Open Journal of Anesthesiology
Vol.4 No.3(2014), Article ID:43409,9 pages DOI:10.4236/ojanes.2014.43012
A Mechanism for Inhaled Anesthetic-Induced Solid Organ Injury: Inflammation
Gary E. Hill1*, Irina Gasanova1, Geoffrey M. Thiele2
1Department of Anesthesiology and Pain Management, University of Texas Southwestern Medical Center, Dallas, USA
2Veterans Administration Alcohol Research Center, Omaha Veterans Administration Medical Center, Omaha, USA
Email: *gary.hill@utsouthwestern.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 5 December 2013; revised 11 February 2014; accepted 26 February 2014
ABSTRACT
Background: Inhaled anesthetics, including halothane, isoand sevoflurane induce proinflammatory cytokine release. Halothane is an inhaled anesthetic agent that is metabolized by the liver into a highly reactive product, trifluoroacetyl chloride, which can react endogenously to form a trifluoroacetyl-adduct (TFA-adduct). The MAA-adduct is formed by acetaldehyde and malondialdehyde reacting with endogenousproteins and is found in both patients and animals post-consumption of alcohol. These TFA and MAA-adducts have been shown to cause the release of proinflammatorycytokines by endogenous inflammatory cells. If both adducts share a similar mechanism of cell activation, receiving general anesthesia following alcohol ingestion could exacerbate the inflammatory response caused by the inhaled general anesthetic halothane and lead to solid organ (including liver and brain) injury. Methods: Control diet and alcohol-fed rats were randomized to receive halothane pretreatments by intraperitoneal injection mixed in sesame oil. Following the intraperitoneal injections, the intact heart was removed, HECs were isolated and stimulated with unmodified bovine serum albumin (Alb), MAA-modified Alb (MAA-Alb), Hexyl-MAA, or lipopolysaccharide (LPS), and supernatant concentrations of TNF-α were determined. Results: Halothane pre-treated rat HECs demonstrated significantly greater TNF-α concentration following MAA-adduct and LPS stimulation than the non-halothane pre-treated in both pair and alcohol-fed rats, but was significantly greater in the alcohol-fed groups. Conclusion: These results demonstrate that halothane and MAA-adduct pre-treatment will increase the inflammatory response (TNF-α release) in rat HECs following LPS and MAA stimulation in vitro. Also, these results suggest that halothane exposure may increase the risk of alcohol-induced solid organ injury secondary to TNF-induced inflammation. Other investigators have reported similar proinflammatory cytokine release with other (isoflurane and sevoflurane) inhaled anesthetic exposure, suggesting that inhaled anesthetics should be used with caution in alcohol consuming humans.
Keywords:Anesthetic-Induced; Solid Organ; Inflammation; Halothane; MAA

1. Introduction
Inhaled anesthetic agents like halothane are commonly used worldwide, including developing countries [1] . Similarly on the rise is per capita alcohol consumption in developing countries [2] . Patients consuming alcohol who are anesthetized with halothane could potentially have inadequate metabolism or adduct formation leading to problems such as cardiovascular disease or liver injury. Hepatic metabolism of halothane and ingested ethanol (ethyl alcohol, alcohol) yields the highly reactive metabolites: trifluoroacetyl chloride (TFA) from halothane and acetaldehyde (AA) and malondialdehyde (MDA) from the oxidation of ethanol [3] [4] . MDA and AA react together with endogenous proteins (most likely the ε-amino group of lysine residues) to form distinctive new adducted proteins [3] [4] . The adduct formed by the combination of MDA and AA has been termed the MAA-adduct by Tuma et al. [4] and has been detected in humans and rats chronically consuming ethanol [5] [6] . In fact, Slatter et al. [7] have recently confirmed that MDA, AA, and lysine react to form a dihydropyridine derivative structurally identical to the MAA-adduct. Similarly, trifluoroacetyl chloride (TFA) will react with amine groups to form a distinctive protein termed the TFA-adduct [8] .
Recent reports by our laboratory have demonstrated that the MAA-adduct will induce the release of the proinflammatory cytokine tumor necrosis factor-alpha (TNF-α) in a purified rat heart endothelial cell culture (HEC) [9] . Trudell et al. [3] reported data suggesting that the TFAadduct may cause cell injury by inducing a similar inflammatory response. Importantly, it has been demonstrated that the TFA-adduct is present in heart tissue obtained from halothane pre-treated rats [10] [11] . If both adducts share a similar mechanism of cell activation, receiving halothane anesthesia following ethanol consumption both acutely and chronically could exacerbate the inflammatory response to halothane. Also of interest is that both halothane and ethanol are metabolized throughcytochrome P450 2E1 (CYP2E1) [12] , possibly providing a shared mechanism. In support of this is data suggesting that acetaldehyde affects ventricular myocyte contraction through mechanisms related to CYP oxidase and lipid peroxidation [13] . This could help explain how ethanol consumption and halothane anesthesia could enhance the sensitization of an individual to halothane and MAA-adducts, thereby increasing their risk of solid organ injury.
