of APXA [18] .
PXAs can have diploid or polyploid karyotype. Chromosomal studies reported loss in chromosome 3, 5, 20 and 22. DNA loss in chromosome 9 has been observed in 50% of PXAs [17] . It has been demonstrated that p53 mutation and EGFR overexpression which had been shown in other glial tumors of WHO grade II had not been present in PXA usually [20] . In contrast BRAF V600E mutation is frequent in PXA. It has been claimed that molecular features of PXA had been similar with those of pilocytic astrocytoma and ganglioglioma instead of WHO grade II glial tumors [8] . APXAs are rare tumors, therefore data about their molecular features are limited [20] . Schindler et al. detected BRAF V600E mutation in 66% of PXA cases. Of these 57% were adult and 43% were pediatric patients. BRAF V600E mutation was seen in 65% of APXA cases. Of these cases 33% were adult and 67% were pediatric cases. BRAF mutation was detected most frequently in tumors located at temporal lobe. It has been suggested that RAS/RAF/MEK/ERK kinase pathway should be investigated in tumors which did not show BRAF mutation [8] . In one of our cases (Case 2), we could not detect BRAF V600E mutation. In the case which did not show BRAF V600E mutation, p53 was also negative. Santago et al. observed BRAF V600E mutation in only 1 of their 6 APXA cases [9] . A comprehensive study about BRAF mutation in APXAs is not present. In addition, distinction between primary and secondary tumors has not been studied yet.
WHO defines PXA as an astrocytic tumor. A number of molecular abnormalities are associated with astrocytomas, and recent studies suggest that most of these abnormalities are related with cell cycle regulation, including the p16 gene. However, Nasuha et al. reported that p16 had been negative in an APXA case in a 10 year-old male patient [10] . They claimed that the p16 gene and LOH on chromosome 10 had not played role in tumorigenesis of PXA [10] . In contrast, we have detected p16 mutation in 1 of our cases. In this case, we have also seen BRAF V600E mutation which supports astrocytic origin of PXAs.
The mechanisms of anaplastic transformation of PXAs are not clear [5] [21] . Secondary APXAs are reported more frequently than primary APXAs [4] -[7] . Vu et al. [17] observed recurrence in 16 primary APXA cases. Mean recurrence time was reported to be 12 months. In Vu et al.’s review 50% of 14 cases showed recurrence after 15 months. In 55.6% of cases which had been underwent subtotal resection recurred after 6 months. In 5 of 16 recurrent cases malignant transformation was observed. Four of these tumors were GBM and the other one was fibrosarcomatous APXA. In APXAs originating from primary PXAs, mean recurrence time is reported to be 29.7 months after total resection, and 37.1 months after subtotal resection. Twelve cases recurred as GBM, 13 cases as small cell GBM, 3 cases as malignant glioma, 2 cases as oligodendroglioma, 1 case as giant cell GBM. Recurrence interval in secondary APXAs is longer than that of primary APXAs’ but it has been reported that secondary APXAs had worse prognosis [17] . It is prognostically important to differentiate APXA from GBM because APXAs are reported to have more favorable prognosis [5] [22] . All three cases in our series were primary APXA. We have observed recurrence in all these cases between 6 months and 1 year time period.
4. Conclusion
As a result, we have presented 3 primary APXA cases all meeting the histopathological criteria suggested by Giannini et al. [3] . APXAs are aggressive tumors displaying frequent recurrences. Two of these cases are immunopositive for p53; therefore we think that this marker may not be so useful in differentiating APXA from GBM. Two of our cases displayed BRAF V600E mutation, compatible with the literature. In addition, we think that p16 may play a role in anaplastic transformation of PXAs.
Conflict of Interest
The authors have no financial conflict of interests.
Cite this paper
NilÇomunoğlu,PınarKarabağlı,ŞebnemBatur,BügeÖz, (2015) Anaplastic Pleomorphic Xanthoastrocytoma: Morphological and Molecular Features of Three Cases. Open Journal of Pathology,05,117-124. doi: 10.4236/ojpathology.2015.54016
References
- 1. Giannini, C., Paulus, W., Louis, D.N. and Liberski, P. (2007) Pleomorphic Xanthoastrocytoma. In: Louis, D.N., Ohgaki, H., Wiestler, O.D. and Cavenee, W.K., Eds, World Health Organization Classification of Tumours, WHO Classification of Tumours of the Central Nervous System, IARC Press, Lyon, 22-24.
