Open Journal of Gastroenterology
Vol.3 No.3(2013), Article ID:34099,13 pages DOI:10.4236/ojgas.2013.33029
Helicobacter infection decreases basal colon inflammation, but increases disease activity in experimental IBD
![]()
1Department of Molecular and Cellular Physiology, LSU Health Shreveport, Shreveport, USA
2Department of Microbiology and Immunology, LSU Health Shreveport, Shreveport, USA
3Department of Gastroenterology and Hepatology, LSU Health Shreveport, Shreveport, USA
4Department of Pathology, LSU Health Shreveport, Shreveport, USA
Email: jalexa@lsuhsc.edu
Copyright © 2013 Christopher P. Monceaux et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 5 April 2013; revised 1 May 2013; accepted 10 May 2013
Keywords: Helicobacter; colon; lymphatics; Crohn’s disease; ulcerative colitis
ABSTRACT
Background: Helicobacter species are best known for their roles in the pathology of gastritis; however, several Helicobacter species also colonize the intestine, and less is known about effects of Helicobacter on the development of intestinal inflammation. To evaluate contributions of Helicobacter in inflammatory bowel disease, we investigated whether and how pre-existing intestinal colonization would affect disease severity and biomarkers of inflammation in experimental IBD. Materials and Methods: Mice were infected with H. muridarum 2 weeks prior to induction of colitis mediated by 3% dextran sulfate (DSS). Disease activity index, stool blood and consistency, colon length, myeloperoxidase, histopathology, blood and lymphatic vessels, and numbers of dilated mucosal crypts were measured in control, DSS-only, H. muridarum-infected, and H. muridarum-infected + DSS mice. Results: Prior to DSS challenge, H. muridarum-infected mice showed little distal gut injury by several indices of colon inflammation with decreased blood vessel density in the submucosa, and lower lymphatic density in the mucosa and submucosa. However, after DSS colitis, H. muridarum-infected mice exhibited significantly greater disease. Weight change, stool bleeding, diarrhea, and angiogenesis were all increased in H. muridarum-infected mice in DSS colitis compared to DSS controls. Conclusions: Our data show that Helicobacter colonization of the intestine, unlike that of the stomach, lowers basal gut inflammatory scores, but increases disease activity and inflammation in an acute colitis model. Intestinal Helicobacter infection may therefore represent a significant sub-clinical factor which predisposes the gut to inflammatory injury.
1. INTRODUCTION
Helicobacter is a genus of Gram-negative bacteria that includes Helicobacter pylori (H. pylori), which is welladapted for colonization of the human stomach via binding to gastric epithelial cells [1]. In less than 30 years since its discovery, H. pylori has been associated with the causation of gastric ulcers and gastric cancer [2-4 ]. In addition to H. pylori, various Helicobacter species have been discovered in other mammalian organ systems, including the intestinal tract [5-7 ].
Inflammatory bowel disease (IBD) includes Crohn’s disease (CD) and ulcerative colitis (UC) [8]. CD is characterized by transmural inflammation/injury at any point along the gut, while ulcerative colitis involves mucosal damage that is limited to the colon [8,9 ]. Despite their differences in topographical distribution, CD and UC share several characteristics, including malabsorption, protein-losing enteropathy, cachexia, edema, and diarrhea. In addition, blood and lymphatic vessel expansion and remodeling have been reported previously in active IBD [10-12 ]. Angiogenic mediators, such as vascular endothelial growth factors (e.g., VEGF-A), fibroblast growth factor, and inflammatory cytokines (e.g., TNF-α), contribute to IBD pathophysiology by triggering activation of vascular endothelial cells and expansion of the microvasculature, which leads to increased lymphocyte infiltration and vascular leakage [13,14 ]. In contrastlymphatic vessels are largely important because of their function to remove inflammatory cells and cytokines. Consequently, the lymphatic expansion seen in IBD is likely a compensatory mechanism allowing clearance of inflammatory substances that accumulate during inflammation. Van Kruiningen and Colombel actually propose that dysfunctions of the intestinal lymphatics, such as lymphocytic and granulomatous lymphangitis have a significant role in IBD pathogenesis. Several animal studies show that obstruction of small intestinal lymphatic vessels results in intestinal dysfunction similar to IBD [13, 15 ]. Therefore, IBD etiology may reflect both increased leukocyte accumulation and cytokine release from activated, angiogenic microvessels but also decreased removal of these substances by dysfunctioning lymphatic networks.
Several models of ileitis and colitis suggest that bacteria are necessary, but insufficient triggers of IBD [16 ]. Several other studies have reported that Helic bacter may modulate IBD. For example, H. macacae has been linked with chronic idiopathic colitis in young rhesus monkeys [17 ]. Further, a study of children with CD reported PCR evidence for Helicobacter infection in 59% of children with CD vs. 9% of healthy controls [18]. Similarly, Laharie et al. found that evidence of H. pullorum or H. canadensis infection was significantly associated with CD in adults [19 ]. Of particular interest, the study showed that enterohepatic Helicobacter species predominated, rather than H. pylori [19 ]. Finally, H. canis, another enterohepatic Helicobacter, has also been detected in duodenal ulcerations in CD [20]. Therefore, it appears that certain Helicobacter species may be related to the pathogenesis of IBD. However, the exact mechanism of Helicobacter involvement is still undiscovered.
In this study, we examined the effect of H. muridarum infection on the development of acute gut injury caused by DSS-treatment. H. muridarum infects the intestines of mice and is typically considered a commensal organism [21 ]. Co-colonization of mice with H. pylori and H. muridarum was found to ameliorate gastric pathology induced by H. pylori alone [22 ]. Intestinal pathology arising from H. muridarum infection has only been documented in H. muridarum mono-associated SCID mice following transfer of certain T cell populations [23,24 ]. We examined differences in general disease activity and histopathology between control, H. muridarum-infected, DSS, and DSS + H. muridarum-infected mice. We also analyzed the change in lymphatic and blood vessel abundance to determine if H. muridarum affects IBD pathogenesis via alteration of the number of these vessels. The results indicate that H. muridarum infection does not cause a greater increase in histopathological disturbance than that found in mice treated with DSS only. However, we also report that changes in disease activity, such as stool blood, weight loss, etc., were significantly increased in mice pre-infected with H. muridarum compared to mice treated with DSS only. Finally, we discovered that initially H. muridarum infection causes a decrease in blood vessel density, but an increase in the density of blood vessels is seen after the stress of the DSS-treatment that is greater than the increase seen in the DSStreated mice treated without H. muridarum pre-infection. Therefore, it appears that H. muridarum may cause changes in blood vessel density that lead to an increase in disease activity.
2. MATERIALS & METHODS
2.1. Isolation and Propagation of H. muridarum Strain TM1
We obtained gastric homogenates from mice that were believed to have been colonized with candidates H. heilmannii (generously provided by Dr. Jani O’Rourke, University of New South Wales, Australia). Scrapings from the frozen homogenate were inoculated into Ham’s F-12 medium supplemented with 1% FBS, β-cyclodextrin, vancomycin, trimethoprim, and 5-fluorocytosine. Once growth of a highly-motile, spiral-shaped organism was apparent, efforts were begun to isolate the organism in pure culture. Aliquots of the culture were inoculated onto Campylobacter blood agar, plates. A small amount of culture was stabbed into F-12 soft agar [25], and new broth cultures were inoculated using medium drawn from the culture surface to minimize transfer of non-motile organisms. Cefsulodin and colistin were added, in addition to the above antimicrobial agents. H. muridarum-strain TM1 was unable to form colonies on any of the solid media tested, but was able to grow in F-12 soft agar containing 4% FBS. Organisms were taken from the edge of the soft agar growth region and re-inoculated into broth. Limiting dilution was also carried out on broth cultures. All organisms obtained from limiting dilution or soft agar were identical in morphology and displayed the same rapid back-and-forth motility as the organism seen in the original culture.
