World Journal of Cardiovascular Diseases
Vol.3 No.2(2013), Article ID:30262,9 pages DOI:10.4236/wjcd.2013.32028
Granulocyte-colony stimulating factor suppresses early inflammatory response of striatum in a cardiopulmonary bypass-circulatory arrest model of ischemic brain injury in newborn piglets*
![]()
1Department of Surgery, University of California, San Diego, USA
2Department of Anesthesiology & Critical Care, Mayo Clinic, Rochester, USA
3Department of Biochemistry & Biophysics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, USA
4Department of Anesthesiology & Critical Care, Children’s Hospital of Philadelphia, Philadelphia, USA
Email: #pastuszk@mail.med.upenn.edu
Copyright © 2013 Peter Pastuszko et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 11 January 2013; revised 2 March 2013; accepted 9 April 2013
Keywords: Brain; Cardiopulmonary Bypass; Inflammatory Response; Animal Model
ABSTRACT
We investigated the effect of Granulocyte-Colony Stimulating Factor (G-CSF) on expression of proand anti-inflammatory proteins in the striatum of newborn piglet brain following cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA). Piglets were placed on CPB, cooled to 18˚C, subjected to 30 min of DHCA and 1 h of low-flow (20 ml/kg/min), rewarmed to 37˚C, separated from CPB circuit and monitored for 2 h. Striatum was then isolated for protein analysis. The levels of proteins are presented relative to the mean in the control group (mean ± SEM, n = 6). DHCA increased the levels of pro-inflammatory proteins: IL-1alpha (158% ( 23%, P = 0.05), IL-6 (152% ( 16%, P = 0.03), TNF-alpha (144% ( 2%, P = 0.003), MIP-3 alpha (148% ( 12.6%, P = 0.03), NAP-3 (216% ( 16%, P = 0.05), GRO (165% ( 19%, P = 0.03) and BLC (140.4 ( 15%, P = 0.05). Compared to DHCA, the G-CSF-treated group had significantly decreased levels of IL-6 (110.8% ( 11% vs. 152% ( 16%, P = 0.05), TNF-alpha (120.6% ( 5.4% vs. 144% ( 2%, P = 0.001), MIP-3 alpha (148% ( 12.6% vs. 104.8% ( 13%, P = 0.02) and NAP-2 (216% ( 16% vs. 122% ( 23%, P = 0.002). The levels of anti-inflammatory proteins did not change in DHCA group compared to control, except for VEGF which decreased to 37.5% ( 9%, P = 0.003. The levels of all protective proteins in the G-CSF group increased versus the DHCA group, but the increases did not attain a P value of 0.05. Conclusions: In an immature brain subjected to circulatory arrest, the early inflammatory response in the striatum is diminished by pretreatment with G-CSF.
1. INTRODUCTION
Numerous studies have shown that cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA) are responsible for long-term sequelae such as neurological, developmental, motor and behavioral deficits [1-3]. The high occurrence of motor abnormalities is a major concern because these can affect between 20% and 60% of patients [2,3]. The severity of these abnormalities can be influenced by many factors, including the type of congenital heart defect, duration of CPB and the degree of inflammatory responses resulting from the release of endotoxin and increased levels of cytokines during the CPB [4-7]. Several studies have shown that the inflammatory cytokines such as IL-6, CRP, IL-10, and IL-8 can significantly increase in infants and children after CPB [8-11] and that these increases can contribute to postoperative morbidity and mortality. It had been suggested that elevation of these proteins may be predicttive of subsequent cognitive dysfunction [12,13]. In particular, increase in the level of IL-6 has been linked to disturbance of motor function [14].
One of the most important subcortical structures in the motor circuit and the main input site in the basal ganglia is the striatum (caudate putamen and nucleus accumbens). This region of the brain has been implicated in movement and memory disorders [see review 15]. The striatum is also involved in the various aspects of behavioral plasticity. Synaptic plasticity forms the basis of learning and memory and involves protein phosphorylation and protein synthesis, which lead to strengthening of preexisting synapses and establishment of new synaptic connections [15].
The goal of this research was to measure the early changes in inflammatory proteins in the striatum of newborn piglets following CPB-DHCA and to determine if pre-treatment with granulocyte-colony stimulating factor (G-CSF) can diminish these changes. G-CSF, a member of the cytokine family of growth factors, is a glycoprotein broadly present within the central nervous system. Exogenous administration of G-CSF has been shown to be neuroprotective in a variety of ischemic models [16-21]. G-CSF is reported to have potent anti-inflammatory [19], anti-excitatory [16] and anti-apoptotic properties. Our earlier study showed that, in a model of CPB-DHCA in newborn piglets, G-CSF treatment decreased pro-apoptotic signaling, particularly in the striatum [22]. This is in agreement with other models of hypoxia, showing that G-CSF can not only reduce hypoxiainduced apoptosis and inflammation but also enhance neurogenesis and angiogenesis [16,23-27].
Our hypothesis was that CPB-DHCA increases the levels of pro-inflammatory proteins in the striatum of newborn piglets and that pretreatment of piglets with G-CSF would suppress this increase and possibly also increase the levels of anti-inflammatory, protective proteins. To test this, we measured the effect of G-CSF pretreatment on the expression of selected proand antiinflammatory proteins in the striatum of newborn piglets following CBP-DHCA. CPB-DHCA is often use to surgical repair of congenital defects in the cardiopulmonary system. Variations in the procedure have been used in attempts to increase the “safe” time available for surgery and/or decrease the negative sequellae, including adding selective cerebral perfusion or periods of low perfusion rates. The negative sequelae are similar, however, consistent with the underlying mechanism(s) for the injury being due to the tissue hypoxia/ischemia and stress inherent in the procedure. We have used a model of CPBDHCA with a period of low flow bypass, in continuity with our earlier studies [28], but the underlying mechanisms and pattern of brain injury should be similar for other variants of the CPB-DHCA procedure.