Therefore, we hypothesize that halothane pre-treatment may potentiate the inflammatory response induced by the MAA-adduct as determined by TNF-α release. Thus, this rat-model study evaluates the effects of halothane pretreatment in combination with an alcohol diet on in vitro HEC TNF-α release following stimulation with the MAAadduct.
2. Methods
2.1. Chemicals and Proteins
Bovine serum albumin (Alb) was purchased from CalBiochem (La Jolla, CA). Acetaldehyde (AA) was obtained from Aldrich Chemical Co. (Milwaukee, WI). Malondialdehyde (MDA) was obtained as the sodium salt (MDA ~Na) by treatment of tetramethoxypropane (Aldrich Chemical Co.) with NaOH, according to the method of Kikugawa and Ido [14] . Lipopolysaccharide and Eschericahia coli 0111:B4 (LPS) was purchased fromSigma Chemical Co. (St. Louis, MO). Halothane was purchased from Halocarbon Laboratories (River Edge, NJ).
2.2. Production of the Malondialdehyde-Acetaldehyde Adduct
MAA-Alb was prepared as described by Tuma et al. [15] Briefly, Alb was modified with 1 mM malondialdehyde (MDA) and 1 mM acetaldehyde (AA) by incubating at 37 degrees for 72 hours. Following incubation, free and reversibly-bound MDA or AA was separated by exhaustive dialysis against a phosphate buffer for 24 hours at 4 degrees. Fluorescence measurements were obtained on post dialysis samples using a Perkin Elmer (Norwalk, CT) LS-5B spectrophotofluorometer attached to a Perkin Elmer GP-100 graphics printer as previously described [15] . Protein concentrations were measured as described by Bradford [16] .
2.3. Animal Preparation
Male Wistar rats purchased from Charles River Laboratories (Willmington, MA) were maintained on a Purina rat chow diet until they reached a weight of 140 - 150 grams andthenwere divided into three groups. These three groups were housed individually and acclimated to the LieberDeCarli liquid control diet (Dyets, Inc., Bethlehem, PA) for 3 days [17] . The rats were paired by weight, one rat was given the ethanol-containing diet ad libitum, and the other rat was fed an isocaloric amount of the control liquid diet as determined by the pair-fed rat from the day before. Pair feeding was continued for 6 weeks. Finally, the ethanol-containing diet consisted of 18% of total calories as protein, 35% as fat, and 36% as ethanol. In the control liquid diet, ethanol was replaced isocalorically with carbohydrates. The final group was given free access to standard laboratory chow and water.
For adduct immunization rats were injected once per week for 3 consecutive weeks beginning on day fourteen with one of the following protocols: 1) An injection of Alb only (25 µg/ml) subcutaneously plus an i.p. injection of an equal volume of sesame oil were given to control animals.; 2) halothane as a 21.5% solution in sesame oil at a dose of 10 mmol/kg intraperitoneally (i.p.); 3) MAA-Alb (25 µg/ml) subcutaneously (s.c.); or 4) MAAAlb and halothane combined in the previously mentioned doses. After one month (day twenty nine) on their respective diets, and 24 hours following the final injection of Alb, halothane (i.p.), MAA-Alb (s.c.), or MAA-Alb (s.c.) plus halothane (i.p.) combined, the rats were sacrificed, and hearts removed for use in in vitro studies as described below. All animals were allowed free access to their food and for water up to 1 hour before sacrifice. All procedures were approved by the animal subcommittee of the Omaha VA Medical Center, and are in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals.
2.4. Transaminase Assay
Animals injected with the above ligands were bled prior to sacrifice and serum transaminase enzymes determined using an (ALT/GPT and AST/GOT) assay kit purchased from Sigma Diagnostics (St. Louis, MO).
2.5. Isolation and Culture of Heart Endothelial Cells (HECs)
Male Wistar rats were anesthetized intraperitoneal with phenobarbital (100 mg/kg) and the intact beating heart was immediately removed under sterile conditions. After mincing and dispase digestion, heart endothelial cells (HECs) were isolated and grown to confluency as previously described [9] [18] . In brief, HECs were separated by centrifugation at 400 × g for 10 min followed by three washes with M199-F12 (GIBCO, Grand Island, NY) containing 10% fetal bovine serum (GIBCO). Cells collected were >90% HECs, verified by staining with mouse anti-rat RECA-1 (Harlan Bioproducts for Science, Indianapolis, IN) and mouse anti-Factor VIII-von Willebrand’s Factor (Cedar Lane Laboratories Limited, Hornby, Ontario, Canada) [9] . Cells were seeded into 24 well tissue culture plates (Becton-Dickinson Labware, Franklin Lakes, NJ) containing fibronectin (20 µg/well) (Sigma Chemical Company, St. Louis, MO) and grown to confluency at 37˚C for 48 - 72 hours.