PCR of 16S ribosomal DNA from pure cultures was carried out using Helicobacter genus-specific primers [26]. The resulting product was sequenced by the Arizona State University DNA Laboratory. The strain was determined to be H. muridarum by comparing the 16S sequence with those of other bacteria using the NCBI Blast search engine. The sequences of the two isolates were identical to each other and 99% identical to the 16S ribosomal RNA gene sequence from H. muridarum ATCC 49282 over the sequenced stretch of 669 nucleotides. The only discrepancy was due to an “N” in our sequence data. There was a single nucleotide mismatch with H. muridarum strain ST1. H. muridarum TM1 was further found to be strongly urease positive using a rapid urease test. We were able to obtain growth at 42˚C, unlike H. muridarum isolates reported by Solnick, et al. [27]. We did not perform electron microscopy or flagella stains, but the strong back-and-forth motion characteristic of strain TM1 is consistent with the presence of bipolar flagella, as reported for other H. muridarum strains [27].
2.2. Infection of Murine Intestines
H. muridarum was grown in 25 cm2 tissue culture flasks containing Ham’s F-12 medium supplemented with 1% FBS under conditions described above. After microscopically verifying appropriate morphology and motility, the culture was centrifuged at 4˚C at 7840 × g for 15 minutes and the pellet was resuspended in 3 ml 0.9% NaCl. 200 ul aliquots of the concentrated H. muridarum suspension were administered orally to each mouse using a gavage needle.
Infection was confirmed by stool culture two weeks post-infection (data not shown). Fresh fecal pellets were collected and placed in microfuge tubes with F12 medium containing 1% fetal bovine serum and antimicrobial agents capable of suppressing most non-Helicobacter fecal organisms. The tubes were spun at 10,000 × g and then incubated at 37˚C for 10 minutes. The upper portion of the supernatant was then removed and added to a well in a 24-well plate containing F12 + 5% FBS and antibiotics, as described above. This procedure enriched for motile bacteria, thus reducing contamination by the bulk of fecal organisms, which are non-motile or weakly motile. Antibiotic selection further reduces growth of unwanted microorganisms. Cultures were monitored daily by phase contrast microscopy for organisms with characteristic shape and motility. A rapid urease test was used to further confirm that recovered organisms were H. muridarum. The presence of urease activity was detected by adding 10 µl of culture to 100 µl of urease detection solution (1 mM sodium phosphate buffer, pH 6, 33 mM urea, 63 µg/ml phenol red). If urease is present, the solution turns from yellow to red within 15 minutes. Our H. muridarumstrain has extremely high urease activity and usually caused a color change within 30 seconds.
2.3. Induction of Experimental Colitis
Four groups of animals were used: control uninfected mice (CU), control H. muridarum-infected mice (MU), DSS-treated uninfected mice (CD), and DSS H. muridarum-infected mice (MD). Experimental colitis was induced in C57B6 mice (n = 5) by supplementing drinking water with 3% DSS as described in Ganta et al. (DSS, MW = 36 - 50 kDa; ICN Biomedical, Costa Mesa, CA); mice in the control groups (n = 5) group received tap water without DSS [28 ]. Mice were inoculated with H. muridarum ≥ 2 weeks prior to initiation of DSS treatment in order to make sure that infection was well-established.
3% DSS administration produces an erosive distal colitis with an initial onset at 3 - 4 days that is characterized by progressive weight loss, diarrhea, occult blood, leukocyte infiltration, colon shortening, loss of intestinal epithelial barrier, and histopathological changes in colon structure [29,30 ]. Mice typically lose ~20% body weight by day 10 with continuous administration of 3% DSS. Over 9 days, mice were given unrestricted access to pellet diet (Purina, St. Louis, MO), and tap water (control), or tap water containing 3% DSS in ‘‘colitis’’ groups. Mouse weight, stool form, and occult blood were recorded daily. On day 9, mice were sacrificed by cardiac puncture while under ketamine/xylazine anesthesia (k/x = 150/60 mg/kg via intraperitoneal injection). Tissue samples for histological studies were fixed in cold 3.7% phosphate-buffered formalin, or frozen at −20˚C for myeloperoxidase (MPO) analysis.
2.4. Disease Activity Index & Tissue Histopathology Evaluation
The “disease activity index” (DAI), is a combinatorial index of disease, including stool blood, stool form, and weight loss [31]. It is used to quantify the extent of induced disease and benefit of experimental therapies. Body weight change is defined as the percentage difference in body weight for each mouse as a fraction of the starting (day = 0) weight.
Tissues were analyzed for histopathological injury using the criteria established by Cooper et al. [29]. This system includes edema, extent of injury, leukocyte infiltration, crypt abscesses, and loss of goblet cells. In this grading system, inflammation severity was scored on a 0 - 3 scale, (0: none; 1: slight; 2: moderate; 3: severe), the extent of injury was scored on a 0 - 3 scale (0: none; 1: mucosal; 2: submucosal; 3: transmural), and crypt damage was scored on 0 - 4 scale (0: none; 1: basal 1/3 damaged; 2: basal 2/3 damaged; 3: only surface epithelium intact; 4: loss of entire crypt and epithelium). Each value was multiplied by an extent index from 1 - 4 that reflects the amount of involvement for each section (1: 0% - 25%, 2: 26% - 50%, 3: 51% - 75%, 4: 76% - 100%). At least 3 sections from each colon were analyzed to produce each score value. A maximum possible histopathological score for this assay is 40.
2.5. Immunohistochemical Analysis
Colon preservation in formalin, sectioning of the tissue, and immunohistochemistry were completed as described in Ganta et al., 2010 ADDIN REFMGR.CITE ADDIN EN.CITE.DATA [28 ]. Staining for vascular endothelial growth factor receptor-3 (lymphatic vessels) and mouse endothelial cell antigen-32 (blood vessels) was performed. Once stained, the vessels in each layer of the entire colon wall were counted.
2.6. Tissue MPO
Tissues were prepared as described previously [32 ]. Briefly, the mouse colons were dissected and homogenized in phosphate buffer (5 mM; pH 6). The samples were centrifuged at 30,000 × g for 30 minutes at 4˚C. The supernatant was discarded, and the pellet was washed again in phosphate buffer. Then the pellet was suspended in 50 mM phosphate buffer (pH 6, 25˚C), which contained 0.5% hexadecyltrimethylammonium bromide. The samples were frozen and thawed 3X with sonication between each cycle. Protein concentrations in samples were determined by BCA assay (Bio-Rad). Samples were frozen until MPO assays were performed. Each sample was sonicated for 6 secs, and 20 μl added to the wells (n = 3). 180 μl of phosphate buffer (50 mM, pH6), 0.0167% o-dianisidinedi HCl (ICN.), and 1% H2O2 was added to each 96-well and incubated at 37˚C. A450nm was measured at 80 min. The activity is expressed as change in absorbance per 60 minutes per mg of protein.
2.7. Ethical Considerations
Animal protocols were approved by the LSU Institutional Animal Care and Use Committee. All mice were monitored daily for disease activity; any animals showing restricted movement or loss of 20% body weight were humanely sacrificed following terminal ketamine/ xylazine anesthesia (k/x = 150/60 mg/kg via intraperitoneal injection) and thoracotomy.
2.8. Statistical Analysis
All statistical analysis was completed using GraphPadInstat 3. Comparisons between the four groups (CU, CD, MU, and MD) were completed using one-way ANOVAs with Tukey-Kramer post-tests. Repeated measures one-way ANOVAs with Dunnett’s post-tests were used on comparisons between day 0 scores and later scores from within the same group. Statistical significance was defined as p < 0.05.
3. RESULTS
3.1. H. muridarum Infection Exacerbates Increases in Disease Activity Index
The Disease Activity Index (DAI) was measured for all four groups of mice (Figure 1(a)). Throughout the 9 days of the study, DAI scores for CU and MU mice were constant at day “0”. The DAI scores for CD and MD mice rose as time passed, and the MD scores were significantly higher than the CU and MU mice scores after day 2. The CD scores became significantly higher than the CU and MU scores on days 6 - 9. Further, DAI scores of the MD mice were significantly higher than the scores of CD mice on days 6 - 8, indicating an earlier worsening of the disease state in MD mice. Finally, the MD mice had a significantly higher DAI score than at day “0” after day 2. MD mice also showed significant disease activity 2 days earlier than CD mice.
