Open Journal of Gastroenterology
Vol. 2 No. 3 (2012) , Article ID: 22037 , 4 pages DOI:10.4236/ojgas.2012.23022
Young patient with juvenile polyposis syndrome: A case report
![]()
1Gastroenterology Department, Fez University Hospital, Fez, Morocco
2Pathology Department, Fez University Hospital, Fez, Morocco
Email: *elyousfimounia2005@yahoo.fr
Received 29 May 2012; revised 3 July 2012; accepted 18 July 2012
Keywords: Hamartomatous Polyp; Polyposis; Juvenile Polyp; Juvenile Polyposis
ABSTRACT
A case is reported here of juvenile polyposis in a 15- year-old girl. She was diagnosed upon the assessment of the dysenteric syndrome. Coloscopy and gastroscopy had shown polyps of variable size whose histological study confirmed the hamartomatous character typical of the youthful non adenomatous polyps free of dysplasia. There was no localization in the small bowel. Coloprotectomy with ileoanal anastomosis was carried out in view of the importance of diarrhoea and the impacts on the general state of health.
1. INTRODUCTION
Juvenile polyposis syndrome (JPS) is a rare autosomal dominant entity in which the patient develops multiple juvenile polyps. An estimate for prevalence in the population is 1 in 100,000 individuals. JPS patients are usually diagnosed by age 30, but the mean age of presentation is 9.5 years. The diagnosis is reached on the basis of the pathological features of the polyps. The most serious complication later in life is colorectal cancer, and the average age for the diagnosis of cancer is 34 years.
The present report concerns a new case of JPS in a young girl patient.
2. CASE PRESENTATION
The case reported here concerns a young girl 15 years of age, born of a nonconsanguineous marriage, the eldest in a family of three children. One year earlier, she presented dysentery with bloody mucoid diarrhoea, sometimes purulent, accompanied by significant weight of loss. Clinical examination revealed that the patient was in poor general health, showing arachnodactyly, clinodactyly of the fifth finger, digital hippocratism and retrognathia (Figures 1(a)-(d)). She also presented with failure to thrive estimated at least 2 standard deviations. Abdominal examination objectified diffuse abdominal sensitivity. Digital rectal examination showed irregular mucosa and allowed to identify several polypoid formations. Laboratory tests showed malabsorption syndrome and hypochromic microcytic anemia of 9 g/dl. Colonoscopy showed the presence of an aspect of polyposis from rectum to caecum, due to multiple polyps of variable size (5 - 10 mm) and irregular surface, which were biopsied (Figure 2). Upper gastrointestinal endoscopy (Figure 3) showed polyposis in fundus and bulbe, biopsies were performed. The papilla was normal. CT enteroclysis (Figure 4) showed a feature of gastric and colonic polyposis in respect of the small bowel. A capsule endoscopy was indicated but not performed lack of means to the patient. Histological study indicated the presence of typical hamartomatous nonadenomatous polyps of juvenile polyposis free of dysplasia (Figures 5 and 6). In view of dysentery and the significant long-term risk of degradation of these polyps, total coloprotectomy with ileoanal anastomosis on reservoir was performed to the patient. Histological examination showed typical hamartomatous polyps of juvenile polyposis without adenomatous contingent and free of dysplasia. An oncogenetic consultation was made with DNA blood sampling; the search for constitutional mutations proved negative. In case of such a presence, a screening of the siblings shall be considered. The patient shall be submitted to endoscopic gastric control on a regular basis adjusted following molecular study.
3. DISCUSSION
JPS is a rare autosomal dominant hereditary disorder characterized by multiple hamartomatous polyps that invade all the gastrointestinal tract, in the colon and rectum (98%), stomach (13.6%), duodenum (2.3%), jejunum and ileum (6%) in order of frequency[1,2]. The frequency of this syndrome is approximately one per 100,000 births. The average of diagnosis is 9 years. It is often established after gastrointestinal exploration for rectal bleeding. Iron
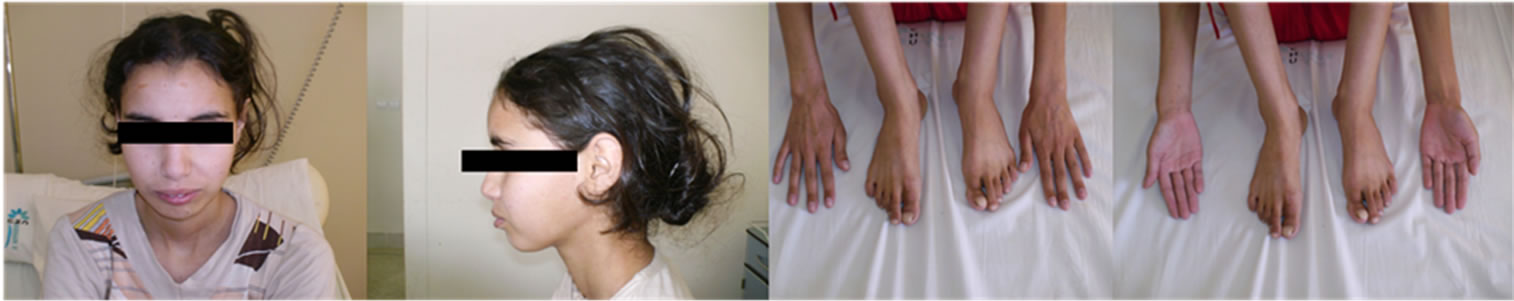 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 1. (a)-(d): arachnodactyly, clinodactyly of fifth finger, digital hippocratism and retrognathia.