2.6. Percentage of Cell Necrosis Determinations
The percentage of cell necrosis (apoptosis or death) of HECs during exposure to MAA-Alb was determined by an enzyme (lactic acid dehydrogenase, LDH) release assay of the HEC supernatant as described by Korzeniewski and Callewaert [19] . Briefly, following stimulation of HECs with 1, 5, 10, and 25 µg/ml MAA-Alb or media only (control) for 3 and 24 hours, the HECs were centrifuged (200 × g, 10 min) and a 100 µl aliquot of the HEC supernatant was transferred to the corresponding wells of flat-bottomed micro-titer plates. Subsequently, 100 µl of a freshly prepared lactic acid dehydrogenase substrate mixture [5.4 × 10−2 lactate (Acros Organics, New Jersey, USA), 6.6 × 10−4 M 2piodophenyl-3p-nitrophenyl tetrazolium chloride (Acros), 2.8 × 10−4 M phenazine methosulfate (Acros), and 1.3 × 10−3 M nicotineanide nucleotide NAD in 0.2 M Tris buffer, pH 8.2 (Sigma)] was added to each well. The plates were incubated in the dark at room temperature for 10 min and the reaction stopped by the addition of 50 µl/well of a 1 M HCl solution. A microtiter plate reader (MR 7000, Dynatech Labs, Inc., Chantilly, VA) was used to monitor the resultant light absorbance at 490 nm while 630 nm was used as reference. LDH activity, expressed as change inabsorbance/min, was calculated with Biolin 2.21 software (Dynatech) on an IBM compatible computer. Percentage· necrosis of the HECs was determined by the following formula: 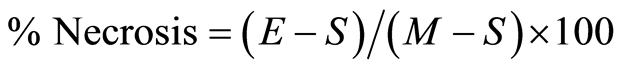 [19] , where E is the optical density (OD) of the experimentally induced release of LDH activity from the HECs incubated in the presence of the various concentrations of MAA-Alb, S is the spontaneous release of LDH activity (OD) from HECs incubated with media only, and M is the maximal release of LDH activity (0D) determined by total HEC necrosis induced by exposure to 10% Triton X-100 (Fisher Scientific, Fair Lawn, NJ) [19] .
[19] , where E is the optical density (OD) of the experimentally induced release of LDH activity from the HECs incubated in the presence of the various concentrations of MAA-Alb, S is the spontaneous release of LDH activity (OD) from HECs incubated with media only, and M is the maximal release of LDH activity (0D) determined by total HEC necrosis induced by exposure to 10% Triton X-100 (Fisher Scientific, Fair Lawn, NJ) [19] .
2.7. Endotoxin Assay for LPS Contamination
Prior to any stimulation all ligands, buffers, and media were tested for LPS content, which could influence the levels of background cytokine secretion. Samples were monitored for endotoxin using a Limulus Amebocyte Lysate assay from BioWhittaker (Walkersville, MD). Those samples contaminated with LPS at concentrations greater than 0.1 ng/ml were not utilized in these studies.
2.8. MAA-Alb Stimulation of HECs
HECs were washed on the day of the experiment with M199-F12 without serum and allowed to incubate for 1 hour to remove excess serum components. Following this incubation period, cells were stimulated with: 5 µg/ml Alb, MAA-Alb, LPS, and 10 µM Hexyl-MAA (a synthetic analog to the MAA-adduct) in serum free M199-F12 for 3 hours. Supernatant was collected and frozen at −70 degrees until assayed using a commercially available TNFα ELISA kit.
2.9. TNF-Alpha ELISA
Quantification of TNF-α levels of the HEC supernatants was performed with a Factor-Test-XTM rat TNF-α ELISA kit (Genzyme Diagnostics, Cambridge, MA), which employs a multiple antibody sandwich principle. The ELISA kit was developed, stopped and read at 450 nm on a MR 7000 plate reader using BIOLINXTM software. Final concentrations of TNF-α is expressed in pg/ml.
2.10. Statistical Analysis
All results are reported as means ± Standard Deviation (SD). Analysis of variance (ANOVA) was used to compare means between treatment groups. Dunnett’s two-tailed ttest was used to determine if any pre-treatment was significantly different when compared to the unpretreated (control) group of similar diet and in vitro stimulant conditions. P values of 0.05 or less were regarded as statistically significant.
3. Results
3.1. Transaminase Release
In an effort to determine liver damage from the administration of halothane and the MAA-adduct, serum from these animals were collected and assayed for the release of the serum transaminases, ALT and AST. Results indicated no difference between the animals injected with Alb, halothane, MAA-Alb, or halothane + MAA-Alb. There was a slight increase in ALT/AST levels in the ethanol-fed animals, yet these results were determined to be statistically insignificant.