2. METHODS
2.1. Animal Model
Eighteen male newborn piglets, 3 - 5 days old (2.0 - 3.0 kg) were anesthetized with 4% isoflurane (Novaplus, Hospira Inc., Lake Forest, IL). Pulse oximetry, ECG and temperature measurements begun immediately after induction of anesthesia. 1.5% lidocaine-HCl solution was used as a local anesthetic. After tracheotomy, pancuronium (1.5 mg/kg) was used for neuromuscular blockade to allow mechanical ventilation. Fentanyl-citrate (30 μg/kg) was injected intravenously, and the animals were mechanically ventilated with a mixture of oxygen (FiO2 21%) and 0.5% isoflurane. The femoral artery and vein were then cannulated and anesthesia was maintained with 0.5% isoflurane and boluses of pancuronium (1 mg/kg/hr). After a 2 h period, cardiopulmonary bypass was initiated. Following bypass, the animals recovered for 2 h under anesthesia and then were euthanized by intravenous administration of saturated KCl.
All animal procedures were in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals and have been approved by the local Animal Care and Use Committee.
2.2. CPB Technique
The circuit was primed with Plasmalyte-A and 25% albumin. Donor whole blood was then added to maintain a hematocrit of 25% - 30%. Heparin (1000 units), Fentanyl (50 µg), Pancuronium (1 mg), CaCl2 (500 mg), Methylprednisolone (60 mg), Cefazolin (100 mg), Furosemide (2 mg), and NaHCO3 (25 meq) were then added to the pump prime. A membrane oxygenator (Lilliput; COBE Cardiovascular, Inc., Arvada, Co), a roller pump system (COBE) and arterial filter (Capiox;Terumo Cardiovascular Systems, Corp, Ann Arbor, Mich) were used. Median sternotomy was performed and, after 500 units of heparin were administered IV, the ascending aorta and the right atrial appendage were cannulated. Full CPB flow rate was set at 150 ml/kg/min. The pH-stat blood gas management was maintained in all experiments.
After cooling to 18˚C, the piglets were introduced to 30 min of DHCA followed by 1h of low-flow CBP at 20 ml/kg/min.
The choice of a DHCA-low flow experimental model in our study reflects changes in the attitude and practice regarding the acceptable duration of DHCA. It is recognized by many clinical centers that DHCA longer than 30 min can be detrimental neurologically. Therefore, it is becoming increasingly common among CV surgeons who treat congenital heart defects such as hypoplastic left heart syndrome to perform the difficult arch repair under DHCA and then complete the remainder of the surgery with the low flow.
Following CPB, all animals were rewarmed for 30 min at full flow (150 ml/kg/min), separated from CPB and recovered for 120 min under anesthesia, and finally euthanized by intravenous injection of saturated KCl. The control animals did not undergo CBP but were anesthetized and mechanically ventilated the same way as other groups of animals. After euthanasia, the striatum was immediately dissected from the brain, frozen in liquid nitrogen and kept at −80˚C for later analysis.
The animals were randomly assigned to one of three groups: 1) CPB with circulatory arrest (n = 6, DHCA group); 2) DHCA with G-CSF at a concentration of 34 mg/kg (n = 6, G-CSF group); and 3) sham-operated (n = 6). G-CSF was given by intravenous injection two hours prior the beginning of bypass.
2.3. Protein Expression
Expression of inflammatory proteins was determined by RayBiotech, Inc. (Norcross, GA) using protein arrays (Sandwich-Based Glass Slide Cytokine Antibody Arrays, G series). To prepare striatal protein samples for the analysis, about 100 mg of the frozen striatum per animal was homogenized on ice in a low salt Cell Lysis Buffer (RayBiotech, Inc.), containing Protease Inhibitors Cocktail, Complete, (Roche Applied Science, Indianapolis, IN) with a broad inhibitory specificity for serine, cysteine and metalloproteases. After homogenization the samples were centrifuged at 10,000 xg for 10 min. Equal amounts of tissue lysates (supernatant), normalized by the total protein content were applied to the arrays. Protein concentration was determined by using BCA (bicinchoninic acid) Protein Assay Kit (Pierce, Rockford, IL).
2.4. Data Analysis
The measurements of individual protein levels are presented in percentage of their mean levels in the control group ± SEM for 6 experiments. The data were tested for significance by one way ANOVA and the p values for comparisons between groups were calculated by a twotailed t-test. In order to aid in distinguishing comparisons among groups, letters are used: a—for comparison between the control and DHCA groups; b—for the comparison between the control group and G-CSF groups; c—for the comparison between DHCA and G-CSF groups. The selected proteins are either pro-inflammatory or antiinflammatory, and members of the same class that change in the same direction as the other members might be expected to augment the response. These are mentioned if the P value for the differences is less than 0.1 (90% tile confidence level).
3. RESULTS
The immunoreactivities of 40 proteins involved in inflammatory responses were determined in striatum of the newborn piglet brain. Only the proteins which are significantly different between experimental groups or proteins which are known to play a role in inflammatory signaling are presented on the graphs.
3.1. Effect of DHCA and G-CSF on Immunoreactivity of Pro-Inflammatory Proteins in Striatum of Newborn Piglets
The effects of DHCA and G-CSF on 11 pro-inflammatory proteins are shown on Figures 1-3. Figure 1 demonstrates that DHCA significantly increased the levels of IL-1alpha, IL-6 and TNF-alpha (158% ± 23%, a:P = 0.05; 152% ± 16%, a:P = 0.03; 144% ± 2%, a:P = 0.003, respectively). The level of IL-1 beta was not significantly different in the DHCA group as compared to control.
