Journal of Materials Science and Chemical Engineering
Vol.1 No.7(2013), Article ID:41578,11 pages DOI:10.4236/msce.2013.17003
Characterization and Evaluation of Bond Strength of Dental Polymer Systems Modified with Hydroxyapatite Nanoparticles
1Department of Dentistry, Federal University of Vales do Jequitinhonha and Mucuri, Diamantina, Brazil
2Advanced Biomaterial Center-BioMat, UFVJM, Diamantina, Brazil
3Institute of Science and Technology, UFVJM, Diamantina, Brazil
Email: *vitorcdumont@gmail.com
Copyright © 2013 Vitor César Dumont et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2013 are reserved for SCIRP and the owner of the intellectual property Vitor César Dumont et al. All Copyright © 2013 are guarded by law and by SCIRP as a guardian.
Received November 18, 2103; revised December 18, 2013; accepted December 26, 2013
Keywords: Nanostructures; Composites; Mechanical Properties; Fracture
ABSTRACT
The study Modifies an adhesive system with hydroxyapatite nanoparticles as load, characterizes it and evaluates the effectiveness of its bond to dental structure. The middle thirds of healthy premolar tooth crowns were obtained, and each crown was sectioned vertically, resulting in two sections. The sections were divided into ten groups (n = 15), in which resin composite restorations were simulated: (G1E and G1D) conventional adhesive system (SAC); (G2E and G2D) SAC modified with HAP; (G3E and G3D) Primer modified with HAP; (G4E and G4D) monocomponent adhesive system; (G5E and G5D) self-etching adhesive system. The specimens were submitted to the microshear test and characterization technique. There was statistically significant difference (Kruskal-Wallis) between the groups (p < 0.01). G3 presented the highest bond strength to enamel (64.40 MPa, ±7.36) and dentin (39.59 MPa, ±21.46). The majority of specimens were found adhesive fractures. Bond strength to enamel and dentin of the primer modified with HAP of SAC showed higher values.
1. Introduction
In Dentistry, one desires a true chemical and micromechanical bond of the restorative material to the tooth structure, even under adverse conditions of humidity and thermal variations in the oral cavity [1]. The development of dental adhesive agents has produced a range of composites classified according to the type of tissue conditioning, and the number of steps for their application [2-4] with direct influence on the bond strength to the dental structure [5].
The bond to dentin is more complex than that to enamel, due to its heterogeneous nature, with a larger organic content and water [6]. Dentin is a tissue that requires a wet bonding technique, since it is formed of hydric components with distinct and variable morphology. This characteristic demands adhesive systems with increasingly hydrophilic formulations, in an attempt to improve the mechanical retention of composites to dentin. There are still few reports on the mechanical performance of amphiphilic adhesive systems which, in addition to a mechanical bond, may promote a strong and lasting chemical bond to dentin [7].
After light activation, the approximation of monomers to establish cross links causes a significant reduction in the volume of the polymer composite. The high coefficient of linear thermal expansion generates polymerization shrinkage which creased internal stresses and causes rupture at the bond interface. This is one of the main causes of gap formation at the tooth-resin interface [5-8]. The type of failure that may occur between the hybrid layer and the tooth structure causes considerable clinical consequences, such as bacterial invasion, recurrent caries development, dentinal sensitivity and pulp irritation [9,10].
Over the last few decades, the search for a polymer material with a capacity to bond to dental structures has become one of the main study projects [11]. Various load particles have been added to dental adhesives to strengthen the link of bond strength to the dental structure, diminishing polymerization shrinking and increasing the modulus of elasticity of the adhesive layer [12]. Recently, load nanoparticles [13-16], especially nanoparticles of ceramic materials [15,16], have been used in the formulation of composites, with the purpose of improving physical and mechanical properties.
Of the many synthetic materials studied as substratesupport for the development of new materials, hydroxyapatite [Ca10(PO4)6(OH)2] has been widely used and considered one of the most biocompatible ceramics, due to the similarity to the mineral constituents of human bones and teeth [17]. Hydroxyapatite nanoparticles have been used to increase the mechanical properties of resin composites, observing the bioactivity and bond of these composites to the dental structure [18]. In reports in the literature [19] the incorporation of hydroxyapatite in polymer adhesives has also significantly improved the degree of monomer conversion and polymerization rate. Thus, hydroxyapatite nanoparticles may be a promising option of material for the preparation of new dental adhesives with superior properties.