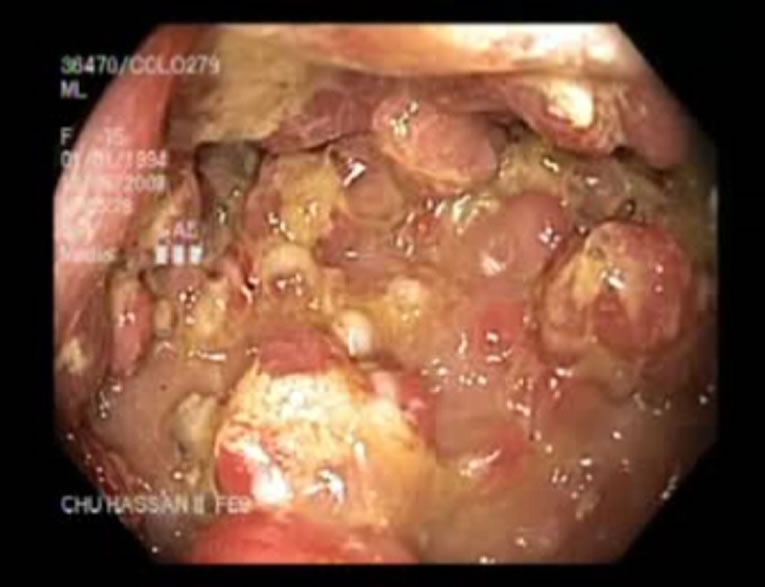
Figure 2. Colonic polyps.
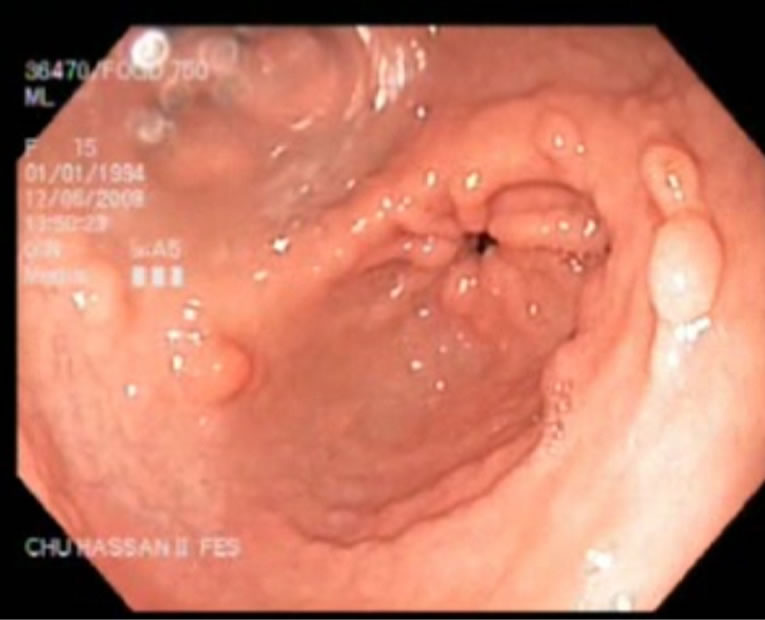
Figure 3. Polyposis in antrum.
deficiency, chronic diarrhea or protein-losing enteropathy can be revealing [3].