The G-CSF group versus control had significant decrease in IL-1beta (44.7% ± 10.7%, b:P = 0.01). The increase in IL-1alpha (128.7% ± 8.6%, b:P = 0.09) and TNF-alpha (120.6% ± 5.4%, b:P = 0.09) were only significant at the 90% tile confidence level.
In the G-CSF-treated group, as compared to DHCA group, a significant decreases in the levels of IL-6 (110.8% ± 11% vs. 152% ± 16%, c:P = 0.05) and TNF-alpha (120.6% ± 5.4% vs. 144% ± 2%, c:P = 0.001) were observed. The levels of IL-1 alpha and IL-1beta also decreased but these differences did not attain a 90 percentile level of confidence.
Changes in other pro-inflammatory proteins are shown in Figure 2. MIP-3 alpha, NAP-2 and GRO are significantly increased in the DHCA group compared to control (148% ± 12.6%, a:P = 0.03; 216% ± 16%, a:P = 0.005; 165% ± 19%, a:P = 0.03, respectively). The G-CSF
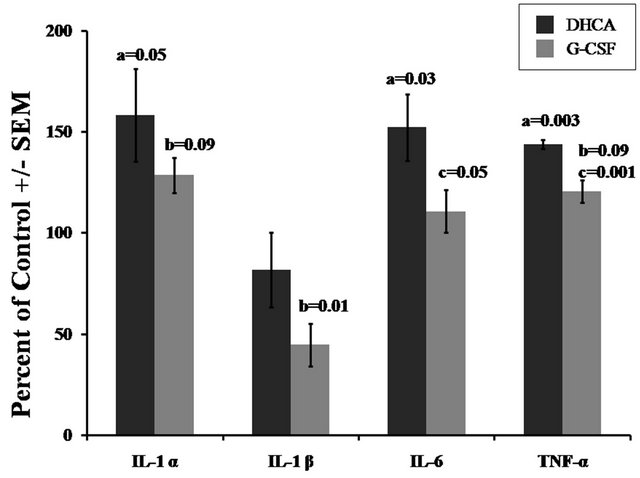
Figure 1. Effect of DHCA and G-CSF on the immunoreactivity of pro-inflammatory proteins (IL-1α, IL-1β, IL-6 and TNF-α) in the striatum of newborn piglets. The results are means from 6 experiments ± SEM. The data are expressed in percentage relative to mean values obtained from a control group of 6 piglets. P-values were determined by the two tailed Student t-test. a—for the comparison between the control group and DHCA; b—for the comparison between the control group and G-CSF; c—for the comparison between DHCA and G-CSF.
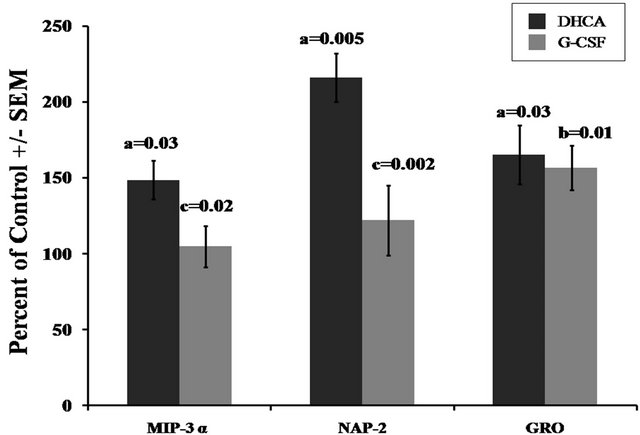
Figure 2. Effect of DHCA and G-CSF on the immunoreactivity of pro-inflammatory proteins (MIP-3α, NAP-2 and GRO) in the striatum of newborn piglets. The presentation is described in the legend of Figure 1.
group versus control had significant increase in GRO (156.7% ± 14.5%, b:P = 0.01). The levels of MIP-3a and NAP-2 in the G-CSF group were not significantly different from the control group.
Compared to DHCA, pretreatment with G-CSF resulted in significantly lower values for MIP-3 alpha (148% ± 12.6% vs. 104.8% ± 13%, c:P = 0.02) and NAP-2 (216% ± 16% vs. 122% ± 23%, c:P = 0.002). No significant differences between these groups of animals were observed in GRO.
Figure 3 shows the changes in BLC, IL-8, IL-12 and ENA-78. The DHCA group had a significant increase in BLC (140.4% ± 15%, a:P = 0.05) compared to control. The increases in IL-8 (122.7% ± 6.3%, a:P = 0.08) and ENA-78 (170% ± 34%, a:P = 0.07) were significant only at the 90%tile confidence level. Level of IL-12 was also increased but the differences did not attain a 90 percentile level of significance. In G-CSF pre-treatment samples, changes in above proteins were not significantly different from control.
Compared to DHCA, pretreatment with G-CSF resulted in significant decrease in the level of IL-12 from 138% ± 17% to 101.9% ± 12%, c:P = 0.06. There was a trend, but not statistically significant, for G-CSF treatment to diminish the DHCA induced increases in BLC, IL-8 and ENA-78.
3.2. Effect of DHCA and G-CSF on Immunoreactivitity of Neuroprotective Proteins in Striatum
Figures 4 and 5 show the responses of proteins selected for their reported protective role in brain injury, to DHCA and G-CSF pre-treatment. In the DHCA group, there was a significant decrease as compared to control in VEGF (37.5% ± 9%, a:P = 0.003) whereas the level of TNF-beta increased significantly only at a 90% tile confidence level (TNF-beta (118.5% ± 6%, a:P = 0.08). The increase in IL-5 was not statistical different from control (Figure 4).