Weight loss was measured in CU, MU, CD, and MD mice (Figure 1(b)). The % of original weight was similar for all groups up until day 3. From day 4 onward, the % original weight of CU mice was significantly lower than MU. The MD mice had significantly greater weight loss on days 7 - 9 than CD mice. Both the CD and MD mice exhibited a large weight loss compared to the CU or MU mice. After day 6, the difference between the MD mice and the CU or MU mice was significant, whereas the % original weight of the CD mice was significantly less only on day 9. The MD mice lost a large portion of their body weight, approximately 23% by day nine. By comparison, the CD mice lost approximately 15% of their body weight by day nine. The MD mice also had significantly less weight than on day “0” by day 6 of measurements. However, CD mice did not have significant weight loss until day 7. Therefore, the weight loss was accelerated by approximately one day.
Diarrhea (stool form) (Figure 1(c)), was evaluated to determine effects of treatments on each group of mice. The stool grade for CU and MU mice remained stable for the duration of the study. Conversely, the grade of the MD and CD mice increased steadily throughout the study. By day 4, scores were significantly greater than the CU or MU mice. On days 7 and 8, the MD mice had a significantly higher stool grade than the CD mice. Furthermore, the MD mice did not have higher stool grade scores any earlier than the CD mice. Therefore, the H. muridarum did not affect the time of onset for changes in stool consistency.
Stool blood measurements (Figure 1(d)) showed that the stool blood in CU and MU mice remained stable at “0” throughout the study. Both CD and MD mice showed significantly increased stool blood after day 5 compared to CU and MU mice. At days 6 - 9, the scores of occult blood were significantly higher in MD mice compared to CD mice. The presence of more blood in the stool of MD mice vs. CD mice suggests that these combined stresses increase tissue injury and/or capillary fragility. MD mice had significantly elevated stool blood by day 4. For the CD mice, this occurred on day 5. Once again shows that colitis may progress more quickly in the H. muridaruminfected mice.
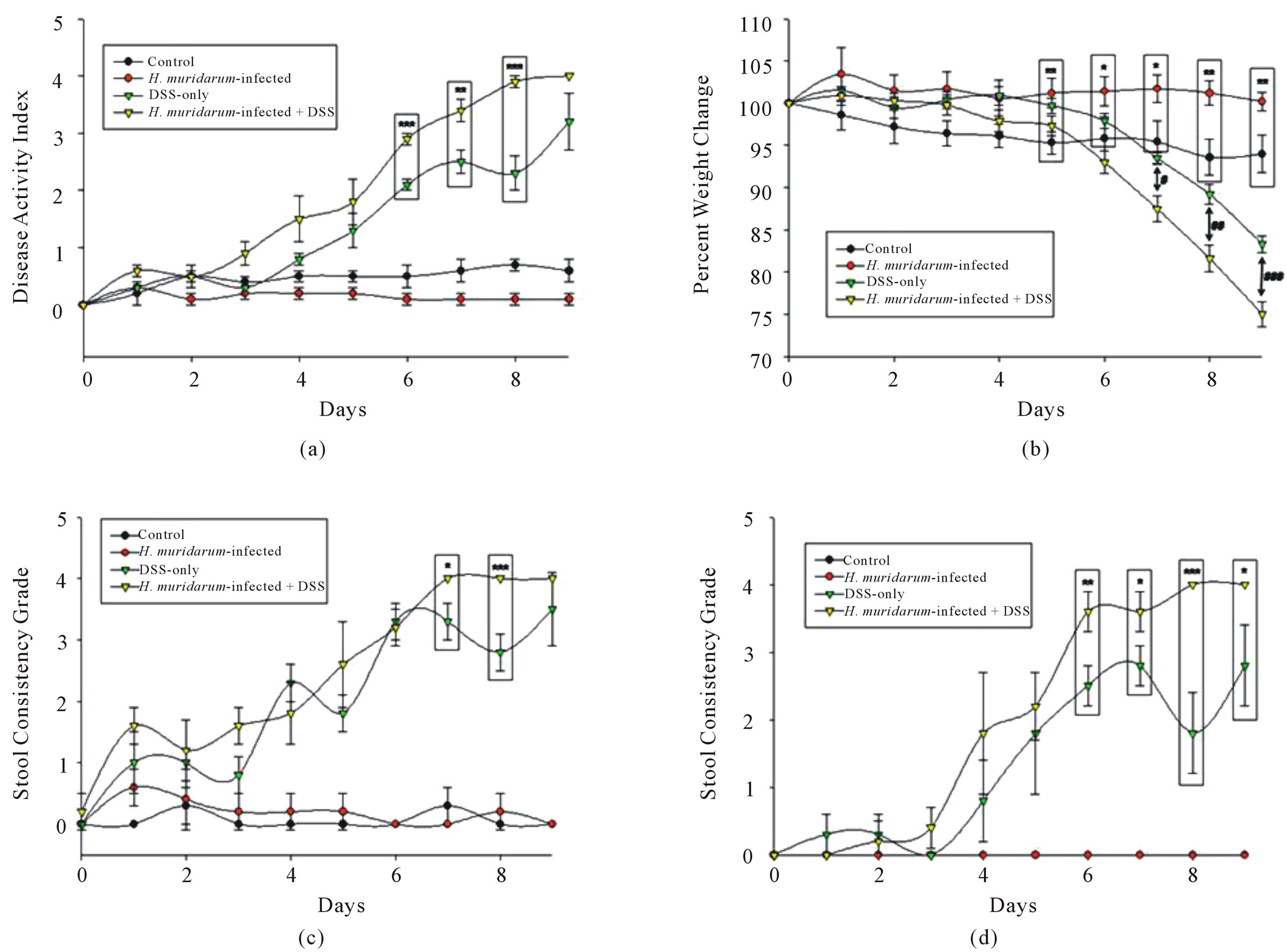
Figure 1. (a) Both the CD and MD mice show a steady increase in DAI (p < 0.001) from CU and MU mice on days 6 - 8. The MD mice also show a higher DAI than the CD mice on days 3 (p < 0.01), 6 (p < 0.001), 7 (p < 0.01), and 8 (p < 0.001). (b) CD mice show a decrease (p < 0.001) in weight after day 7, and MD mice show a decrease (p < 0.001) in weight after day 6. MD mice also showed a significantly larger decrease in weight than the CD mice on days 7 - 9. (c) CD and MD mice had higher stool grades than CU and MU mice beyond day 3. MD mice have a higher stool grade than CD mice on days 7 and 8. (d) Both CD and MD mice had higher amounts of blood in their stool compared to the CU and MU mice beyond day 5. The MD mice had a significantly higher amount of blood in the stool compared to CD mice on days 6 - 9.
3.2. H. muridarum Infection Has Several Pathological Effects on Colitis Mice
A small decrease (non-significant) difference in colon length was seen in the MU mice compared to the CU mice. Both CD and MD mice showed a significant reduction in colon length compared to the CU and MU mice (Figure 2(a)). The MD mice also had a shorter colon than the CD mice, but the difference was not significant.
Histological analysis of the murine intestinal tissue sections showed that a significantly higher amount of dilated mucosal crypts was present in the CD and MD mice when compared to the CU and MU mice. Interestingly, the MD mice also had approximately 2.5 times the number of the abnormal crypts than the CD mice. Therefore, the MD mice show a great increase in what appears to be a histological marker of normal colitis (Figure 2(b)).
MPO activity was assayed in colon samples of all 4 subsets of mice and expressed as avg ± SEM. The change in absorbance at 450 nm/60 min/mg of sample from CD mice (0.058 ± 0.009) was significantly higher (p < 0.05) than the absorbance levels of the CU mice (0.035 ± 0.008). The change in absorbance of samples from MU (0.034 ± 0.006) and MD (0.03012 ± 0.015) mice were not higher than the absorbance of the control samples. Furthermore, the absorbances of the CU, MU, and MD samples were very similar. Therefore, MPO activity is not increased in either type of H. muridarum-infected mouse, but the activity is increased in CD mice (Figure 2(c)).