The aim of this study was to modify a polymer dental adhesive system with hydroxyapatite nanoparticles as load, characterize it by means of light microscopy, scanning electron microscopy, Energy dispersive X-ray spectroscopy, X-ray diffraction and Fourier Transformed infrared spectroscopy technique (ATR-FTIR) and evaluate the effectiveness of its bond to dental structure.
2. Experimental Procedure
The study was submitted to the Research Ethics Committee of the Federal University of Vales do Jequitinhonha e Mucuri, under protocol No.186/10.
2.1. Preparation of Experimental Units
The sample size calculation was made from a pilot study, considering the degree of confidence of the sample of 95% and statistical power of 95% desired and estimated. The tolerable error of sampling was 4% [20], stipulating a sample size of fifteen.
Sixty-six human pre-molar teeth, whole and recently extracted due to orthodontic indication in subjects under the age of 21 years were selected, well cleaned and autoclave sterilized (Cristófoli Vitale, Cristófoli, Brazil) at 120˚C, working pressure of 1 kgf/cm2, for 20 min [21]. Each of the teeth was section in its horizontal direction, using a diamond disc in a metallographic cutter (ELSAW, Elquip, Brazil), thus obtaining the middle third of the tooth crown. Two sections for test were obtained by sectioning each part of the tooth crown in its vertical direction (Figure 1(a)). The sections were embedded in a chlorinated polyvinyl chloride tube (CPVC), (AMANCO, Brazil) 2 cm in diameter and 1.5 cm high, and filled with polyester resin. In half of the embedded sections, enamel surfaces were exposed (Figure 1(b)) and in the other half, dentin surfaces (Figure 1(c)). Initially, the exposed surfaces were polished with 400 grain abrasive paper in a polishing machine and metallographic grinder (PLF-DV, Fortel, Brazil) with constant cooling. After this, 600 grain abrasive paper was used to simulate the smearlayer. The test specimens (tsps) were washed and stored in distilled water at a temperature of 37˚C ± 1˚C in an oven (Fanem, Brazil), and were then randomly distributed into ten groups (n = 15), according to Table 1.
With the exception of Groups G5E and G5D, enamel
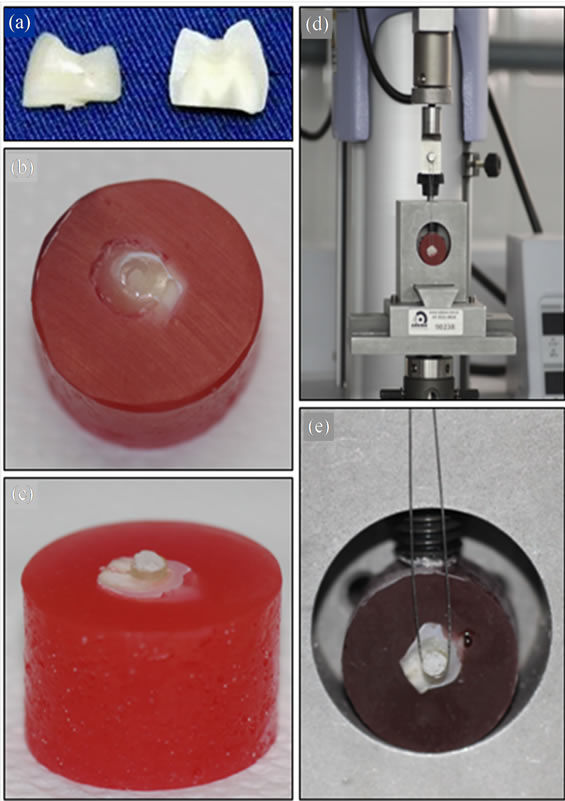
Figure 1. Tooth sections (a) embedded in CPVC, showing exposure of enamel (b) and dentin (c) surfaces, with simulation of resin composite restoration, device used for fixating the tooth-polyester resin sets to the universal test machine for the microtensile test, with orthodontic wire tied to the device and the load cell at the upper extremity of the machine (d) to involve the area closest to the interface of the restoration and dental structure during the test (e).

Table 1. Identification of the study groups and materials used, with their respective commercial brands, manufacturers and organic and inorganic constitution.
*E-Enamel; **D-Dentin.
and dentin surface etching of the other groups was performed with 37% phosphoric acid for 30 and 15 s, respectively, and was followed by washing for the same length of time.