Our patient had other abnormalities including arachnodactyly, clinodactyly of the fifth finger clubbed fingers and retrognathia. in fact, patients with juvenile polyposis can have many extra-digestive abnormalities [3]: cutaneous (telangiectasia, nevus, alopecia), bone (hypertelorism, bone cysts, congenital cleft lip and palate), brain
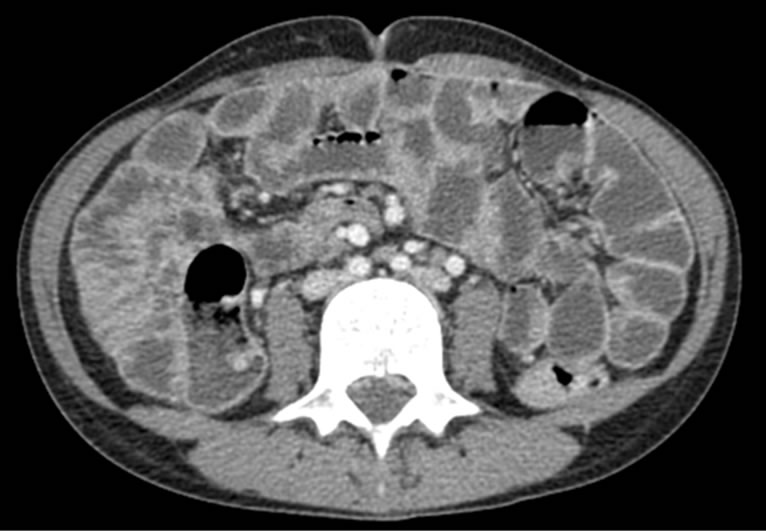
Figure 4. CT-enteroclysis: normal aspect of small bowel.
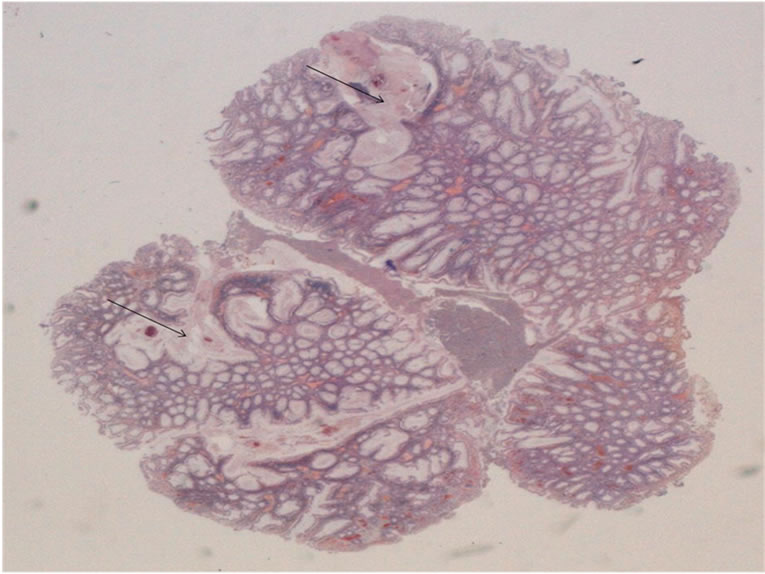
Figure 5. HES × 10: juvenile polyp showing branching and cystic change (arrows).
(hydrocephalus, macrocephaly), cardiovascular (ventricular septal defect or arteriovenous malformation in a RenduOsler), urinary tract as well as mental retardation [4].
Polyps have macroscopic and histological aspects which usually correspond to those of solitary juvenile polyps [5]. and are rounded, smooth-surfaced with a short pedicle. Histologically, the lamina propria is abundant, without smooth muscle [5]. These polyps may be the seat of focus of dysplasia and, in some cases, true adenomas were described in their vicinity. This variant, called hereditary
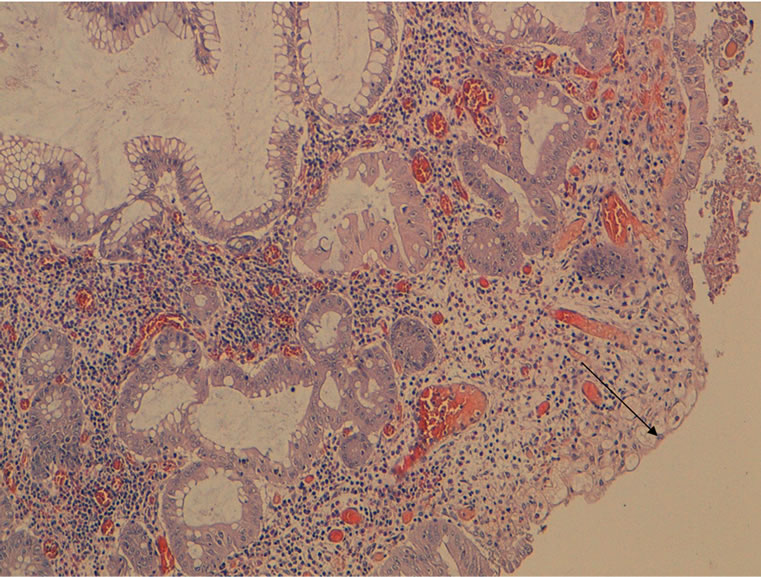
Figure 6. HES × 100: juvenile polyp showing surface ulceration (arrow).