Histopathological score of the colon tissue was measured for all four different groups of mice after they were
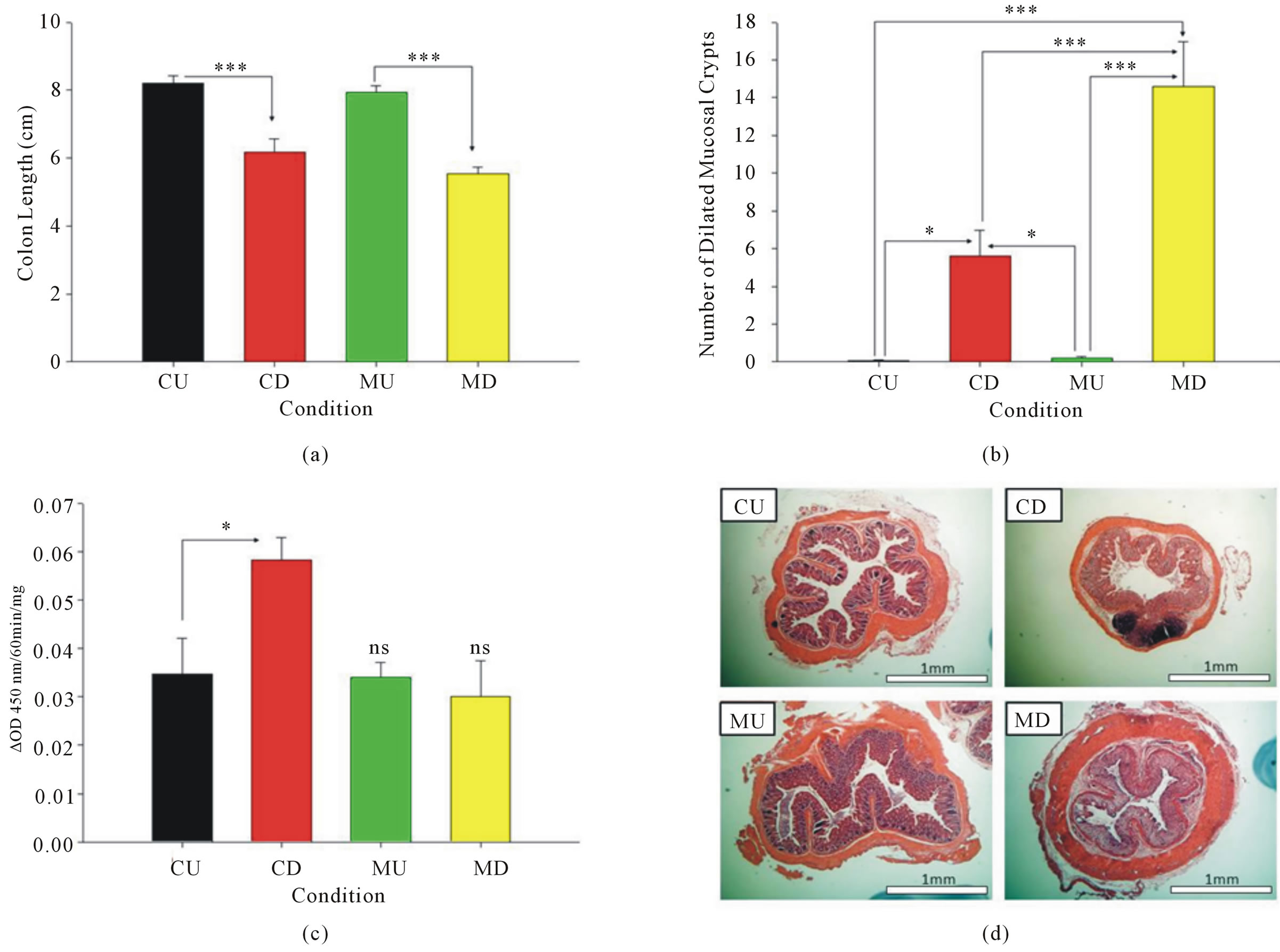
Figure 2. (a) The CD mice had shorter colons than CU mice and the MD mice shorter colons than the MU mice (p < 0.001). (b) Both CD (p < 0.05) and MD (p < 0.001) mice had higher amounts of dilated mucosal crypts than CU and MU mice. In addition, MD mice had more dilated mucosal crypts than CD mice (p < 0.001). (c) MD and MU mice did not show a difference in MPO activity from the CU mice. However, the CD mice had an increase in activity compared to the CU mice (p < 0.05). (d) H&E stains of colons from CU, CD, MU, and MD mice used for histopathology scoring.
sacrificed on day 9 using H&E-stained tissue sections (Figure 2(d)). The CD and MD mice had significantly higher scores than the CU or MU mice (p < 0.001). However, there was no significant difference between the CU and MU mice or the CD and MD mice (data not shown). Furthermore, no significant differences were seen between these mice on any of the individual subscales: severity of inflammation, extent of injury, and crypt damage (data not shown).
3.3. H. muridarum Infection Alters Lymphatic Vessels
Lymphatic vessels were visualized in the mucosa, submucosa, and serosal layers of the colon using VEGFreceptor 3 (VR3) immunohistochemistry (Figure 3(a)). The stained slides were used to determine the abundance of lymphatic vessels in the mucosa (Figure 3(b)), submucosa (Figure 3(c))), and serosa (Figure 3(d)) of each mouse. In the mucosa, MU mice had a significantly lower level of lymphatic vessels than the CU mice (p < 0.05). The MU mice also displayed significantly lower levels of lymphatic vessels than both the CD and MD mice (p < 0.001). Therefore, the infection greatly decreased the amount of lymphatic vessels whenever no disease is induced. The MD mice illustrated a significant increase in lymphatics compared to the CU mice (p < 0.001). This shows that H. muridarum infection increases the amount of lymphatic vessels whenever disease is induced via DSS. The increase is possibly a compensatory mechanism for the greatly reduced number of lymphatics that is induced by the infection when disease is not present. An increase in lymphatics in CD mice compared to CU mice was also observed but was not significant. The amount of lymphatic vessels was also greater in MD mice than in CD mice, but the difference was not significant.
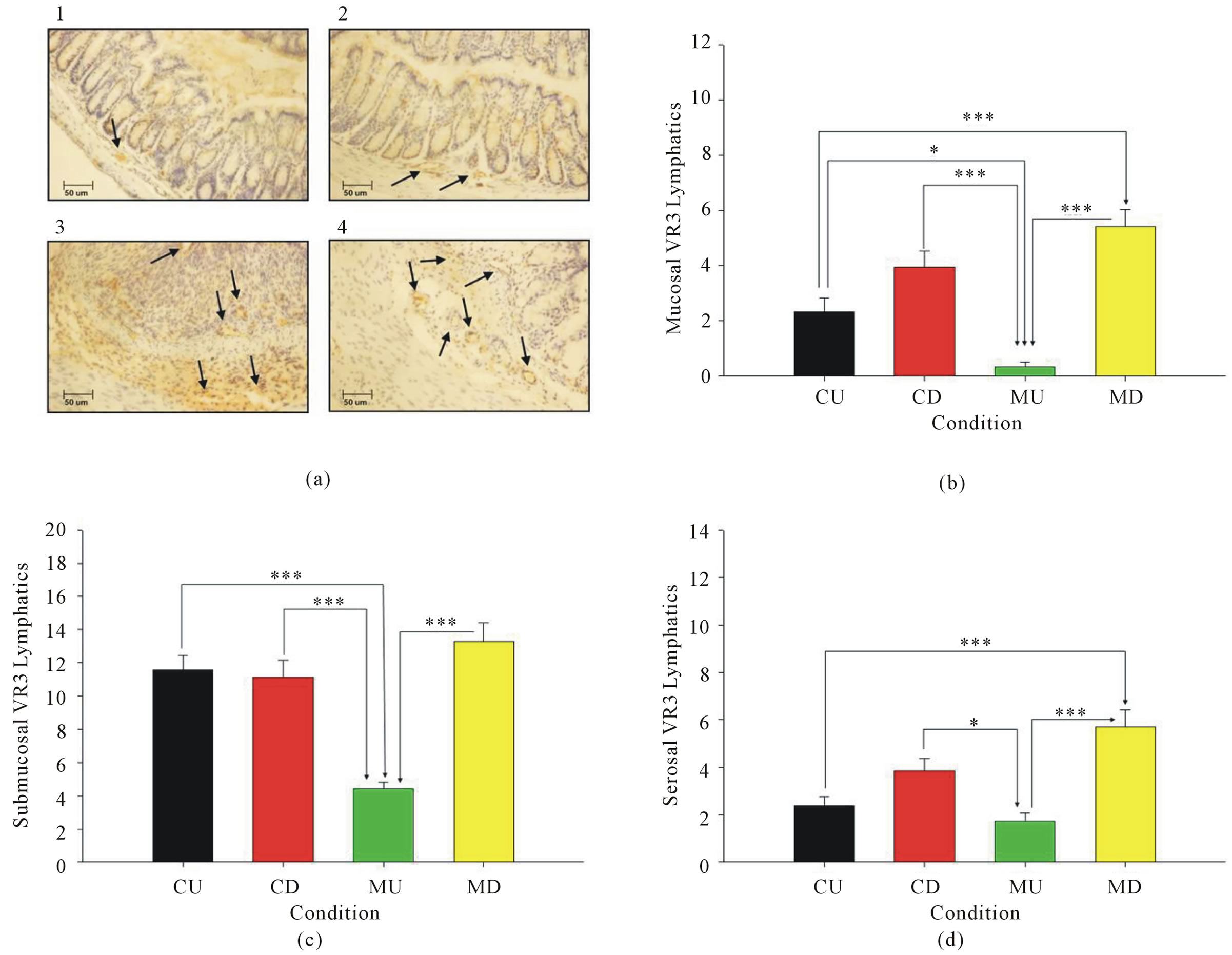
Figure 3. (a) Immunohistochemical staining for VEGFR3 (lymphatic marker) was performed in 1) CU, 2) MU, 3) CD, and 4) MD mice. Arrows denote lymphatic vessels. B) A lower lymphatic vessels are present in mucosa of MU compared to CU (p < 0.05), CD (p < 0.001), and MD (p < 0.001) mice. MD mice also had more lymphatics than the CU mice (p < 0.001). C) In the submucosa, MU mice have less lymphatic vessels than the CU, CD, and MD mice (p < 0.001). (d) The serosa of the MU mice had less lymphatic vessels than the CD (p < 0.05) and MD mice (p < 0.001). The MD mice also had more lymphatics than the CU mice (p < 0.001).