3.2. Effects of Increasing Concentrations of MAA-Alb on in Vitro HEC Cell Death
In order to determine what concentrations of MAA-Alb would result in cell death of HECs, cells were isolated from chow-fed rats and stimulated with increasing doses of the antigen MAA-Alb. As shown in Table 1, HECs incubated with media alone, 1 and 5 µg/ml of MAA-Alb had little effect on % cell death after 3 and 24 hours of incubation. However, both 10 and 25 µg/ml of MAA-Alb caused a significant increase in cell death over the control and lower concentrations of the same antigen. There was a statistically significant difference in the amount of cell death observed when HECs were exposed to10 µg/ml for 24 hours as compared to the 3 hour stimulation period. However, these differences were not observed with 25 µg/ml of MAA-Alb. For these rea

Table 1. The percentage of cell death of heart endothelial cells (HECs) after stimulation with MAA-Alb as determined by LDH release.
Results are expressed as means +/− SD for 6 determinations in each group. Values different from the media control are indicated (*) at P < 0.05.
sons, 5 µg/ml of MAA-Alb was chosen as the optimum concentration for use in the remainder of the experiments described in this manuscript.
3.3. Effects of Pre Treatment with Halothane on TNF-α Release by HECs
In order to determine the effects of halothane pre-treatmenton the release of TNF-α by HECs, the cells were isolated from pair and ethanol-fed rats that had been injected as previously described in Materials and Methods with one of the following; Alb, halothane, MAA-Alb, or both halothane and MAA-Alb. The isolated HECs were stimulated in vitro with Alb, MAA-Alb, LPS, or Hexyl-MAA (a synthetic analog of MAA). As shown in Table 2, HECs from ethanol-fed rats injected with MAA-Alb + halothane and stimulated with 5 µg/ml of Alb significantly (P < 0.01)increased the amount of TNF-α release when compared to animals injected with Alb. Increases in TNF-α release were observed in Alb, halothane, and MAA-Alb + halothane injected ethanol-fed animals in comparison to the pair-fed controls (P < 0.05). The most significant increase was demonstrated in the MAA + halothane injected ethanol fed rats when compared to the halothane or MAA-Alb injected control liquid fed animals (P < 0.001). As a positive control, LPS was used as the stimulating antigen, (Table 3) and found to increase TNF-α secretion in the ethanol-fed animals as previously shown by others [20] . There was an increase over the Alb control in animals injected with halothane, MAA-Alb, and MAA + halothane (P < 0.05).
Pair (control liquid) and ethanol-fed rats injected with the above antigens increased TNF-α release 3 fold in response to stimulation with MAA-Alb in comparison to the Alb or LPS stimulated HECs. As shown in Table 4, increases in TNF-α release was demonstrated in the halothane, MAA-Alb, and halothane + MAA-Alb rat HECs after stimulation with MAA-Alb ascompared to the Alb injected rat HECs (P < 0.001). When comparing the four groups of rats, TNF-α release was increased in HECs from ethanol-fed rats over the pair-fed controls (P < 0.001). Ethanol-fed rats injected with halothane or MAA-Alb had similar effects on HEC secretion of TNF-α, while MAA-Alb + halothane together had a 3 fold increase over halothane or MAA-Alb alone (P <0.001). This synergistic response was also observed in the pair-fed controls for the MAA-Alb + halothane injected rat HECs. As demonstrated in Table 5, Hexyl-MAA (the synthetic analog of MAA) stimulated HECs similar to that found with MAAAlb stimulation. Increases in TNF-α secretion are witnessed in the ethanol-fed rats for all the experimental conditions (P < 0.001). There is a 2 - 3 fold increase in groups injected with halothane, MAA-Alb, or halothane + MAA-Alb over the Alb injected control (P < 0.001). The most significant increase is the 2 fold increase in the MAA-Alb + halothane injected ethanol-fed rats (P < 0.001). These data demonstrate an additive effect using the combination of both antigens.
4. Discussion
Tumor necrosis factor-alpha (TNF-α), the most proximalpro-inflammatory cytokine mediator released following LPS (endotoxin) stimulation, induces an inflammatory response through several mechanisms, including increased neutrophil-endothelial cell adherence, increased endogenous nitric oxide production [21] , and the stimulation of the production and release of other pro-inflammatory cytokines, including interleukin-8 (IL-8) [22] . Recently, the role of TNF-α in the process of apoptosis (cell death) has been demonstrated [23] . For example, while apoptosis occurs naturally in the liver at a low and controlled rate, an increased rate of apoptosis is observed following increased cellular TNF-α concentration. The direct correlation between TNF-α concentrations and the rate of apoptosis has been described in several types of liver diseases characterized by cell necrosis and death [24] . Apoptosis has also been demonstrated in heart endothelial cells in response to TNF-α and Interleukin-18 [25] [26] . Cell death experiments demonstrate that the MAA-adduct caused cell death only at concentrations of 10 µg/ml or greater (Table 1). Since TNF-α may cause cell death, [24] the significant increase in the
Table 2. Alb-stimulated TNF-α release (pg/ml) by HECs.
Results are mean values ± SD for 6 experiments. Values different from control (pair) fed rats are indicated (*) at P < 0.05. Values different from Halothane or MAA-Alb injected rats (same feeding) are indicated (˚) P < 0.01.