- 2. Burger, P.C. and Scheithauer, B.W. (2007) Pleomorphic Xanthoastrocytoma. In: Burger, P.C. and Scheithauer, B.W., Eds, Tumors of the Central Nervous System, AFIP, Washington DC, 106-113.
- 3. Giannini, C., Scheithauer, B.W., Burger, P.C., Brat, D.J., Wollan, P.C., Lach, B. and O’Neill, B.P. (1999) Pleomorphic Xanthoastrocytoma: What Do We Really Know about It? Cancer, 85, 2033-2045.
http://dx.doi.org/10.1002/(SICI)1097-0142(19990501)85:9<2033::AID-CNCR22>3.0.CO;2-Z - 4. Marton, E., Feletti, A., Orvieto, E. and Longatti, P. (2007) Malignant Progression in Pleomorphic Xanthoastrocytoma: Personal Experience and Review of the Literature. Journal of the Neurological Sciences, 252, 144-153.
http://dx.doi.org/10.1016/j.jns.2006.11.008 - 5. Hirose, T., Ishizawa, K., Sugiyama, K., Kageji, T., Ueki, K. and Kannuki, S. (2008) Pleomorphic Xanthoastrocytoma: A Comparative Pathological Study between Conventional and Anaplastic Types. Histopathology, 52, 183-193.
http://dx.doi.org/10.1111/j.1365-2559.2007.02926.x - 6. Sharma, A., Nand Sharma, D., Kumar Julka, P. and Kishor Rath, G. (2011) Pleomorphic Xantho-astrocytoma—A Clinico-Pathological Review. Neurologia i Neurochirurgia Polska, 45, 379-386.
http://dx.doi.org/10.1016/S0028-3843(14)60109-2 - 7. Schmidt, Y., Kleinschmidt-DeMasters, B.K., Aisner, D.L., Lillehei, K.O. and Damek, D. (2013) Anaplastic PXA in Adults: Case Series with Clinicopathologic and Molecular Features. Journal of Neuro-Oncology, 111, 59-69.
http://dx.doi.org/10.1007/s11060-012-0991-4 - 8. Schindler, G., Capper, D., Meyer, J., Janzarik, W., Omran, H., Herold-Mende, C., Schmieder, K., Wesseling, P., Mawrin, C., Hasselblatt, M., Louis, D.N., Korshunov, A., Pfister, S., Hartmann, C., Paulus, W., Reifenberger, G. and von Deimling, A. (2011) Analysis of BRAF V600E Mutation in 1320 Nervous System Tumors Reveals High Mutation Frequencies in Pleomorphic Xanthoastrocytoma, Ganglioglioma and Extra-Cerebellar Pilocytic Astrocytoma. Acta Neuropathologica, 121, 397-405.
http://dx.doi.org/10.1007/s00401-011-0802-6 - 9. Dias-Santagata, D., Lam, Q., Vernovsky, K., Vena, N., Lennerz, J.K, Borger, D.R., Batchelor, T.T., Ligon, K.L., Iafrate, A.J., Ligon, A.H., Louis, D.N. and Santagata, S. (2011) BRAF V600E Mutations Are Common in Pleomorphic Xanthoastrocytoma: Diagnostic and Therapeutic Implications. PLoS ONE, 29, e17948.
http://dx.doi.org/10.1371/journal.pone.0017948 - 10. Nasuha, N.A., Daud, A.H., Ghazali, M.M., Yusoff, A.A., Zainuddin, N., Abdullah, J.M., Mutum, S.S., Biswal, B.M., Ariff, A.R., Sulong, S. and Isa, M.N. (2003) Molecular Genetic Analysis of Anaplastic Pleomorphic Xanthoastrocytoma. Asian Journal of Surgery, 26, 120-125.
http://dx.doi.org/10.1016/S1015-9584(09)60233-5 - 11. Kepes, J.J., Rubinstein, L.J. and Eng, L.F. (1979) Pleomorphic Xanthoastrocytoma: A Distinctive Meningocerebral Glioma of Young Subjects with Relatively Favorable Prognosis: A Study of 12 Cases. Cancer, 44, 1839-1852.
http://dx.doi.org/10.1002/1097-0142(197911)44:5<1839::AID-CNCR2820440543>3.0.CO;2-0 - 12. Gelpi, E., Popovic, M., Preusser, M., Budka, H. and Hainfellner, J. (2005) Pleomorphic Xanthoa-strocytoma with Anaplastic Features Presenting without GFAP Immunoreactivity: Implications for Differential Diagnosis. Neuropathology, 25, 241-246.