Compared to controls, the G-CSF group had higher levels of IL-5 (166% ± 26%, b:P = 0.05) and TNF-beta (132.1% ± 4%, b:P = 0.01). The VEGF level was higher than in DHCA group but was still below control level (52% ± 16.7%, b:P = 0.04).
There was a trend for higher levels of protective proteins, presented on Figure 4, in the G-CSF group than in the DHCA group, with TNF-beta reaching statistical significant increase only at the 90% tile (132.6% ± 3.9% vs. 118.5% ± 6%, c:P = 0.09 (Figure 4).
The levels of BDNF, IL-10 and IL-15 did not change significantly in DHCA group as compared to control (Figure 5). In G-CSF group as compared to control the levels of all three proteins were higher but statistical significant increase was shown only for BDNF (121.7% ±

Figure 3. Effect of DHCA and G-CSF on the immunoreactivity of pro-inflammatory proteins (BLC, IL-8, IL-12 and ENA-78) in striatum of newborn piglets. The presentation is described in the legend of Figure 1.
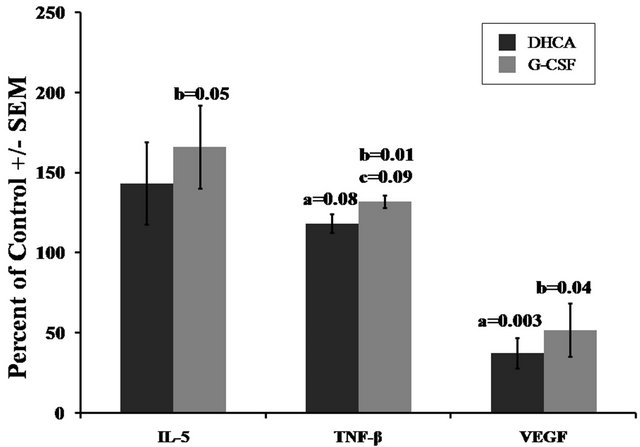
Figure 4. Effect of DHCA and G-CSF on the immunoreactivity of anti-inflammatory proteins (IL-5, TNF-β and VEGF) in striatum of newborn piglets. The presentation is described in the legend of Figure 1.
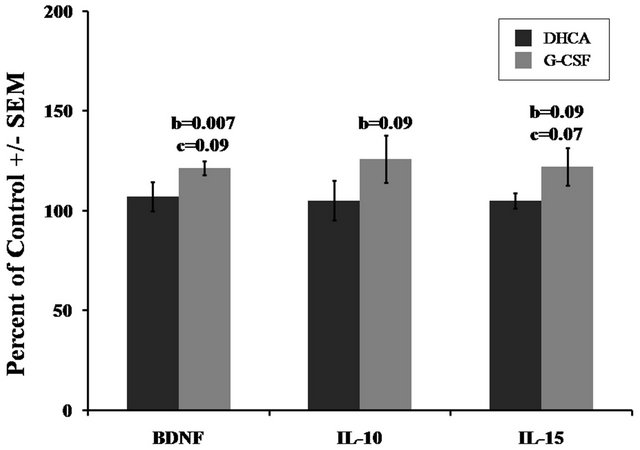
Figure 5. Effect of DHCA and G-CSF on the immunoreactivity of anti-inflammatory proteins (BDNF, IL-10 and IL-15) in striatum of newborn piglets. The presentation is described in the legend of Figure 1.
3.4% of control, b:P = 0.007). The increases in IL-10 (126.2% ± 11.8%, b:P = 0.09) and IL-15 (122.4% ± 9.5%, b:P = 0.09) were significant only at the 90% confidence level.
The G-CSF group, compared to DHCA, show significant increases, but only if calculated by one tailed t-test, in BDNF (107.3% ± 7.3% vs. 121.7% ± 3.4%, c:P = 0.09) and IL-15 (105% ± 4% vs.122.4% ± 9.5%, c:P = 0.07). The increase in IL-10 was not statistically significant (P value > 0.1).
4. DISCUSSION
The purpose of this study was to test the hypothesis that cardiopulmonary bypass (CBP) and deep hypothermic circulatory arrest (DHCA), in a newborn piglet, induces an pro-inflammatory response in the striatum and that response can be suppressed by pre-treatment with G-CSF. To achieve this goal, we measured the levels of proand anti-inflammatory proteins in the striatum. There are several reasons for focusing our study on G-CSF modulation of inflammatory response in the striatum: 1) Cardiac surgery is associated with variable degrees of neurologic injury, ranging from relatively minor neuro-cognitive deficits to severe cerebral infarction [29,30]; 2) DHCA had been shown to increase pro-inflammatory activity; 3) Motor abnormalities following CPB-DHCA affect a significant percent of patients; (4) The striatum plays a major role in controlling motor function; 5) Our earlier study [22] showed that the striatum can be more sensitive than other brain regions to apoptotic injury following CPB-DHCA; 6) Pro-apoptotic signaling in striatum has been reported to be diminished by pre-treatment with G-CSF [22].
The results show that DHCA significantly increased levels of 9 pro-inflammatory proteins, IL-1α, IL-6, TNF- α, MIP-3α, NAP-2, GRO, BLC, IL-8 and ENA-78. These results are in agreement with reports showing that CPB increases inflammatory responses in the brain [4,7,31- 35].