In the submucosa, MU mice had significantly lower levels of lymphatic vessels than the CU, CD, and MD mice (p < 0.001). The CU, CD, and MU mice had roughly equivalent numbers of lymphatic vessels in the submucosa. In the serosa, MU mice had significantly lower levels of lymphatics than the CD (p < 0.001) and MD (p < 0.05). The CD mice had increased lymphatic vessels compared to the CU mice, but the difference was not significant. The MD mice had a greater amount of lymphatics than the CD mice, but this difference was also not significant. Finally, the MD mice showed significantly greater levels of lymphatic vessels than the CU mice (p < 0.001).
3.4. H. muridarum Infection Increases Blood Vessel Abundance in DSS Mice
Blood vessels were visualized in the mucosa, submucosa, and serosal layers of the colon using mouse endothelial cell antigen-32 (MECA-32) immunohistochemistry (Figure 4(a)). The stained slides were used to quantify the abundance of blood vessels in the mucosa (Figure 4(b)), submucosa (Figure 4(c)), and serosa (Figure 4(d)) of the colon. In MD mice, the quantity of blood vessels in the mucosa was significantly greater than in the CU, CD, and MU mice (p < 0.001). The quantity of vessels in the mucosa of CU, CD, and MU mice was similar. In the submucosa, MD mice had a significantly greater number of blood vessels than the CU, CD, and MU mice (p < 0.001). Furthermore, the MU mice had significantly lower amounts of blood vessels than the CU mice (p < 0.05). As before with lymphatics, the submucosa also contained the most blood vessels out of the three layers of intestinal tissue.
In the serosa, the CU (p < 0.001) and MU (p < 0.01)

Figure 4. (a) Results of immunohistochemical staining for MECA-32+ vessels in the 1) CU, 2) MU, 3) CD, and 4) MD mice are pictured. The arrows point to the MECA-32+ vessels, which are stained brown. (b) A higher amount of blood vessels was present in the mucosa of MD mice compared to the CU, CD, and MU mice (p < 0.001). (c) A higher amount of blood vessels was seen in the submucosa of MD compared to the CU, CD, and MU mice (p < 0.001). A lower amount of blood vessels is seen in the MU mice compared to the CU mice (p < 0.05). (d) A higher quantity of blood vessels was seen in the MD mice compared to the CU (p < 0.001) and MU (p < 0.01) mice.
mice had significantly fewer blood vessels than the MD. The CD mice also had fewer blood vessels than the MU mice, but the difference was not significant. Moreover, the CD mice exhibited an increase (not significant) in blood vessels from the CU and MU mice.
4. DISCUSSION
Since the H. pylori—gastritis connection was discovered, many studies have described the role of this bacterium in gastric mucosal injury. In addition to epithelial injury, another important proposed target of H. pylori pathogenesis is the gastric mucosal microcirculation [33]. Kalia et al. described that toxigenic H. pylori increased leukocyte adhesion, platelet aggregation, post-venular capillary vasoconstriction and leakage of labeled albumin [34,35 ]. Several other studies have demonstrated that H. pylori-induced microvascular disturbances may involve platelet activating factor and nitric oxide (NO), since hexanolamine-PAF and L-arginine prevented increased platelet emboli and thrombi in mucosal capillaries and post-capillary venules [36 ]. P-selectin also participates in these forms of platelet leukocyte binding [37 ], and aggregation of the platelets is known to participate in the recruitment of leukocytes [38].
Additionally, mast cells and mast cell-derived histamine may contribute to microcirculatory disturbances associated with H. pylori. Atuma et al. showed that mast cell stabilization (with ketotifen) or the PAF antagonist WEB2086 blocked H. pylori extract induced vasoconstriction [39]. Kalia et al. showed that ketotifen and pyrilamine, an H1 receptor antagonist, prevent increased macromolecular leakage of labeled albumin in response to H. pylori extracts ADDIN REFMGR.CITE ADDIN EN.CITE.DATA [36 ]. Kurose et al. found that albumin leakage produced by H. pylori was decreased by mast cell stabilization, or blocking antibodies against CD11b/ CD18, ICAM-1 or P-selectin [37 ].
While Helicobacter colonizes the gastric mucosa and causes local injury, Helicobacter has also been found in other areas of the gastrointestinal tract including the liver, gallbladder, and colon [40]. Despite the large amount of research on H. pylori-induced gastric mucosal injury, much less has been accomplished on the extragastric effects of H. pylori. However, those few studies have illustrated that Helicobacters (pylori, etc.) also disturb microcirculatory integrity in acute pancreatitis, and increase the severity of ischemia-induced pancreatitis [33]. In addition, lipopolysaccharide released by H. pylori triggers high rates of NO production through iNOS-induction [41 ], which has been associated with increased vascular permeability in the heart, kidney, liver, and lung [42 ]. Some investigators have also studied the effects of H. pylori in coronary artery disease. Pellicano et al. have shown a higher occurrence of H. pylori infection in patients with acute myocardial infarction [43]. Prasad et al. studied the effect of H. pylori pathogen burden on endothelial dysfunction. They discovered that an elevated pathogen burden, (which included H. pylori infection plus normal commensal bacteria and viruses), was a significant risk factor for endothelial dysfunction and coronary artery disease [44 ].
4.1. Does H. pylori Infection Attenuate Gut Inflammation? Effects in IBD
Extra-gastric effects of H. pylori have also been studied in the large intestine where it apparently provokes different responses than in the stomach. For example, in a DSS colitis model, Luther et al. have shown that H. pylori DNA decreases dendritic cell production of proinflammatory cytokines, (IFN-γ and IL-12), which might lessen IBD severity [45 ]. However, E. coli DNA produced an intense inflammatory edema, irritation, bleeding and diarrhea. This reaction to E. coli DNA was mediated by toll-like receptor (TLR) 9 expressed on dendritic cells which produced Type 1 IFNs, which in this study were described as “pro-inflammatory” cytokines. By comparison, there was a reduced systemic level of type I IFN in mice treated with H. pylori DNA, which might explain decreased IBD symptoms in these mice during experimental colitis. Conversely, Katakura found that production of type I IFN mediated by TLR 9 was protective in the same colitis model (DSS) [46 ]. Therefore, TLR 9 responses have been shown to both intensify and reduce gut inflammation. A possible explanation for this difference is that TLR 9 bound by gut floral DNA on the epithelial apical membrane decreased NF-kB dependent genes and suppressed inflammation. Conversely, binding of bacterial antigens to TLR-9 expressed on the basolateral surface of gut epithelial cells, stimulated NF-kB and led to a greater immune and inflammatory response [47]. In this sense, TLR-9 may act as a “positional” sensor of bacterial antigens which either increases or suppresses inflammation depending on the integrity of the epithelial barrier.