Table 3. LPS-stimulated TNF-α release (pg/ml) by HECs.
Results are expressed as mean ± SD for 6 experiments. Values different from control fed (pair fed) are indicated (*) at P < 0.05. Values different from halothane or MAA injected rats (same feedings) are indicated (˚) at P < 0.05. Values different from Alb-injected same feeding group are indicated (+) at P < 0.05.
Table 4. MAA-Alb stimulated TNF-α release (pg/ml) by HECs.
Results are expressed as means ± SD for 6 experiments. Values different from control (pair) fed group are indicated at (*) P < 0.001. Values different from halothane or MAA-injected rats (same feedings) are indicated (˚) P < 0.001. Values different from control fed Alb-injected rats are indicated (+) P < 0.01.
Table 5. Hexyl-MAA-Alb stimulated release of TNF (pg/ml) by HECs.
Results are expressed as means ± SD for 6 experiments. Values different from the control fed same group (pair fed) are indicated (*) P < 0.001. Values different from Halothane or MAA-Alb injected rats (same feedings) are indicated at (˚) P < 0.001. Values different from control fed Alb-injected rats are indicated (+) at P < 0.001.
percentage of HEC cell death following stimulation with I 0 and 25 µg/ml of MAA-Alb may be due to the direct toxicity of the MAA-adduct or indirectly due to MAA-adduct-induced TNF-α release. Further studies will be required to clarify this issue. Ohki et al. [27] reported, consistent with the results of our study, that ethanol fed rats demonstrate greater TNF-α release when exposed to LPS, with the increased TNF-α release following ethanol feeding caused increased neutrophil-endothelial adherence. Other investigators have similarly demonstrated increased neutrophil-endothelial adherence induced by ethanol, suggesting ethanol ingestion induces an inflammatory injury [28] . Since the MAA-adduct is a primary metabolic end product of ethanol metabolism, our demonstration that this adduct induces TNF-α release and that alcohol feeding potentiates endotoxin-induced TNF-α release is consistent with the understanding that alcohol adduct induced TNF-α release may play a significant role in alcohol induced solid organ injury [20] .
MDA has been detected in guinea pig heart tissue following exposure to halothane [29] . Also of interestis studies showing circulating antibodies to cardiac protein-acetaldehyde adducts have been found in alcoholic heart muscle disease [30] . If AA from alcohol metabolism and MDA from halothane are present in heart tissue, the possibility of MAA-adduct formation is likely. The majority of adducts formed when acetaldehyde reacts with proteins for short time periods are unstable AA-protein adducts. With time, unstable AA-adducts stabilize and can form an irreversible adduct. This irreversible, stable adduct has been demonstrated to be the MAA-adduct [15] . These MAA-adducts have also been found in atherosclerotic human aortic heart tissue [9] , providing a mechanism of heart muscle injury. Heavy alcohol consumption can accelerate human atherosclerotic heart disease [31] , making MAAadducts a possible candidate for this process.
Increased TNF-α levels have similarly been implicated in other disease states, like end-stage heart disease and intractable end stage cardiac failure [32] . Since TFA-adduct production is induced with a single dose of halothane and persists in measurable concentrations in rat heart for greater than 90 hours (but less than 10 days), [8] [33] the possibility for cross-reaction with MAA-adductsis plausible. These experiments demonstrated that halothane and the MAA adduct-induced HEC TNF-α release in a synergistic manner. In support of this data, Trudell et al. [3] demonstrated that antibodies raised against AA-adducts and TFAadducts cross-react, suggesting that the immunologic properties of both adducts may be similar. This data suggests that similar immunologic mechanisms may be shared by both halothane hepatitis and ethanol-induced hepatitis. The data reported in this study demonstrates that halothane pre-treatment doespotentiate the MAAadduct induced TNF-α release in vitro of HECs. This gives support to the Trudell et al. [3] demonstration that the AAadducts and TFA-adducts induce organ injury by the release of chemoattractants during the metabolism of ethanol and halothane resulting in the recruitment of inflammatory cells as the initial step in the initiation of solid organ injury. TNF-α production and release is a primary biologic mechanism in inflammatory cell recruitmentsince TNF-α induces the production of IL-8 [34] . Interleukin-8 is the primary cytokine responsible for the promotion of inflammatory cell (neutrophils and monocytes) chemotaxis (migration) and recruitment toward an inflammatory site [34] . Further studies need to be done in order to prove that the TFA-adduct and MAA-adduct crossreact.