The increases in pro-inflammatory signaling are consistent with tissue inflammation contributing to the adverse effects on learning, memory, and other cognitive domains by altering brain function [36] and may be predictive of cognitive decline. The elevation of some proinflammatory proteins such as IL-6 [13] and TNF-α are especially indicative of systemic inflammation and are predictive of subsequent cognitive dysfunction [12]. Gessler et al. [14] reported that an increased plasma concentration of IL-6 level in children after CPB was associated with poorer neuromotor performance, and therefore possible damage in striatum. Allan et al. [37] reported that in infants undergoing cardiopulmonary bypass, higher postoperative IL-6 and IL-8 plasma concentrations were associated with longer ICU stay and higher 24-hour lactate and correlated with greater postoperative severity of illness. Other clinical studies on different ischemic conditions such stroke also reported early (<24 h) and sustained (up to 7 days) increases in plasma levels of IL-6 and these correlated with neurocognitive outcome [38- 42]. Consistent with this, decreased level of TNF-α, has been reported to correlate with improved efficacy of protective strategies after ischemia [43].
Our study shows that both IL-6 and TNF-α in the striatum were significantly elevated following CPBDHCA. Other proteins, such as IL-1α and other chemokines, were also significantly increased. The role of some of these proteins in brain injury is poorly understood but, for example, IL-1 receptor antagonists has been reported to decrease hypoxic-ischemic injury [44,45], and neonatal mice deficient in IL-1 converting enzyme are resistant to hypoxic-ischemic insults [46]. Inhibition of endogenous IL-1 activity with IL-1 receptor antagonist (IL-1RA) protects against ischemic injury [47]. Similarly, inhibittion of IL-1β converting enzyme [48,49] or deletion of IL-1β and IL-1α [50] results in markedly reduced ischemic damage and neuronal cell death.
In general, increased levels of many chemokines seem to play a critical role in ischemic brain injury and conversely, a broad spectrum of antagonists of chemokine receptors offer protection against brain injury [51].
CPB-DHCA also caused significantly decreased levels of VEGF. The VEGF has been reported to have two opposing roles in the brain. It can have pro-inflammatory properties and increase brain edema and tissue damage following ischemia [52]. VEGF increases the permeability of the blood brain barrier, and its over-expression in the mouse brain leads to intra-cerebral hemorrhage via expression of MMP-9 [53]. On the other hand, VEGF can also induce angiogenesis and increase vascular permeability [54,55]. These changes would be expected to be neuro-protective by inducing proliferation of astrocytes and preventing death of dopaminergic neurons [56]. VEGF has been reported to have anti-apoptotic properties, through up-regulation of Survivin, Akt and Bcl-2, which are key anti-apoptotic proteins [57-60]. A decrease of VEGF below the level seen in sham-operated animals can, therefore, have a negative effect and increase striatal injury caused by CPB-DHCA.
The changes in inflammatory proteins that we found were determined at one time point, 2 h following DHCA. Earlier transient changes and those that might occur after longer recovery times would have been missed. Different pro-inflammatory proteins are upor down-regulated at different time points following a variety of hypoxicischemic insults. For example, IL-6 has a short half-life in plasma [61] and has been reported to peak at 2 h after CPB [31]. Hedtjärn et al. [62] measured expression of 148 genes related to the immune-inflammatory response in immature brain following hypoxia-ischemia (HI). The authors reported that, at 2 h after HI, only three genes were upregulated, but at 8 h the number of significantly upregulated genes had increased to 49. At 24 hours after HI, 77 genes were upregulated, and at 72 hours after HI as many as 119 genes were upregulated.
Homi et al. [31] reported that, in a model of middle cerebral artery occlusion (MCAO) with CPB in rats, serum levels of TNF-α and IL-1β increased at the end of CPB, achieved peak values 2 h after CPB and then rapidly decreased to normal levels at 6 h following hypoxia. The IL-10 showed a similar pattern but reached its maximum concentration at the end of CPB and then decayed slowly to below a detectable threshold 24 h after CPB. The IL-6 level, however, was increased 2 h after CPB in the MCAO plus CPB group. Similarly, a clinical study on children reported that the plasma concentrations of IL-6 and IL-8 were strongly increased at 3 h after CPB [14].
A major question of this study was whether injection of G-CSF prior to bypass ameliorates inflammatory signaling in the striatum of newborn piglets. Our results show that G-CSF treatment abolished the DHCA-dependent increases in IL-6 and TNF-α, two proteins for which an increase is correlated with increased brain injury. This result is consistent with the work of Pollari et al. [63] who reported that, in vitro, exposure of cell cultures to G-CSF reduced their TNF-α production. In our study treatment with G-CSF also decreased the levels of MIP-3 and NAP-2, two cytokines believed to play a role in neuronal cell injury.
When compared to the control group, G-CSF treatment resulted in decreased level of IL-1β, a protein reported to be a marker of brain injury, and increased levels of IL-5, TNF-β, VEGF and BDNF, proteins with well described protective roles in model of hypoxic brain injury.
Our results strongly suggest that injection of G-CSF prior to bypass protects the striatum of newborn piglets from inflammation-dependent injury. At this early time of recovery it seems that major protective effect of G-CSF is by decreasing signaling proteins that exacerbate tissue inflammation rather than increasing those which suppress inflammation. When the sums of the increases in all of pro-inflammatory proteins in DHCA and G-CSF groups were compared it can been seen that DHCA increased the immunoreactivities to 146.8% ± 18% of control values whereas G-CSF diminishes this increase to 105% ± 13% of control. Changes in proteins which suppress inflammation were much smaller and the summation increased from 101% ± 10.3% in DHCA group to 116.2% ± 10.6% in G-CSF group compared to control.
A protective role of G-CSF following CPB that we found in this study is consistent with similar properties of this compound in other hypoxic-ischemic models. A protective effect of G-CSF has been reported in animal models of stroke, spinal cord injury and such neurodegenerative diseases as Alzheimer’s and Parkinson’s [64-67].