mixed polyposis, is now considered as a distinct juvenile polyposis [6]. Accordingly, we can distinguish the adenomatous polyposes and their phenotypic variants, PeutzJeghers and juvenile polyposis [7,8]. In our patient, histology showed neither a contingent of adenomas nor dysplasia.
Due to its genetic heterogeneity, juvenile polyposis may result from the alteration of different genes. Germline mutations of tumor suppressor gene SMAD4/MADH4, localized on chromosome 18q21.1 [9-11], reported for the first time in 1998, were found in about 20% to 50% of families affected by juvenile polyposis [12,13]. The SMAD4 gene encodes an enzyme involved in the transduction pathway of TGF. Germline mutations inactivate the protein SMAD4, which, most often, is no longer detectable in juvenile polyps.
For many years, hamartomatous polyposes were considered benign. In reality, the risk of cancer in juvenile polyposis was first described in 1966 [14]. This risk is estimated at 50% for the colon and rectum [15,16], 15% for the stomach, 2% for the pancreas and 1% for the duodenum. Given the risk of gastrointestinal cancer associated with juvenile polyposis, molecular diagnostics optimize management at the family level within a genetics consultation. When the causal mutation of SMAD 4 is evidenced in the family, based on one member affected, research may be extended to relatives at risk, especially to siblings and children whose theoretical risk of being carriers is 50%. This analysis allows to record monitoring by suggesting endoscopy only to carrier subjects. A routine screening colonoscopy is recommended from the age of 10 - 12 years in case of family history. The occurrence of symptoms may require colonoscopy before this age. It must be performed every two years until age 40 or beyond if a genetic diagnosis has been established. Gastroscopy is also recommended at diagnosis because of the possible presence of gastric polyps and the risk of gastric or duodenal adenocarcinoma. Gastric endoscopic surveillance every 3 years thereafter is recommended. The impact of small bowel exploration by capsule remains to be defined. When the mutation is not identified, monitoring should be considered in all relatives at high risk [17-19].
No therapeutic management guidance has been codified. It depends on the polyps count and their histological characteristics. Few and non-dysplastic polyps can be treated by endoscopic polypectomy provided that regular monitoring can be ensured. The occurrence of complications, impossibility to remove all polyps, or the presence of a large number of adenomatous polyps or double contingent, are indications that require colectomy with ileorectal anastomosis or coloprotectomy [20]. This is the case of our patient whose significant rectal bleeding made it imperative to perform radical surgery of the colon and rectum.
In other respects, partial or total gastrectomy, depending on the seat of lesions, may also be discussed [20].
In conclusion, the present report presents another case of juvenile polyposis, and highlights the place of surgery in case of hemorrhagic evolution and the impact on the patient’s general condition.
REFERENCES
- Brosens, L.A.A., Langeveld, D., Van Hattem, W.A., Giardiello, F.M. and Offerhaus, G.J.A. (2011) Juvenile polyposis syndrome. World Journal of Gastroenterology, 17, 4839-4844.
- Watanabe, A., Nagashima, H., Motoi, M. and Ogawa, K. (1979) Familial juvenile polyposis of the stomach. Gastroenterology, 77, 148-151.
- Brosens, L.A., van Hattem, W.A., Jansen, M., de Leng, W.W., Giardiello, F.M. and Offerhaus, G.J. (2007) Gastrointestinal polyposis syndromes. Current Molecular Medicine, 7, 29-46. doi:10.2174/156652407779940404
- Gallione, C.J., Repetto, G.M., Legius, E., Rustgi, A.K., Schelley, S.L., Tejpar, S., Mitchell, G., Drouin, E., Westermann, C.J. and Marchuk, D.A. (2004) A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet, 363, 852-859. doi:10.1016/S0140-6736(04)15732-2
- Aaltonen, L.A., Jass, J.R. and Howe, J.R. (2000) Juvenile polyposis. In: Hamilton, S.R. and Aaltonen, L.A., Eds., Pathology and Genetics of Tumours of the Digestive System, IARC Press, Lyon, 130-132.