These results are consistent with several studies demonstrating that other inhaled anesthetic agents induce proinflammatory cytokine release. The more commonly currently administered inhaled anesthetic isoflurane has been demonstrated to increase TNF-α release in rodent brain [35] , whileother studies [36] also using rat brain preparations demonstrated increased IL-1 release following isoflurane exposure. Similar proinflammatory cytokine release (IL-6) and subsequent activation of nuclear factor kappa B resulting in (cytotoxic injury) neuroinflammation in rat brainneuroglioma cells have been reported following sevoflurane exposure [37] . Other investigators [38] have linked inhaled anesthetic-induced proinflammatory cytokine release and the resulting cytotoxicity (apoptosis) in human brain neuroglioma cells to post operative cognitive dysfunction (delirium) and dementia. Our results demonstrate a similar mechanism of solid organ injury shared by other inhaled anesthetics: inflammation secondary to proinflammatory cytokine (TNF-α) generation and release resulting in cell death (apoptosis) and subsequent solid organ injury.
5. Conclusion
In conclusion, the current study demonstrates that halothane or MAA-Alb pre-treatment potentiates the HEC TNFα release following MAA-adduct stimulation of ethanolfed rats when compared to control pair-fed rats. This suggests that the TFA-adduct resulting from the metabolism of halothane increases the inflammatory response, as measured by TNF-α release, following LPS and alcohol (MAA-Alb) adduct stimulation, and that this TNF-α release may contribute to post-halothane exposure solid organ injury. The data also suggest that solid organ injury following halothane administration may be enhanced by prior ethanol consumption. This may also be an important mechanism for the increased risk of cardiovascular disease following excessive alcohol consumption. Finally, these results demonstrate that the combination of ethanol consumption and inhaled anesthetic exposure may enhance the inflammatory response (proinflammatory cytokine generation and release) to surgical stress [39] and therefore suggest that inhaled anesthetic agents should be used with caution in patients with a history of acute or chronic ethanol abuse.
References
- Oropeza-Hernandez, L.F., Quintanilla-Vega, B., Reyes-Mejia, R.A., Serrano, C.J., Garcia-Latorre, E.A., Dekant, W., Manno, M. and Albores, A. (2003) Trifluoroacetylated Adducts in Spermatozoa, Testes, Liver and Plasma and CYP2EI Induction in Rats after Subchronic Inhalatory Exposure to Halothane. Toxicology Letters, 144, 105-116. http://dx.doi.org/10.1016/S0378-4274(02)00335-1
- Boutayeb, A. and Boutayeb, S. (2005) The Burden of Non Communicable Diseases in Developing Countries. International Journal of Equity Health, 4, 2.
- Trudell, J.R., Ardies, C.M. and Anderson, W.R. (1991) The Effect of Alcohol Andanesthetic Metabolites on Cell Membranes. A Possible Direct Immune Mechanism. Annals of the New York Academy of Sciences, 625, 806-817. http://dx.doi.org/10.1111/j.1749-6632.1991.tb33923.x
- Tuma, D.J., Hoffman, T. and Sorrell, M.F. (1991) The Chemistry of AcetaldehydeProtein Adducts. Alcohol AlcoholSuppl, 1, 271-276.
- Xu, D., Thiele, G.M., Beckenhauer, J.L., Klassen, L.W., Sorrell, M.F. and Tuma, D.J. (1998) Detection of Circulating Antibodies to MalondialdehydeAcetaldehyde Adducts in Ethanol-Fed Rats. Gastroenterology, 115, 686-692. http://dx.doi.org/10.1016/S0016-5085(98)70148-9
- Rolla, R., Vay, D., Mottaran, E., Parodi, M., Traverso, N., Arico, S., Sartori, M., Bellomo, G., Klassen, L.W., Thiele, G.M., Tuma, D.J. and Albano, E. (2000) Detection of Circulating Antibodies against Malondialdehyde-Acetaldehyde Adducts in Patients with Alcohol-Induced Liver Disease. Hepatology, 31, 878-884. http://dx.doi.org/10.1053/he.2000.5373
- Slatter, D.A., Murray, M. and Bailey, A.J. (1998) Formation of a Dihydropyridine Derivative as a Potential Cross-Link Derived from Malondialdehyde in Physiological Systems. FEBS Letters, 421, 180-184. http://dx.doi.org/10.1016/S0014-5793(97)01554-8
- Christen, U., Burgin, M. and Gut, J. (1991) Halothane Metabolism: Immunochemical Evidence for Molecular Mimicry of Trifluoroacetylated Liver Protein Adducts by Constitutive Polypeptides. Molecular Pharmacology, 40, 390-400.