In conclusion, the results presented in this paper show that CPB results in potent stimulation of an inflammatory response measured in the striatum. Administration of G-CSF prior to CPB diminishes this stimulation and is consistent with decrease the neuromotor and cognitive deficits associated with this procedure and improved outcome.
REFERENCES
- Ballweg, J.A., Wernovsky, G. and Gaynor, J.W. (2007) Neurodevelopmental outcomes following congenital heart surgery. Pediatric Cardiology, 28,126-133. doi:10.1007/s00246-006-1450-9
- Bellinger, D.C., Wypij, D. and duPlessis, A.J., et al. (2003) Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: The Boston circulatory arrest trial. Journal of Thoracic Cardiovascular Surgery, 126, 1385-1396. doi:10.1016/S0022-5223(03)00711-6
- Limperopoulos, C., Majnemer, A., Shevell, M.I., et al. (2002) Predictors of developmental disabilities after openheart surgery in young children with congenital heart defects. Journal of Pediatrics, 141, 51-58. doi:10.1067/mpd.2002.125227
- Paparella, D., Yau, T.M. and Young, E. (2002) Cardiopulmonary bypass induced inflammation: Pathophysiology and treatment. An update. European Journal CardioThoracic Surgery, 21, 232-244. doi:10.1016/S1010-7940(01)01099-5
- Ben-Abraham, R., Weinbroum, A.A., Dekel, B. and Paret, G. (2003) Chemokines and the inflammatory response following cardiopulmonary bypass—A new target for therapeutic intervention? Paediatric Anaesthesiology, 13, 655-661. doi:10.1046/j.1460-9592.2003.01069.x
- Butler, J., Rocker, G.M. and Westaby, S. (1993) Inflammatory response to cardiopulmonary bypass. Annals of Thoracic Surgery, 55, 552-559. doi:10.1016/0003-4975(93)91048-R
- Levy, J.H. and Tanaka, K.A. (2003) Inflammatory response to cardiopulmonary bypass. The Annals of Thoracic Surgery, 75, S715-S720. doi:10.1016/S0003-4975(02)04701-X
- Hövels-Gürich, H.H., Schumacher, K., Vazquez-Jimenez, J.F., et al. (2002) Cytokine balance in infants undergoing cardiac operation. The Annals of Thoracic Surgery, 73, 601-608. doi:10.1016/S0003-4975(01)03391-4
- Arkader, R., Troster, E.J., Abellan, D.M., et al. (2004) Procalcitonin and C-reactive protein kinetics in postoperative pediatric cardiac surgical patients. Journal of Cardiothoracic Vascular Anesthesiology, 18, 160-165. doi:10.1053/j.jvca.2004.01.021
- Beghetti, M., Rimensberger, P.C., Kalangos, A., et al. (2003) Kinetics of procalcitonin, interleukin 6 and C-reactive protein after cardiopulmonary-bypass in children. Cardiology in the Young, 13, 161-167. doi:10.1017/S1047951103000301
- Butler, J., Pathi, V.L., Paton, R.D., et al. (1996) Acutephase responses to cardiopulmonary bypass in children weighing less than 10 kilograms. Annals of Thoracic Surgery, 62, 538-542. doi:10.1016/0003-4975(96)00325-6
- Harris, T.B., Ferrucci, L., Tracy, R.P., et al. (1999) Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. American Journal of Medicine, 106, 506-512. doi:10.1016/S0002-9343(99)00066-2
- Weaver, J.D., Huang, M.H., Albert, M., et al. (2002) Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology, 59, 371-378. doi:10.1212/WNL.59.3.371
- Gessler, P., Schmitt, B., Prètre, R. and Latal, B. (2009) Inflammatory response and neurodevelopmental outcome after open-heart surgery in children. Pediatrics Cardiology, 30, 301-305. doi:10.1007/s00246-008-9354-5
- Pastuszko, A. (2007) Dopamine, its role in regulation newborn brain metabolism and neuronal injury during hypoxia and posthypoxic reoxygenation. In: Mishra, Om P., Ed., Mechanisms of Hypoxic Brain Injury and Potential Strategies for Neuroprotection, Transworld Research Network, Trivandrum, 113-155.
- Schneider, A., Kruger, C., Steigleder, T., et al. (2005) The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. Journal of Clinical Investigation, 115, 2083- 2098. doi:10.1172/JCI23559
- Schabitz, W.R., Kollmar, R., Schwaninger, M., et al. (2003) Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke, 34, 745-751. doi:10.1161/01.STR.0000057814.70180.17
- Gibson, C.L., Bath, P.M. and Murphy, S.P. (2005) G-CSF reduces infarct volume and improves functional outcome after transient focal cerebral ischemia in mice. Journal of Cerebral Blood Flow and Metabolism, 25, 431-439. doi:10.1038/sj.jcbfm.9600033
- Hartung, T., Von Aulock, S., Schneider, C. and Faist, E. (2003) How to leverage an endogenous immune defense mechanism: The example of granulocyte colony-stimulating factor. Critical Care Medicine, 31, 65-75. doi:10.1097/00003246-200301001-00010
- Solaroglu, I., Tsubokawa, T., Cahill, J. and Zhang, J.H. (2006) Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience, 143, 965-974. doi:10.1016/j.neuroscience.2006.09.014
- Xiao, B.G., Lu, C.Z. and Link, H. (2007) Cell biology and clinical promise of G-CSF: Immunomodulation and neuroprotection. Journal of Cellular Molecular Medicine, 11, 1272-1290. doi:10.1111/j.1582-4934.2007.00101.x
- Pastuszko, P., Schears, G.J., Pirzadeh, A., et al. (2012) Effect of granulocyte colony stimulating factor (G-CSF) on expression of select proteins involved in apoptosis in a neonatal piglet brain following cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA). Journal of Thoracic Cardiovascular Surgery, 143, 1436- 1442. doi:10.1016/j.jtcvs.2012.01.018
- Lee, S.T., Chu, K., Jung, K.H., et al. (2005) Granulocyte colony-stimulating factor enhances angiogenesis after focal cerebral ischemia. Brain Research, 1058, 120-128. doi:10.1016/j.brainres.2005.07.076
- Shyu, W.C., Lin, S.Z., Yang, H.I., et al. (2004) Functional recovery of stroke rats induced by granulocyte colony-stimulating factor-stimulated stem cells. Circulation, 110, 1847-1854. doi:10.1161/01.CIR.0000142616.07367.66
- Kawada, H., Takizawa, S., Takanashi, T., et al. (2006) Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation, 113, 701-710. doi:10.1161/CIRCULATIONAHA.105.563668
- Boneberg, E.M., Hareng, L., Gantner, F., et al. (2000) Human monocytes express functional receptors for granulocyte colony stimulating factor that mediate suppression of monokines and interferon-gamma. Blood, 95, 270-276.