The surfaces of G1E and G1D were dried with absorbent paper. A thin coat of hydrophilic monomer (Table 1) was applied with a microbrush (Vigodent, Brazil), restrictedly on the etched surfaces, which were softly dried for 5 s to allow complete solvent evaporation. Two consecutive coats of hydrophobic monomer (Table 1) were applied with a microbrush, without excess, and submitted to a light jet of air to favor flow over the dental structure. The hydrophobic monomer was light activated for 10 s. After this a metal matrix (1 mm in diameter and 2 mm deep) was placed over the enamel and dentin surfaces of the tsps, in which restorations were simulated (Figures 1(b) and (c)) with nanoparticulate resin composite FILTEK Z350 (3M Espe, Brazil) (Table 1) and light activated for 40 s.
The nanoparticles were obtained by means of hydroxyapatite synthesis by the wet process based on the precipitation route, with calcium hydroxide and orthophosphoric acid as precursors, with a molar Ca/P ratio fixed at 1.67 [22]. The HAP nanoparticles were weighed on an analytical scale (Al 204, Mettler Toledo, Brazil) and mixed with the hydrophobic and hydrophilic monomers at a concentration of 0.10 g/ml, using a magnetic agitator (ARE, Velp Scientifica, Brazil), at a speed of 360 rpm for 30 s.
In Groups G2E and G2D and in G3E and G3D the same procedures were performed as for the previously mentioned groups, however, using the hydrophobic and hydrophilic monomers, modified with HAP, respectively.
In G4E and G4D two consecutive coats of monocomponent adhesive system (Table 1) were applied with a microbrush on the etched enamel and dentin surfaces. The adhesive system was gently dried for 5 s and light activated for 10 s.
On the etched surfaces of G5E and G5D the excess water was removed with absorbent paper. One coat of self-etching adhesive system (Table 1) was applied with a microbrush, restrictedly on the surfaces, actively for 20 s. After this, the adhesive was dried with a jet of air at high pressure for 5 s, for complete solvent evaporation. The adhesive was light activated for 10 s.
Nanoparticulate composite resin Z350 was used to perform the restorations in all the other groups, in the same way as performed in G1E and G1D. The materials were used strictly in accordance with all the respective manufacturers’ instructions. The materials were light activated using a light emitting diode appliance (LED) Optilight LD Max (Gnatus, Brazil) at the interval of 800 mW/cm², monitored by a radiometer.
The tsps were stored in distilled water at 37˚C (±1˚C) for 14 days.
2.2. Bond Strength Test
After drying at ambient temperature for 12 h, the tsps were submitted to the microshear bond strength test in a Universal test machine EZ Test (Shimadzu, Japan) with a load cell of 200 Kgf at a speed of 0.5 mm/min to measure the bond strength of the materials. For this test, a circular metal device was used to hold the specimen and fixate the set. An orthodontic wire 2 mm thick was tied to a fixed base on the load cell placed at the top part of the test machine (Figure 1(d)), involving the area closest to the base of the restoration (Figure 1(e)), which was subjected to traction until rupture. The bond strength was calculated automatically by the software program (Trapezium, version 1.1.5, Shimadzu, Japan) of the equipment, which is generally measured with the apparent failure load divided by the surface area. The results obtained were transformed into MPa.
2.3. Fracture Pattern Analysis
After analysis of the fracture areas of all the tsps, by means of light microscopy (LM) under a stereomicroscope (Stemi 2000C, Carl Zeiss, Canada), at 15X magnification, the type of failure that occurred at the tooth/ adhesive system/restoration interface was classified according to Fowler [23], with modifications: 1 - adhesive, fracture of the adhesive system, hydrophobic monomer present in the restorative resin composite or in the tooth structure or in both; 2 - cohesive in resin, fracture in the restorative resin composite, resin present on both sides of the test specimen; 3 - cohesive on dental structure, fracture in enamel or dentin, dental tissue present on one of the two remaining sides of the test specimen; 4 - mixed, presence of two or more types of fractures described above.
2.4. Characterization of Materials
2.4.1. Scanning Electron Microscopy and X-Ray Dispersive Energy Spectroscopy
The fracture interfaces representative of each group were sputter-coated with goldpalladium, and analyzed by scanning electron microscopy (SEM), in equipment CS- 3500 (Shimadzu, Japan). Qualitative element analysis of the tsps was performed by energy dispersive X-Ray spectroscopy (EDX), in a spectrophotometer CS3200 (Oxford, England).