- Hill, G.E., Miller, J.A., Baxter, B.T., Klassen, L.W., Duryee, M.J., Tuma, D.J. and Thiele, G.M. (1998) Association of Malondialdehyde-Acetaldehyde (MAA) Adducted Proteins with Atherosclerotic-Induced Vascular Inflammatory Injury. Atherosclerosis, 141, 107-116. http://dx.doi.org/10.1016/S0021-9150(98)00153-1
- Pumford, N.R., Martin, B.M., Thomassen, D., Burris, J.A., Kenna, J.G., Martin, J.L., Pohlm L.R. (1993) Serum Antibodies from Halothane Hepatitis Patients React with the Rat Endoplasmic Reticulum Protein ERp72. Chemical Research in Toxicology, 6, 609-615. http://dx.doi.org/10.1021/tx00035a004
- Huwyler, J. and Gut, J. (1992) Exposure to the Chlorofluorocarbon Substitute 2,2-Dichloro-1,1,1-trifluoroethane and the Anesthetic Agent Halothane Is Associated with Transient Protein Adduct Formation in the Heart. Biochemical and Biophysical Research Communications, 184, 1344-1349. http://dx.doi.org/10.1016/S0006-291X(05)80030-0
- Spracklin, D.K., Hankins, D.C., Fisher, J.M., Thummel, K.E. and Kharasch, E.D. (1997) Cytochrome P450 2EI Is the Principal Catalyst of Human Oxidative Halothane Metabolism in Vitro. Journal of Pharmacology and Experimental Therapeutics, 281, 400-411.
- Meskar, A., Plee-Gautier, E., Amet, Y., Berthou, F. and Lucas, D. (2001) AlcoholXenobiotic Interactions. Role of Cytochrome P450 2EI. Pathological Biology (Paris), 49, 696-702. http://dx.doi.org/10.1016/S0369-8114(01)00235-8
- Kikugawa, K., Kosugi, H. and Asakura, T. (1984) Effect of Malondialdehyde, Aproduct of Lipid Peroxidation, on the Function and Stability of Hemoglobin. Archives of Biochemistry Biophysics, 229, 7-14.
- Tuma, D.J., Thiele, G.M., XuD, D., Klassen, L.W. and Sorrell, M.F. (1996) Acetaldehyde and Malondialdehyde React Together to Generate Distinctprotein Adducts in the Liver during Long-Term Ethanol Administration. Hepatology, 23, 872-880. http://dx.doi.org/10.1002/hep.510230431
- Bradford, M.M. (1976) A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemistry, 72, 248-254.
- Lieber, C.S. and DeCarli, L.M. (1986) The Feeding of Ethanol in Liquid Diets. Alcoholism: Clinical and Experimental Research, 10, 550-553. http://dx.doi.org/10.1111/j.1530-0277.1986.tb05140.x
- Ferry, B., Halttunen, J., Leszczynski, D., Schellekens, H., Meide, P.H. and Hayry, P. (1987) Impact of Class II Major Histocompatibility Complex Antigen Expression on the Immunogenic Potential of Isolated Rat Vascular Endothelial Cells. Transplantation, 44, 499-503. http://dx.doi.org/10.1097/00007890-198710000-00008
- Korzeniewski, C. and Callewaert, D.M. (1983) An Enzyme-Release Assay for Natural Cytotoxicity. Journal of Immunological Methods, 64, 313-320.
- Niemela, O., Parkkila, S., Yla-Herttuala, S., Villanueva, J., Ruebner, B. and Halsted, C.H. (1995) Sequential Acetaldehyde Production, Lipid Peroxidation, and Fibrogenesis in Micropig Model of Alcohol-Induced Liver Disease. Hepatology, 22, 1208-1214. http://dx.doi.org/10.1016/0270-9139(95)90630-4
- Jorens, P.G., Van Overveld, F.J., Bult, H., Vermeire, P.A. and Herman, A.G. (1991) LArginine-Dependent Production of Nitrogen Oxides by Rat Pulmonary Macrophages. European Journal of Pharmacology, 200, 205-209. http://dx.doi.org/10.1016/0014-2999(91)90573-9
- Pober, J.S. (1988) TNF as an Activator of Vascular Endothelium. Annales de l’Institut Pasteur Immunology, 139, 317- 323. http://dx.doi.org/10.1016/0769-2625(88)90149-3
- Ferrari, R., Bachetti, T., Agnoletti, L., Comini, L. and CurelloS, S. (1998) Endothelial Function and Dysfunction in Heart Failure. European Heart Journal, 19, G41-47.
- Bour, E.S., Ward, L.K., Cornman, G.A. and Lsom, H.C. (1996) Tumor Necrosis Factor-Alpha-Induced Apoptosis in Hepatocytes in Long-Term Culture. American Journal of Pathology, 148, 485-495.