- Saito, M., Kiyokawa, N., Taguchi, T., et al. (2002) Granulocyte colony-stimulating factor directly affects human monocytes and modulates cytokine secretion. Experimental Hematology, 30, 1115-1123. doi:10.1016/S0301-472X(02)00889-5
- Pastuszko, P., Liu, H., Mendoza, A., et al. (2007) Regulatory pathways to neuronal injury or survival are dependent on the rate of low flow cardiopulmonary bypass following circulatory arrest in newborn piglets. European Journal of Cardio-Thoracic Surgery, 31, 899-905. doi:10.1016/j.ejcts.2007.01.049
- Newman, M.F., Kirchner, J.L., Phillips-Bute. B., et al. (2001) Longitudinal assessment of neurocognitive function after coronary artery bypass surgery. New England Journal of Medicine, 344, 395-402. doi:10.1056/NEJM200102083440601
- Hindman, B.J. and Todd, M.M. (1999) Improving neurologic outcome after cardiac surgery. Anesthesiology, 90, 1243-1247. doi:10.1097/00000542-199905000-00002
- Homi, H.M., Jones, W.L., de Lange, F., et al. (2010) Exacerbation of systemic inflammation and increased cerebral infarct volume with cardiopulmonary bypass after focal cerebral ischemia in the rat. The Journal of Thoracic and Cardiovascular Surgery, 410, 660-666. doi:10.1016/j.jtcvs.2009.10.063
- Rothenburger, M., Soeparwata, R., Deng, M.C., et al. (2001) Prediction of clinical outcome after cardiac surgery: The role of cytokines, endotoxin, and anti-endotoxin core antibodies. Shock, 16, 44-50. doi:10.1097/00024382-200116001-00009
- Cremer, J., Marti, M., Redl, H., et al. (1996) Systemic inflammatory response syndrome after cardiac operations. Annals of Thoracic Surgery, 61, 1714-1720. doi:10.1016/0003-4975(96)00055-0
- Deng, M.C., Dasch, B., Erren, M., et al. (1996) Impact of left ventricular dysfunction on cytokines, hemodynamics, and outcome in bypass grafting. Annals of Thoracic Surgery, 62, 184-190. doi:10.1016/0003-4975(96)00230-5
- Hennein, H.A., Ebba, H., Rodriguez, J.L., et al. (1994) Relationship of the proinflammatory cytokines to myocardial ischemia and dysfunction after uncomplicated coronary revascularization. Journal of Thoracic Cardiovascular Surgery, 108, 626-635.
- Dantzer, R. and Kelley, K.W. (2007) Twenty years of research on cytokine induced sickness behavior. Brain Behavior and Immunity, 21,153-160. doi:10.1016/j.bbi.2006.09.006
- Allan, C.K., Newburger, J.W., McGrath, E., et al. (2010) The relationship between inflammatory activation and clinical outcome after infant cardiopulmonary bypass. Anesthesia Analgesia, 111, 1244-1251. doi:10.1213/ANE.0b013e3181f333aa
- Denes, A., Thornton, P., Rothwell, N.J. and Allan, S.M. (2010) Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain, Behavior and Immunity, 24, 708-723. doi:10.1016/j.bbi.2009.09.010
- Basic, K.V., Simundic, A.M., Nikolac, N., et al. (2008) Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clinical Biochemistry, 41, 1330-1334. doi:10.1016/j.clinbiochem.2008.08.080
- Emsley, H.C., Smith, C.J., Gavin, C.M., et al. (2003) An early and sustained peripheral inflammatory response in acute ischaemic stroke: Relationships with infection and atherosclerosis. Journal of Neuroimmunology, 139, 93- 101. doi:10.1016/S0165-5728(03)00134-6
- Smith, C.J., Emsley, H.C., Gavin, C.M., et al. (2004) Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurology, 4, 2.