- Jaeger, E., Leedham, S., Lewis, A., Segditsas, S., Becker, M., Cuadrado, P.R., Davis, H., Kaur, K., Heinimann, K., Howarth, K., East, J., Taylor, J., Thomas, H. and Tomlinson, I. (2012) Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nature Genetics, 44, 699-703. doi:10.1038/ng.2263
- van Hattem, W.A., Langeveld, D., De Leng, W.W., Morsink, F.H., van Diest, P.J., Iacobuzio-Donahue, C.A., Giardiello, F.M., Offerhaus, G.J. and Brosens, L.A. (2011) Histological variations in juvenile polyp phenotype correlate with genetic defect underlying juvenile polyposis. The American Journal of Surgical Pathology, 35, 530- 536. doi:10.1097/PAS.0b013e318211cae1
- Lam-Himlin, D., Park, J.Y., Cornish, T.C., Shi, C. and Montgomery, E. (2010) Morphologic characterization of syndromic gastric polyps. The American Journal of Surgical Pathology, 34, 1656-1662.
- Calva-Cerqueira, D., Chinnathambi, S., Pechman, B., Bair, J., Larsen-Haidle, J. and Howe, J.R. (2009) The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clinical Genetics, 75, 79-85. doi:10.1111/j.1399-0004.2008.01091.x
- Delnatte, C., Sanlaville, D., Mougenot, J.F., Vermeesch, J.R., Houdayer, C., Blois, M.C., Genevieve, D., Goulet, O., Fryns, J.P., Jaubert, F., et al. (2006) Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. The American Journal of Human Genetics, 78, 1066-1074. doi:10.1086/504301
- Menko, F.H., Kneepkens, C.M., de Leeuw, N., Peeters, E.A., Van Maldergem, L., Kamsteeg, E.J., Davidson, R., Rozendaal, L., Lasham, C.A., Peeters-Scholte, C.M., et al. (2008) Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clinical Genetics, 74, 145-154. doi:10.1111/j.1399-0004.2008.01026.x
- van Hattem, W.A., Brosens, L.A., De Leng, W.W., Morsink, F.H., Lens, S., Carvalho, R., Giardiello, F.M. and Offerhaus, G.J. (2008) Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut, 57, 623-627. doi:10.1136/gut.2007.142927
- Aretz, S., Stienen, D., Uhlhaas, S., Stolte, M., Entius, M.M., Loff, S., Back, W., Kaufmann, A., Keller, K.M., Blaas, S.H., et al. (2007) High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. Journal of Medical Genetics, 44, 702-709. doi:10.1136/jmg.2007.052506
- Giardiello, F.M., Hamilton, S.R., Kern, S.E., Offerhaus, G.J., Green, P.A., Celano, P., Krush, A.J. and Booker, S.V. (1991) Colorectal neoplasia in juvenile polyposis or juvenile polyps. Archives of Disease in Childhood, 66, 971-975.
- Brosens, L.A., van Hattem, A., Hylind, L.M., IacobuzioDonahue, C., Romans, K.E., Axilbund, J., Cruz-Correa, M., Tersmette, A.C., Offerhaus, G.J. and Giardiello, F.M. (2007) Risk of colorectal cancer in juvenile polyposis. Gut, 56, 965-967. doi:10.1136/gut.2006.116913
- Howe, J.R., Mitros, F.A. and Summers, R.W. (1998) The risk of gastrointestinal carcinoma in familial juvenile polyposis. Annals of Surgical Oncology, 5, 751-756. doi:10.1007/BF02303487
- Howe, J.R., Mitros, F.A. and Summers, R.W. (1998) The risk of gastrointestinal carcinoma in familial juvenile polyposis. Annals of Surgical Oncology, 5, 751-756. doi:10.1007/BF02303487
- Howe, J.R., Ringold, J.C., Hughes, J.H. and Summers, R.W. (1999) Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery, 126, 162-170. doi:10.1016/S0039-6060(99)70150-9
- Brosens, L.A., van Hattem, W.A., Jansen, M., de Leng, W.W., Giardiello, F.M. and Offerhaus, G.J. (2007) Gastrointestinal polyposis syndromes. Current Molecular Medicine, 7, 29-46. doi:10.2174/156652407779940404
- Oncel, M., Church, J.M., Remzi, F.H. and Fazio, V.W. (2005) Colonic surgery in patients with juvenile polyposis syndrome: A case series. Diseases of the Colon & Rectum, Discussion 55-56, 48, 49-55. doi:10.1007/s10350-004-0749-y
NOTES
*Corresponding author.