- Rossig, L., Hoffmann, J., Hugel, B., Mallat, Z., Haase, A., Freyssinet, J.M., Tedgui, A., Aicher, A., Zeiher, A.M. and Dimmeler, S. (2001) Vitamin C Inhibits Endothelial Cell Apoptosis in Congestive Heart Failure. Circulation, 104, 2182-2187. http://dx.doi.org/10.1161/hc4301.098284
- Chandrasekar, B., Vemula, K., Surabhi, R.M., Li-Weber, M., Owen-Schaub, L.B., Jensen, L.E. and Mummidi, S. (2004) Activation of Intrinsic and Extrinsic Proapoptotic Signaling Pathways in Interleukin-18-Mediatedhuman Cardiac Endothelial Cell Death. Journal of Biological Chemistry, 279, 20221-20233. http://dx.doi.org/10.1074/jbc.M313980200
- Ohki, E., Kato, S., Horie, Y., Mizukami, T., Tarnai, H., Yokoyama, H., Ito, E., Fukuda, M., Suzuki, H., Kurose, I. and Ishii, H. (1996) Chronic Ethanol Consumption Enhances Endotoxin Induced Hepatic Sinusoidal Leukocyte Adhesion. Alcoholism: Clinical and Experimental Research, 20, 350A-355A. http://dx.doi.org/10.1111/j.1530-0277.1996.tb01806.x
- Takaishi, M., Kurose, I., Higuchi, H., Watanabe, N., Nakamura, T., Zeki, A., Nishida, J., Kato, S., Miura, S., Mizuno, Y., Kvietys, P.R., Granger, D.N. and Ishii, H. (1996) Ethanol-Induced Leukocyte Adherence and Albumin Leakage in Rat Mesenteric Venules: Role of CD18/Intercellular Adhesion Molecule-1. Alcoholism: Clinical and Experimental Research, 20, 347A-349A. http://dx.doi.org/10.1111/j.1530-0277.1996.tb01805.x
- Durak, I., Kurtipek, O., Ozturk, H.S., Birey, M., Guven, T., Kavutcu, M., Kacmaz, M., Dikmen, B., Yel, M. and Canbolat, O. (1997) Impaired Antioxidant Defence in Guinea Pig Heart Tissues Treated with Halothane. Canadian Journal of Anaesthetics, 44, 1014-1020. http://dx.doi.org/10.1007/BF03011975
- Harcombe, A.A., Ramsay, L., Kenna, J.G., Koskinas, J., Why, H.J., Richardson, P.J., Weissberg, P.L. and Alexander, G.J. (1995) Circulating Antibodies to Cardiac Protein-Acetaldehyde Adducts in Alcoholic Heart Muscle Disease. Clinical Science (London), 88, 263-268.
- Hanna, E.Z., Chou, S.P. and Grant, B.F. (1997) The Relationship between Drinking and Heart Disease Morbidity in the United States: Results from the National Health Interview Survey. Alcoholism: Clinical and Experimental Research, 21, 111-118.
- Torre-Amione, G., Kapadia, S., Lee, J., Durand, J.B., Bies, R.D., Young, J.B. and Mann, D.L. (1996) Tumor Necrosis Factor-Alpha and Tumor Necrosisfactor Receptors in the Failing Human Heart. Circulation, 93, 704-711. http://dx.doi.org/10.1161/01.CIR.93.4.704
- Christen, U., Burgin, M. and Gut, J. (1991) Halothane Metabolism: Kupffer Cells Carry and Partially Process Trifluoroacetylated Protein Adducts. Biochemical and Biophysical Research Communications, 175, 256-262. http://dx.doi.org/10.1016/S0006-291X(05)81228-8
- Smart, S.J. and Casale, T.B. (1994) TNF-Alpha-Induced Transendothelial Neutrophil Migration Is IL-8 Dependent. American Journal of Physiology, 266, L238-L245.
- Wu, X., Lu, Y., Dong, Y., Zhang, G., Zhang, Y., Zhipeng, X., Culley, D., Crosby, G., Marcontonio, E., Tanzi, R. and Xie, Z. (2012) The Inhalation Anesthetic Isoflurane Increased Levels of Proinflammatory TNF-α, IL-6, and IL-18. Neurobiological Aging, 33, 1364-1378. http://dx.doi.org/10.1016/j.neurobiolaging.2010.11.002
- Lin, D. and Zuo, Z. (2011) Isoflurane Induces Hippocampal Cell Injury and Cognitive Impairments in Adult Rats. Neuropharmacology, 61, 1354-1359. http://dx.doi.org/10.1016/j.neuropharm.2011.08.011
- Zhang, L., Zhang, J., Yang, L., Dong, Y., Zhang, Y. and Xie, Z. (2013) Isoflurane and Sevoflurane Increase Interleukin-6 Levels through the Nuclear Factor-Kappa B Pathway in Neuroglioma Cells. British Journal of Anaesthetics, 110, i82-i91. http://dx.doi.org/10.1093/bja/aet115
- Xie, Z., Dong, Y., Maeda, U., Moir, R., Inouye, S., Culley, D., Crosby, G. and Tanzi, R. (2006) Isoflurane-Induced Apoptosis: A Potential Pathogenic Link between Delirium and Dementia. Journal of Gerontology, 61A, 1300-1306. http://dx.doi.org/10.1093/gerona/61.12.1300
- Lin, E., Calvano, S. and Lowry, S. (2000) Inflammatory Cytokines and Cell Response in Surgery. Surgery, 127, 117- 126. http://dx.doi.org/10.1067/msy.2000.101584
Abbreviation List
Trifluroacetyl-adduct (TFA-adduct);
Acetaldehyde and malondialdehyde-adduct (MAA-adduct);
Heart endothelial cells (HECs);
Albumin (Alb);
Lipopolysaccharide (LPS);
NOTES

*Corresponding author.