Vijay-Kumar et al. reported anti-inflammatory effects of TLR-3 activation mediated by binding of synthetic viral RNA which reduces gut injury in experimental colitis [48 ]. Because Helicobacter clearly may activate several types of TLRs (TLR-3, -9), the roles of TLRs in gut inflammation are complex. Luther et al. revealed that the microbial DNA of H. pylori has the ability to block inflammatory stimulation produced by E. coli DNA in the colon. Although Luther et al. showed that H. pylori DNA has anti-inflammatory effects, their findings exclude several alternate mechanisms and mediators active during Helicobacter colonization and do not completely address how Helicobacter colonization might affect the initiation and progression of colitis.
4.2. Non-Pylori Helicobacter Infection May Increase Gut Injury in IBD
There appears to be some association between Helicobacter and gut injury in IBD. Kaakoush et al. found that Helicobacteriaciae were present in 41.5% of children with CD, while only present in 22.5% of normal children (p = 0.0062). Interestingly, these Helicobacter species were not gastric H. pylori, but rather other Helicobacter strains found in the liver, gall bladder, and intestine [49 ]. Enterohepatic Helicobacter has also been found in the intestines of animals [20]. Using a Helicobacter-specific PCR, Helicobacter canis, [a species of Helicobacter that colonizes the colon of canines] was recovered from superficial ulcerations of the superior and descending duodenum in human CD [20]. This discovery indicates that Helicobacter colonization of the gut may correlate with increased susceptibility to IBD. In another model, Helicobacter colonization of the liver was associated with lower production of Th1 cytokines (IL-1β, IFN-γ, TNF-α) and Th17 (IL-17A) [40]. In our hands, H. muridarumdecreased vascular density in intestinal wall layers under basal conditions, but increased it during active inflammation (DSS colitis). Helicobacter contributions to intestinal injury may be complex and state-dependent.
Because Helicobacter within and outside the gastrointestinal tract appear to alter microcirculatory physiology, changes in microvascular structure and function could contribute to gut pathophysiology as is suggested in Helicobacter-associated atherosclerosis and cardiovascular risk. In this study, we found an increase in blood vessels within the mucosa and submucosa of MD mice that was even higher than the increase seen in CD mice. Furthermore, we discovered that the mucosa and submucosa of MU mice had less lymphatic vessels than the CU mice. This decrease in lymphatic vessels could have played a role in worsening of colitis in the infected mice due to a lessened initial capability to evacuate lymph and immune cells from the area of disease. Therefore, it appears that alterations in microcirculation do play a role in contributing to the gut pathophysiology of Helicobacter infected mice.
Our study has shown that the DAI in MD mice is increased early on when compared to CD mice. However, by the end of the 9-day study, the difference seemed to have disappeared, and both groups of mice had similar levels of disease activity. Furthermore, when the mice colons were scored for histopathological changes after sacrifice at day 9, there was no difference in the scores of MD and CD mice. This seems to indicate rapid disease progression in MD mice that culminates in a similar level of disease as the CD mice by the end of disease development. Further studies need to be performed to determine if the histopathological disease state of MD mice is actually worse on earlier days in the study.
Despite evidence from both our study and previous studies that Helicobacter does contribute to IBD gut injury, we saw no increase in the amount of MPO activity in the colon of either the MD or the MU mice. On the other hand, the CD mice saw approximately a doubling of activity. Interestingly, this suggests that neutrophil infiltration of the diseased colon in the colitis mice infected with H. muridarumis less than the infiltration in regular colitis mice. This change may be related to the decreased submucosal blood vessel abundance seen in the MU mice. The decrease in blood vessels could lead to a decrease in the ability to recruit neutrophils to the disease area. Therefore, the gut would be unprepared to handle the type of corrosive injury that occurs when DSS is administered. Even without the increase in neutrophils, greater disease activity index scores are seen in the MD mice than in the CU, CD, and MU mice. Therefore, future studies are needed to determine which immune cells are actively involved in the immune responses in these MD mice.
4.3. Is Helicobacter Infections a Risk Factor for Colorectal Cancer?
In MD mice, the colon shows significantly more dilated mucosal crypts than were observed in CD colons. Crypt dilation is not uncommon in mouse models of colitis, but the number of abnormal crypts was significantly greater than that in the other three groups of mice, including the CD mice. Yoshizawa et al. have previously shown that H. pylori infection alters gland morphology in the stomach, including the dilation of mucous glands [50]. Furthermore, H. pylori has been convincingly shown to play a role in the development of gastric cancer [50]. This is particularly relevant because abnormal mucosal crypts, such as those seen in high numbers in the MD mice, have been identified as precursor markers of colon carcinogenesis [51]. Since the high numbers of abnormal crypts are not seen in the MU mice, it is possible that the additional stress induced by DSS colitis when added to H. muridarum provokes these pre-cancerous histological changes in the colon mucosa. Consequently, H. pylori infection, like H. muridarum, may significantly exacerbate the formation of intestinal cancers in IBD-associated colon cancer, and the appearance of these dilated mucosal crypts may be a warning sign that precedes the onset of colon cancers. However, much more work must be done before we can know if this is actually the case.
Sugimoto et al. have also found an interesting link between H. pylori and carcinogenesis wherein angiotensin II type I receptor mRNA levels were linked to the extent of inflammatory cell infiltration into the gastric mucosa. They also found a significant increase in the amount of angiotensin II type I and type II receptor mRNA levels in gerbils with gastric ulcers. These findings show a possible link between the renin-angiotensin system (RAS) and H. pylori infection. This is important because the RAS system is involved in angiogenesis required for carcinogenesis to proliferation. Therefore, there may be a link between the RAS and gastric (or possibly intestinal) carcinogenesis related to H. pylori infection [52 ]. Interestingly, chronic H. pylori infection of the stomach was also associated with increased rates of intestinal metaplasia [50]. This finding strongly suggests that gastric colonization with H. pylori produces changes in the intestine, particularly in response to gut injury as we see here. Because H. muridarum-colonization significantly increased crypt dilation, a prominent feature of intestinal metaplasia, the intensification of angiogenesis and metaplastic changes seen may represent an important combination of risk factors for development of subsequent CRC. Because Helicobacter might directly affect the intestine through gut colonization and indirectly influence CRC development through induction of gastrin secretion, (a stimulus for intestinal mucosal epithelial growth and a risk factor for CRC), Helicobacter infection might represent an important risk for hyper intense gut injury and malignancy and needs to be evaluated by prospective correlative trials [53].
5. CONCLUSION
In this study, we infected mice with H. muridarum to study its effects on the onset and severity of IBD. We found that H. muridarum increased disease activity and caused earlier disease onset. We also discovered differences in lymphatic and blood vessel densities between the infected and non-infected mice. All of these observations point to the possibility of an active and important role for Helicobacter colonization in the onset and worsening of IBD that involve microvasculature alterations.
6. ACKNOWLEDGEMENTS
This work was supported by the Department of Defense, PR100451, and the Department of Medicine, Division of Gastroenterology & Hepatology, LSU Health-Shreveport. The authors would like to thank Erica Glidewell for her help in the preparation of this manuscript.