http://dx.doi.org/10.1111/j.1440-1789.2005.00612.x - 13. Giannini, C., Scheithauer, B.W., Lopes, M.B., Hirose, T., Kros, J.M. and Vanden Berg, S.R. (2002) Immunophenotype of Pleomorphic Xanthoastrocytoma. The American Journal of Surgical Pathology, 26, 479-485.
http://dx.doi.org/10.1097/00000478-200204000-00010 - 14. Hirose, T., Giannini, C. and Scheithauer, B.W. (2001) Ultrastructural Features of Pleomorphic Xanthoastrocytoma: A Comparative Study with Glioblastoma Multiforme. Ultrastructural Pathology, 25, 469-478.
http://dx.doi.org/10.1080/019131201753343502 - 15. Weldon-Linne, C.M., Victor, T.A., Groothuis, D.R. and Vick, N.A. (1983) Pleomorphic Xanthoa-strocytoma. Ultrastructural and Immunohistochemical Study of a Case with a Rapidly Fatal Outcome Following Surgery. Cancer, 52, 2055-2063.
http://dx.doi.org/10.1002/1097-0142(19831201)52:11<2055::AID-CNCR2820521115>3.0.CO;2-W - 16. Tsutsumi, S., Abe, Y., Yasumoto, Y. and Ito, M. (2010) Anaplastic Pleomorphic Xanthoastrocytoma with a Component of Anaplastic Astrocytoma Presenting as Skull Base Tumor Followed by Downward Extracranial Extension. Case Report. Neurologia Medico-Chirurgica, 50, 1108-1112.
http://dx.doi.org/10.2176/nmc.50.1108 - 17. Vu, T.M., Liubinas, S.V., Gonzales, M. and Drummond, K.J. (2012) Malignant Potential of Pleomorphic Xanthoastrocytoma. Journal of Clinical Neuroscience, 19, 12-20.
http://dx.doi.org/10.1016/j.jocn.2011.07.015 - 18. Giannini, C., Hebrink, D., Scheithauer, B.W., Dei Tos, A.P. and James, C.D. (2001) Analysis of p53 Mutation and Expression in Pleomorphic Xanthoastrocytoma. Neurogenetics, 3, 159-162.
http://dx.doi.org/10.1007/s100480100116 - 19. Perry, A., Giannini, C., Scheithauer, B.W., Rojiani, A.M., Yachnis, A.T., Seo, I.S., Johnson, P.C., Kho, J. and Shapiro, S. (1997) Composite Pleomorphic Xanthoastrocytoma and Ganglioglioma: Report of Four Cases and Review of the Literature. The American Journal of Surgical Pathology, 21, 763-771.
http://dx.doi.org/10.1097/00000478-199707000-00004 - 20. Marko, N.F. and Weil, R.J. (2013) The Molecular Biology of WHO Grade II Gliomas. Neurosurgical Focus, 34, E1.
http://dx.doi.org/10.3171/2012.12.FOCUS12283 - 21. Marucci, G. and Morandi, L. (2011) Assessment of MGMT Promoter Methylation Status in Pleomorphic Xanthoastrocytoma. Journal of Neuro-Oncology, 105, 397-400.
http://dx.doi.org/10.1007/s11060-011-0605-6 - 22. Gallo, P., Cecchi, P.C., Locatelli, F., Rizzo, P., Ghimenton, C., Gerosa, M. and Pinna, G. (2013) Pleomo-rphic Xanthoastrocytoma: Long-Term Results of Surgical Treatment and Analysis of Prognostic Factors. British Journal of Neurosurgery, 27, 759-764.
http://dx.doi.org/10.3109/02688697.2013.776666
Abbreviations and Acronyms
PXA: Pleomorphic xanthoastrocytoma
FISH: Fluorescence in situ hybridization
APXA: Anaplastic pleomorphic xanthoastrocytoma
GBM: Glioblastoma
EGB: Eosinophilic granular bodies
HPF: High power fields
CT: Computerized tomography
MRI: Magnetic resonance imaging
FISH: Fluorescence in situ hybridization
PCR: Polymerase chain reaction
PCR-SSCP: Single strand conformational polymorphism
VEP: Vascular endothelial proliferation
GFAP: Glial fibrillary acidic protein
NOTES

*Corresponding author.