- Waje-Andreassen, U., Krakenes, J., Ulvestad, E., et al. (2005) IL-6: An early marker for outcome in acute ischemic stroke. Acta Neurologica Scandinavica, 111, 360-365. doi:10.1111/j.1600-0404.2005.00416.x
- Yang, G.Y., Gong. C., Qin. Z., et al. (1998) Inhibition of TNF alpha attenuates infarct volume and ICAM-1 expression in ischemic mouse brain. NeuroReport, 9, 2131- 2134. doi:10.1097/00001756-199806220-00041
- Hagberg, H., Gilland, E., Bona, E., et al. (1996) Enhanced expression of interleukin (IL)-1 and IL-6 mRNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatrics Research, 40, 603-609. doi:10.1203/00006450-199610000-00015
- Martin, D., Chinookoswong, N. and Miller, G. (1994) The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxiaischemia. Experimental Neurology, 130, 362-367. doi:10.1006/exnr.1994.1215
- Liu, X.H., Kwon, D., Yang, G.Y., et al. (1998) Neonatal mice deficient in interleukin-1 converting enzyme are resistant to cerebral hypoxic-ischemic injury. Pediatric Research, 43, 320A. doi:10.1203/00006450-199804001-01903
- Relton, J.K. and Rothwell, N.J. (1992) Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Research Bulletin, 29, 243-246. doi:10.1016/0361-9230(92)90033-T
- Friedlander, R.M., Gagliardini, V., Hara, H., et al. (1997) Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. Journal of Experimental Medicine, 185, 933-940. doi:10.1084/jem.185.5.933
- Hara, H., Friedlander, R.M., Gagliardini, V., et al. (1997) Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proceedings of the National Academy of Sciences of the United States of America, 94, 2007-2012. doi:10.1073/pnas.94.5.2007
- Boutin, H., LeFeuvre, R.A., Horai, R., et al. (2001) Role of IL-1alpha and IL-1beta in ischemic brain damage. Journal of Neuroscience, 21, 5528-5534.
- Minami, M. and Satoh, M. (2003) Chemokines and their receptors in the brain: Pathophysiological roles in ischemic brain injury. Life Science, 74, 321-327. doi:10.1016/j.lfs.2003.09.019
- Croll, S.D., Goodman, J.H. and Scharfman, H.E. (2004) Vascular endothelial growth factor (VEGF) in seizures: A double-edged sword. Advance Experimental Medicine and Biology, 548, 57-68. doi:10.1007/978-1-4757-6376-8_4
- Lee, C.Z., Xue, Z., Zhu, Y., et al. (2007) Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke, 38, 2563-2568. doi:10.1161/STROKEAHA.106.481515
- Gluzman-Poltorak, Z., Cohen, T., Herzog, Y., et al. (2000) Nuropilin-2 is a receptor for the Vascular Endothelial Growth Factor (VEGF) forms VEGF-145 and VEGF-165. Journal of Biological Chemistry, 275, 18040-18045. doi:10.1074/jbc.M909259199
- Saito, Y., Uppal, A., Byfield, G., et al. (2008) Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Investigative Ophthalmology & Visual Science, 49, 1591-1598. doi:10.1167/iovs.07-1356
- Temburni, M.K. and Jacob, M.H. (2001) New functions for glia in the brain. Proceedings of the National Academy of Sciences of the United States of America, 98, 3631-3632. doi:10.1073/pnas.081073198
- O’Connor, D.S., Schechner, J.S., Adida, C., et al. (2000) Control of apoptosis during angiogenesis by survivin expression in endothelial cells. American Journal of Pathology, 156, 393-398. doi:10.1016/S0002-9440(10)64742-6
- Dvorak, H.F. (2002) Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. Journal of Clinical Oncology, 20, 4368- 4380. doi:10.1200/JCO.2002.10.088
- Tran, J., Rak, J., Sheehan, C., et al. (1999) Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochemical Biophysical Research Communication, 264, 781-788. doi:10.1006/bbrc.1999.1589
- [61] Zhu, W.H., MacIntyre, A. and Nicosia, R.F. (2002) Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model: Distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. American Journal of Pathology, 161, 823-830. doi:10.1016/S0002-9440(10)64242-3
- [62] McBride, W.T., Armstrong, M.A., Crockard, A.D., et al. (1995) Cytokine balance and immunosuppressive changes at cardiac surgery: Contrasting response between patients and isolated CPB circuits. British Journal of Anaesthesiology, 75, 724-733. doi:10.1093/bja/75.6.724
- [63] Hedtjärn, M., Mallard, C. and Hagberg, H. (2004) Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. Journal of Cerebral Blood Flow and Metabolism, 24, 1333-1351. doi:10.1097/01.WCB.0000141559.17620.36
- [64] Pollari, E., Savchenko, E., Jaronen, M., et al. (2011) Granulocyte colony stimulating factor attenuates inflamemation in a mouse model of amyotrophic lateral sclerosis. Journal of Neuroinflammation, 8, 74-87. doi:10.1186/1742-2094-8-74
- [65] Diederich, K., Schabitz, W.R. and Minnerup, J. (2012) Seeing old friends from a different angle: Novel properties of hematopoietic growth factors in the healthy and diseased brain. Hippocampus, 22, 1051-1057. doi:10.1002/hipo.20904
- [66] Nishio,Y., Koda, M., Kamada, T., et al. (2007) Granulocyte colony-stimulating factor attenuates neuronal death and promotes functional recovery after spinal cord injury in mice. Journal of Neuropathology and Experimental Neurology, 66, 724-731. doi:10.1097/nen.0b013e3181257176
- [67] Pitzer, C., Klussmann, S., Kruger, C., et al. (2010) The hematopoietic factor granulocyte-colony stimulating factor improves outcome in experimental spinal cord injury. Journal of Neurochemistry, 113, 930-942. doi:10.1111/j.1471-4159.2010.06659.x
- [68] Sanli, A.M., Serbes, G., Caliskan, M., et al. (2010) Effect of granulocyte-colony stimulating factor on spinal cord tissue after experimental contusion injury. Journal of Clinical Neuroscience, 17, 1548-1552. doi:10.1016/j.jocn.2010.03.043
NOTES
*The research was supported by grant HL-58669 from the US National Institutes of Health (NIH).
#Corresponding author.