REFERENCES
- Wessler, S., Gimona, M. and Rieder, G. (2011) Regulation of the actin cytoskeleton in helicobacter pylori-induced migration and invasive growth of gastric epithelial cells. Cell Communication and Signaling, 9, 27. doi:10.1186/1478-811X-9-27
- Bashinskaya, B., Nahed, B.V., Redjal, N., Kahle, K.T. and Walcott, B.P. (2011) Trends in peptic ulcer disease and the identification of helicobacter pylori as a causative organism: Population-based estimates from the US nationwide inpatient sample. Journal of Global Infectious Diseases, 3, 366-370. doi:10.4103/0974-777X.91061
- Correa, P. and Piazuelo, M.B. (2012) Evolutionary history of the Helicobacter pylori genome: Implications for gastric carcinogenesis. Gut and Liver, 6, 21-28. doi:10.5009/gnl.2012.6.1.21
- Schottker, B., Adamu, M.A., Weck, M.N. and Brenner, H. (2012) Helicobacter pylori infection is strongly associated with gastric and duodenal ulcers in a large prospective study. Clinical Gastroenterology and Hepatology, Vol. 10, No. 5, 2012, pp. 487-493. doi:10.1016/j.cgh.2011.12.036
- Contreras, M., Salazar, V., Garcia-Amado, M.A., Reyes, N., Aparcero, M., Silva, O., Castro, D., Romero, R., Gueneau, P. and Michelangeli, F. (2012) High frequency of helicobacter pylori in the esophageal mucosa of dyspeptic patients and its possible association with histopathological alterations. International Journal of Infectious Diseases, Vol. 16, No. 5, 2012, pp. e364-e370. doi:10.1016/j.ijid.2012.01.007
- Koshiol, J., Flores, R., Lam, T.K., Taylor, P.R., Weinstein, S.J., Virtamo, J., Albanes, D., Perez-Perez, G., Caporaso, N.E. and Blaser, M.J. (2012) Helicobacter pylori seropositivity and risk of lung cancer. PLoS One, 7, e32106. doi:10.1371/journal.pone.0032106
- Marini, R.P., Muthupalani, S., Shen, Z., Buckley, E.M., Alvarado, C., Taylor, N.S., Dewhirst, F.E., Whary, M.T., Patterson, M.M. and Fox, J.G. (2010) Persistent infection of rhesus monkeys with 'Helicobacter macacae’ and its isolation from an animal with intestinal adenocarcinoma. Journal of Medical Microbiology, 59, 961-969. doi:10.1099/jmm.0.019117-0
- Abraham, C. and Cho, J.H. (2009) Inflammatory bowel disease. New England Journal of Medicine, 361, 2066- 2078. doi:10.1056/NEJMra0804647
- Casellas, F., Lopez-Vivancos, J., Badia, X., Vilaseca, J. and Malagelada, J.R. (2001) Influence of inflammatory bowel disease on different dimensions of quality of life. European Journal of Gastroenterology Hepatology, 13, 567-572. doi:10.1097/00042737-200105000-00017
- Chidlow Jr., J.H., Langston, W., Greer, J.J., Ostanin, D., Abdelbaqi, M., Houghton, J., Senthilkumar, A., Shukla, D., Mazar, A.P., Grisham, M.B. and Kevil, C.G. (2006) Differential angiogenic regulation of experimental colitis. The American Journal of Pathology, 169, 2014-2030. doi:10.2353/ajpath.2006.051021
- Geleff, S., Schoppmann, S.F. and Oberhuber, G. (2003) Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Archiv, 442, 231-237.
- Kaiserling, E., Krober, S. and Geleff, S. (2003) Lymphatic vessels in the colonic mucosa in ulcerative colitis. Lymphology, 36, 52-61.
- Danese, S. (2011) Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: “Brothers in arms”. Gut, 60, 998-1008. doi:10.1136/gut.2010.207480
- Scaldaferri, F., Vetrano, S., Sans, M., Arena, V., Straface, G., Stigliano, E., Repici, A., Sturm, A., Malesci, A., Panes, J., Yla-Herttuala, S., Fiocchi, C. and Danese, S. (2009) VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology, 136, 585-595. doi:10.1053/j.gastro.2008.09.064
- Van Kruiningen, H.J. and Colombel, J.F. (2008) The forgotten role of lymphangitis in Crohn’s disease. Gut, 57, 1-4. doi:10.1136/gut.2007.123166
- Jantchou, P., Monnet, E. and Carbonnel, F. (2006) Environmental risk factors in Crohn’s disease and ulcerative colitis (excluding tobacco and appendicectomy). Gastroenterologie Clinique et Biologique, 30, 859-867. doi:10.1016/S0399-8320(06)73333-4
- Fox, J.G., Boutin, S.R., Handt, L.K., Taylor, N.S., Xu, S., Rickman, B., Marini, R.P., Dewhirst, F.E., Paster, B.J., Motzel, S. and Klein, H.J. (2007) Isolation and characterization of a novel helicobacter species, “Helicobacter macacae,” from rhesus monkeys with and without chronic idiopathic colitis. Journal of Clinical Microbiology, 45, 4061-4063. doi:10.1128/JCM.01100-07
- Man, S.M., Zhang, L., Day, A.S., Leach, S. and Mitchell, H. (2008) Detection of enterohepatic and gastric heliconbacter species in fecal specimens of children with Crohn’s disease. Helicobacter, 13, 234-238. doi:10.1111/j.1523-5378.2008.00607.x
- Laharie, D., Asencio, C., Asselineau, J., Bulois, P., Bourreille, A., Moreau, J., Bonjean, P., Lamarque, D., Pariente, A., Soule, J.C., Charachon, A., Coffin, B., Perez, P., Megraud, F. and Zerbib, F. (2009) Association between entero-hepatic Helicobacter species and Crohn’s disease: A prospective cross-sectional study. Alimentary Pharmacology & Therapeutics, 30, 283-293. doi:10.1111/j.1365-2036.2009.04034.x
- Tankovic, J., Smati, M., Lamarque, D. and Delchier, J.C. (2011) First detection of Helicobacter canis in chronic duodenal ulcerations from a patient with Crohn’s disease. Inflammatory Bowel Diseases, 17, 1830-1831. doi:10.1002/ibd.21610
- Chaouche-Drider, N., Kaparakis, M., Karrar, A., Fernandez, M.I., Carneiro, L.A., Viala, J., Boneca, I.G., Moran, A.P., Philpott, D.J. and Ferrero, R.L. (2009) A commensal Helicobacter sp. of the rodent intestinal flora activates TLR2 and NOD1 responses in epithelial cells. PLoS One, 4, e5396. doi:10.1371/journal.pone.0005396
- Ge, Z., Feng, Y., Muthupalani, S., Eurell, L.L., Taylor, N.S., Whary, M.T. and Fox, J.G. (2011) Coinfection with enterohepatic Helicobacter species can ameliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 mice. Infection and Immunity, 79, 3861-3871. doi:10.1128/IAI.05357-11
- Dijkstra, G., Yuvaraj, S., Jiang, H.Q., Bun, J.C., Moshage, H., Kushnir, N., Peppelenbosch, M.P., Cebra, J.J. and Bos, N.A. (2007) Early bacterial dependent induction of inducible nitric oxide synthase (iNOS) in epithelial cells upon transfer of CD45RB (high) CD4(+) T cells in a model for experimental colitis. Inflammatory Bowel Diseases, 13, 1467-1474. doi:10.1002/ibd.20262
- Jiang, H.Q., Kushnir, N., Thurnheer, M.C., Bos, N.A. and Cebra, J.J. (2002) Monoassociation of SCID mice with Helicobacter muridarum, but not four other enterics, provokes IBD upon receipt of T cells. Gastroenterology, 122, 1346-1354. doi:10.1053/gast.2002.32959
- Testerman, T.L., McGee, D.J. and Mobley, H.L. (2001) Helicobacter pylori growth and urease detection in the chemically defined medium Ham’s F-12 nutrient mixture. Journal of Clinical Microbiology, 39, 3842-3850. doi:10.1128/JCM.39.11.3842-3850.2001
- Riley, L.K., Franklin, C.L., Hook Jr., R.R., and BeschWilliford, C. (1996) Identification of murine helicobacters by PCR and restriction enzyme analyses. Journal of Clinical Microbiology, 34, 942-946.
- Solnick, J.V. and Schauer, D.B. (2001) Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clinical Microbiology Reviews, 14, 59-97. doi:10.1128/CMR.14.1.59-97.2001
- Ganta, V.C., Cromer, W., Mills, G.L., Traylor, J., Jennings, M., Daley, S., Clark, B., Mathis, J.M., Bernas, M., Boktor, M., Jordan, P., Witte, M. and Alexander, J.S. (2010) Angiopoietin-2 in experimental colitis. Inflammatory Bowel Diseases, 16, 1029-1039. doi:10.1002/ibd.21150
- Cooper, H.S., Murthy, S.N., Shah, R.S. and Sedergran, D.J. (1993) Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Laboratory Investigation, 69, 238-249.