2.4.2. X-Ray Diffraction
The composites developed were submitted to X-ray diffraction (DRX) in a diffractometer RXD6000 (Shimadzu, Japan), with monochromatic KαCu radiation (1.5406 A˚) and operational tube with a voltage of 40 kV and current of 30 mA.
2.4.3. Fourier Transform Infrared Spectroscopy
The samples of the composites developed were analyzed by means of Fourier Transform Infrared Spectroscopy (FTIR), by the attenuated total reflectance technique (ATR), (Nicolet 6700, Thermo Electron, USA). Spectra were obtained with 32 scans in the interval between 675 and 4000 cm−1, with a resolution of 4 cm−1 and units of absorbance (abs).
2.5. Statistical Analysis
After performing the normality test (Shapiro-Wilk), a non parametric statistical test (Kruskal-Wallis) was applied to verify differences between the groups, And after this, the Mann-Whitney test to verify the intragroup differences, using the software program Statistical Package for Social Sciences (SPSS for Windows, version 17.0, SPSS Inc., USA).
To evaluate the type of failure that occurred between the different groups, descriptive data analysis was applied, represented by means of graphs.
3. Results
The statistical results showed that there was significant difference between the groups (p < 0.05), as regards bond strength after mechanical testing in enamel and dentin (Table 2).
After microanalysis under stereomicroscope, adhesive fractures were observed in the majority of the specimens on both tooth surfaces. A larger number of cohesive
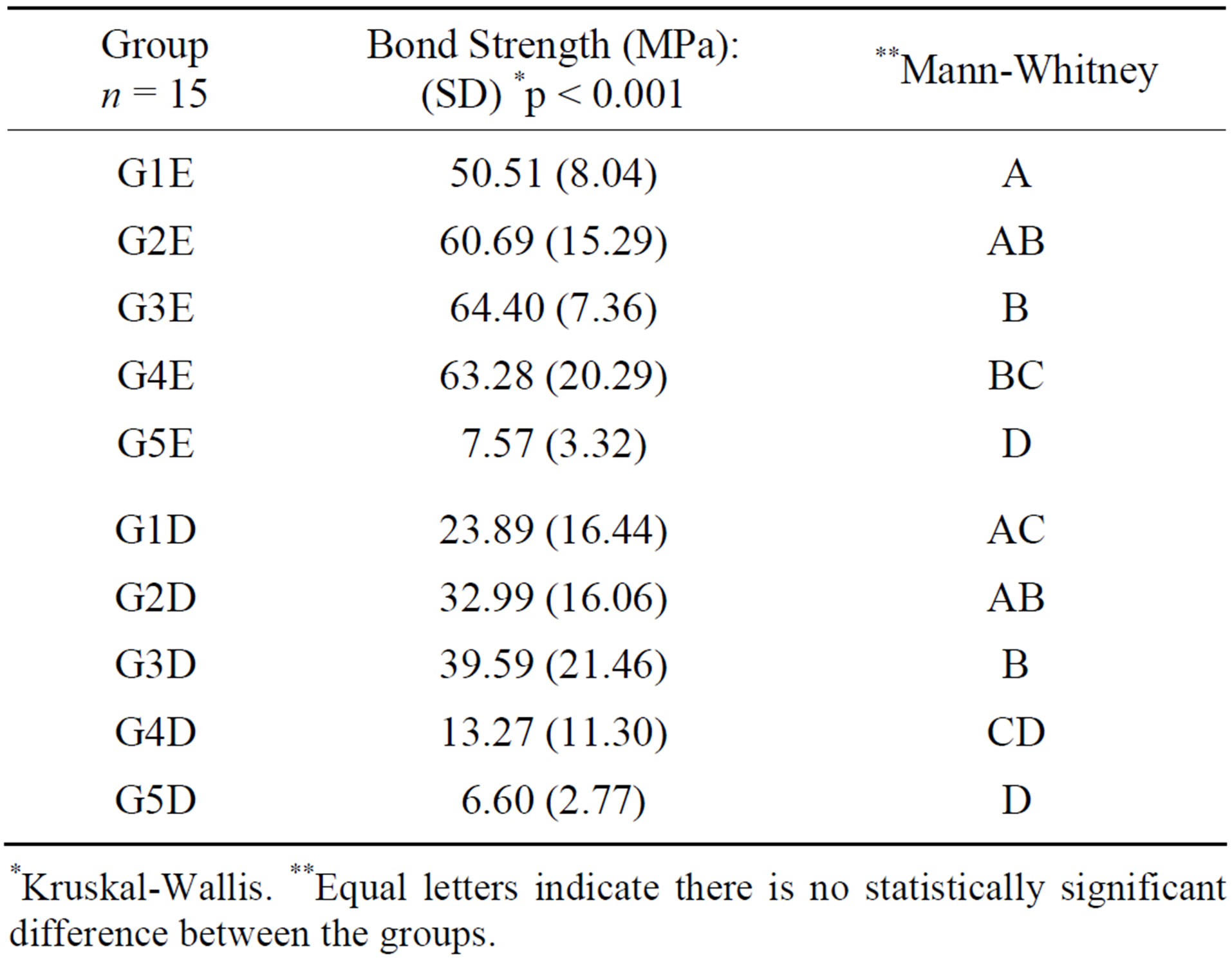
Table 2. Values of bond strength to enamel and dentin, in the groups of materials studied, and respective statistical analyses.