- Sasaki, M., Bharwani, S., Jordan, P., Elrod, J.W., Grisham, M.B., Jackson, T.H., Lefer, D.J. and Alexander, J.S. (2003) Increased disease activity in eNOS-deficient mice in experimental colitis. Free Radical Biology & Medicine, 35, 1679-1687. doi:10.1016/j.freeradbiomed.2003.09.016
- Dieleman, L.A., Palmen, M.J., Akol, H., Bloemena, E., Pena, A.S., Meuwissen, S.G. and Van Rees, E.P. (1998) Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clinical & Experimental Immunology, 114, 385-391. doi:10.1046/j.1365-2249.1998.00728.x
- Murikinati, S., Juttler, E., Keinert, T., Ridder, D.A., Muhammad, S., Waibler, Z., Ledent, C., Zimmer, A., Kalinke, U. and Schwaninger, M. (2010) Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB, 24, 788-798. doi:10.1096/fj.09-141275
- Suzuki, H., Suzuki, M., Imaeda, H. and Hibi, T. (2009) Helicobacter pylori and microcirculation. Microcirculation, 16, 547-558. doi:10.1080/10739680902949953
- Kalia, N., Jacob, S., Brown, N.J., Reed, M.W., Morton, D. and Bardhan, K.D. (1997) Studies on the gastric mucosal microcirculation. 2. Helicobacter pylori water soluble extracts induce platelet aggregation in the gastric mucosal microcirculation in vivo. Gut, 41, 748-752. doi:10.1136/gut.41.6.748
- Kalia, N., Bardhan, K.D., Atherton, J.C. and Brown, N.J. (2002) Toxigenic Helicobacter pylori induces changes in the gastric mucosal microcirculation in rats. Gut, 51, 641- 647. doi:10.1136/gut.51.5.641
- Kalia, N., Bardhan, K.D., Reed, M.W., Jacob, S. and Brown, N.J. (2000) Mechanisms of Helicobacter pyloriinduced rat gastric mucosal microcirculatory disturbances in vivo. Digestive Diseases and Sciences, 45, 763- 772. doi:10.1023/A:1005456029396
- Kurose, I., Granger, D.N., Evans Jr., D.J., Evans, D.G., Graham, D.Y., Miyasaka, M., Anderson, D.C., Wolf, R.E., Cepinskas, G. and Kvietys, P.R. (1994) Helicobacter pylori-induced microvascular protein leakage in rats: Role of neutrophils, mast cells, and platelets. Gastroenterology, 107, 70-79.
- Kalia, N., Bardhan, K.D., Reed, M.W., Jacob, S. and Brown, N.J. (2000) Effects of chronic administration of Helicobacter pylori extracts on rat gastric mucosal microcirculation in vivo. Digestive Diseases and Sciences, 45, 1343-1351. doi:10.1023/A:1005504019868
- Atuma, C., Engstrand, L. and Holm, L. (1999) Helicobacter pylori extracts reduce gastric mucosal blood flow by a nitric oxide-independent but mast celland plateletactivating factor receptor-dependent pathway in rats. Scandinavian Journal of Gastroenterology, 34, 1183- 1189. doi:10.1080/003655299750024689
- Fox, J.G., Ge, Z., Whary, M.T., Erdman, S.E. and Horwitz, B.H. (2011) Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunology, 4, 22-30. doi:10.1038/mi.2010.61
- Brzozowski, T., Konturek, P.C., Sliwowski, Z., Drozdowicz, D., Pajdo, R., Stachura, J., Hahn, E.G. and Konturek, S.J. (1997) Lipopolysaccharide of Helicobacter pylori protects gastric mucosa via generation of nitric oxide. Journal of Physiology and Pharmacology, 48, 699-717.
- Whittle, B.J., Morschl, E., Pozsar, J., Moran, A.P. and Laszlo, F. (2001) Helicobacter pylori lipopolysaccharide provokes iNOS-mediated acute systemic microvascular inflammatory responses in rat cardiac, hepatic, renal and pulmonary tissues. Journal of Physiology, Paris, 95, 257- 259.
- Pellicano, R., Mazzarello, M.G., Morelloni, S., Allegri, M., Arena, V., Ferrari, M., Rizzetto, M. and Ponzetto, A. (1999) Acute myocardial infarction and Helicobacter pylori seropositivity. International Journal of Clinical and Laboratory Research, 29, 141-144. doi:10.1007/s005990050080
- Prasad, A., Zhu, J., Halcox, J.P., Waclawiw, M.A., Epstein, S.E. and Quyyumi, AA. (2002) Predisposition to atherosclerosis by infections: Role of endothelial dysfunction. Circulation, 106, 184-190. doi:10.1161/01.CIR.0000021125.83697.21
- Luther, J., Owyang, S.Y., Takeuchi, T., Cole, T.S., Zhang, M., Liu, M., Erb-Downward, J., Rubenstein, J.H., Chen, C.C., Pierzchala, A.V., Paul, J.A. and Kao, J.Y. (2011) Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut, 60, 1479-1486. doi:10.1136/gut.2010.220087
- Katakura, K., Lee, J., Rachmilewitz, D., Li, G., Eckmann, L. and Raz, E. (2005) Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. The Journal of Clinical Investigation, 115, 695-702.
- Lee, J., Gonzales-Navajas, J.M. and Raz, E. (2008) The “polarizing-tolerizing” mechanism of intestinal epithetlium: Its relevance to colonic homeostasis. Seminars in Immunopathology, 30, 3-9. doi:10.1007/s00281-007-0099-7
- Vijay-Kumar, M., Wu, H., Aitken, J., Kolachala, V.L., Neish, A.S., Sitaraman, S.V. and Gewirtz, A.T. (2007) Activation of toll-like receptor 3 protects against DSSinduced acute colitis. Inflammatory Bowel Diseases, 13, 856-864. doi:10.1002/ibd.20142
- Kaakoush, N.O., Holmes, J., Octavia, S., Man, S.M., Zhang, L., Castano-Rodriguez, N., Day, A.S., Leach, S.T., Lemberg, D.A., Dutt, S., Stormon, M., O’Loughlin, E.V., Magoffin, A. and Mitchell, H. (2010) Detection of Helicobacteraceae in intestinal biopsies of children with Crohn’s disease. Helicobacter, 15, 549-557. doi:10.1111/j.1523-5378.2010.00792.x
- Yoshizawa, N., Takenaka, Y., Yamaguchi, H., Tetsuya, T., Tanaka, H., Tatematsu, M., Nomura, S., Goldenring, J.R. and Kaminishi, M. (2007) Emergence of spasmolytic polypeptide-expressing metaplasia in Mongolian gerbils infected with Helicobacter pylori. Laboratory Investigation, 87, 1265-1276. doi:10.1038/labinvest.3700682
- Bui, H.X., Rosario, A.D., Sonbati, H., Lee, C.Y., George, M. and Ross, J.S. (1991) Helicobacter pylori affects the quality of experimental gastric ulcer healing in a new animal model. Experimental and Molecular Pathology, 55, 261-268. doi:10.1016/0014-4800(91)90006-J
- Sugimoto, M., Ohno, T. and Yamaoka, Y. (2011) Expression of angiotensin II type 1 and type 2 receptor mRNAs in the gastric mucosa of Helicobacter pylori-infected Mongolian gerbils. Journal of Gastroenterology, 46, 1177-1186. doi:10.1007/s00535-011-0433-7
- Burnett-Hartman, A.N., Newcomb, P.A. and Potter, J.D. (2008) Infectious agents and colorectal cancer: A review of Helicobacter pylori, Streptococcus bovis, JC virus, and human papillomavirus. Cancer Epidemiology, Biomarkers & Prevention, 17, 2970-2979. doi:10.1158/1055-9965.EPI-08-0571
ABBREVIATIONS
Dextran Sodium Sulfate (DSS), Inflammatory Bowel Disease (IBD), Myeloperoxidase (MPO), Disease Activity Index (DAI), Control Uninfected Mice (CU), Control H. muridarum-infected Mice (MU), DSS-treated Uninfected Mice (CD), and DSS H. muridarum-infected Mice (MD)