*Kruskal-Wallis. **Equal letters indicate there is no statistically significant difference between the groups.
fractures in enamel were observed in the groups with the adhesive systems modified with HAP (G2E and G3E). In the monocomponent adhesive system (G4E), in addition to the presence of cohesive fractures in enamel, mixed fractures were also observed. Cohesive fractures in the dentinal structure were observed only in Group G3D (Figure 2).
The SEM micrographs of samples representative of the specimens analyzed by LM showed a thin layer of adhesive on the enamel (Figure 3(a)) and dentin surfaces (Figure 3(b)) in the majority of fracture areas of the dental remainders, suggesting the adhesive type of fracture. Spectra of EDS of these surfaces in enamel (Figure 3(a)) and dentin surfaces (Figure 3(b)) showed high peaks of Calcium (Ca) and Phosphorous (P) corresponding to the dental structure, and peaks of low intensity of Silicon (Si) corresponding to the inorganic load of the monocomponent adhesive system. Cohesive fractures in enamel (Figure 3(c)), dentin (Figure 3(d)), restorative resin (Figure 3(e)) and mixed fractures (Figure 3(f)) were also observed at the interfaces. The spectra of EDS of cohesive fractures in enamel (Figure 3(c)), and in dentin (Figure 3(d)) presented peaks of high intensity of Ca and P, while the spectra of EDS of cohesive fracture in the restorative resin (Figure 3(e)) showed peaks of high intensity of Si and Zirconia (Zr) and low intensity of Ca. Mixed fracture in enamel (Figure 3(f)) presented spectra with peaks of Ca, P, Zr and Si. Peaks of Gold and Palladium from the sputter-coating of the materials could also be visualized; however, they were not identified in the spectra.
The crystallography of the HAP nanoparticles was analyzed by the spectra generated by DRX. The information obtained was compared with standardized records from the database of the International Center for Diffraction (JCPDS—International Centre for Diffraction Data, USA) for the calcium phosphate materials, and it was possible to identify the material as being hydroxyapatite. In the qualitative DRX analysis of the HAP nanoparticles, a high degree of crystallinity was observed. In the diffractograms in Figure 4(a), the peaks of higher intensity of the Ca10(PO4)6(OH)2 phase, characteristic of HAP may be visualized, and the presence of a large quantity of amorphous phase on the adhesive of the conventional adhesive system (G1) and in the adhesive with the addition of HAP (G2). In the diffractograms in Figure 4(b) HAP is shown with the presence of crystalline peaks, hydrophobic monomer of the conventional adhesive system with characteristics of amorphous material (G1) and the hydrophilic monomer modified with HAP (G2) with an amorphous phase and peaks characteristic of HAP. A small displacement of HAP peaks (a) may be observed in the diffractograms of Group G3 (Figure 4(b)).
In the FTIR spectra (Figure 5) it was observed that the absorption bands with reference to the 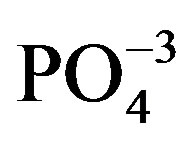 ions of HAP appeared in the spectra in the vibrational mode of stretching (ѵ3) and were evident at 1088, 1039 and 1032 cm−1; the band with reference to
ions of HAP appeared in the spectra in the vibrational mode of stretching (ѵ3) and were evident at 1088, 1039 and 1032 cm−1; the band with reference to 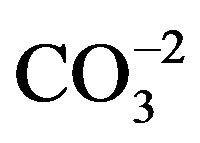 was shown at 864 cm−1. The presence of a narrow band at 3594 cm−1 was observed in the region corresponding to the vibrations of stretching of structural OH-ions. The FTIR spectra of the hydrophobic monomer of Group G1 presented absorption bands with reference to Bis-GMA at 1250, 1297, 1442, 1507, 1635, 1721 and 2962 cm−1 and HEMA at 1639 cm−1 corresponding to the methacrylate group (C=C), 1721 cm−1 to C=O and 1182 cm−1 to C-O. The spectra of the hydrophobic monomer modified with HAP (G2) showed absorption bands similar to those of
was shown at 864 cm−1. The presence of a narrow band at 3594 cm−1 was observed in the region corresponding to the vibrations of stretching of structural OH-ions. The FTIR spectra of the hydrophobic monomer of Group G1 presented absorption bands with reference to Bis-GMA at 1250, 1297, 1442, 1507, 1635, 1721 and 2962 cm−1 and HEMA at 1639 cm−1 corresponding to the methacrylate group (C=C), 1721 cm−1 to C=O and 1182 cm−1 to C-O. The spectra of the hydrophobic monomer modified with HAP (G2) showed absorption bands similar to those of
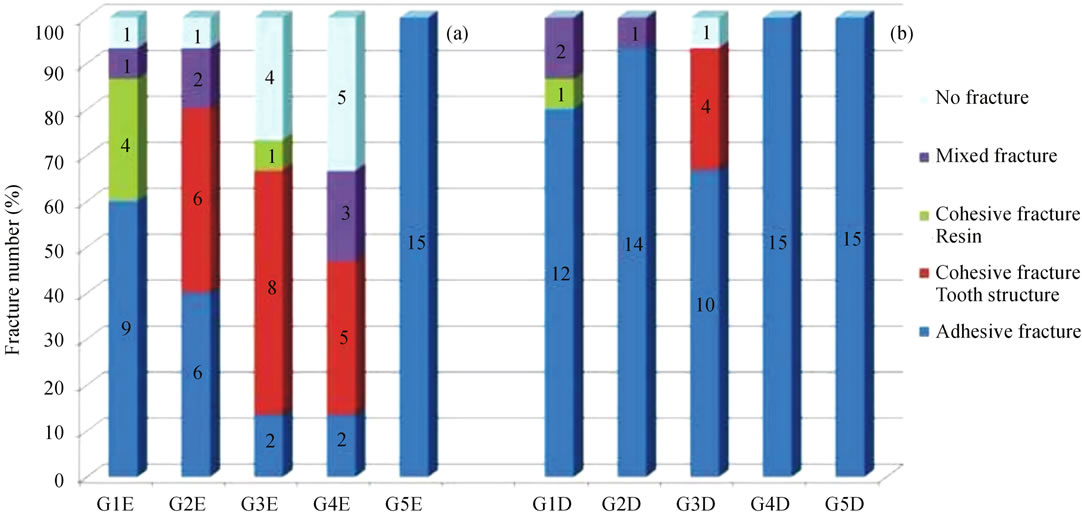
Figure 2. Graphic representation of the number of fractures (%) of the specimens in the Enamel (E) and Dentin (D) groups, after microanalysis by LM.

Figure 3. SEM Micrographs and respective EDX spectra of samples representative of the experimental groups, showing adhesive fracture areas in enamel: G2 (a) and in dentin: G3 (b); cohesive fracture areas in enamel: G2 (c) and in dentin: G3 (b); cohesive fracture areas in resin: G1 (e); and mixed fracture: G4 (f).
the adhesive in G1, with little intensity of absorption of the phosphate bands of HAP. The hydrophilic monomers of G1, which contain HEMA, presented the same bands as previously described. In G3, the hydrophilic monomer modified with HAP showed evident bands of HAP, free OH groups at 3243 cm−1 and wide absorption bands from 3374 cm−1 corresponding to the OH groups of HAP and HEMA.
4. Discussion
Premolar teeth extracted from individuals under the age of 21 years were used with the purpose of avoiding the interference of variables in the bond strength values. Physiological alterations resulting from aging of the dentinal tissue increase the degree of dentin mineralization, increasing its thickness and reducing its permeability [24].
The insertion of inorganic particles promoted improvements in the mechanical properties of the polymers, considering that the composites modified with HAP nanoparticles (G2 and G3) presented the highest bond strength values in comparison with the precursor material G1, to both dental surfaces. As the volumetric fraction of the inorganic matrix increases, the link of bond force is strengthened, because the diminishment of the organic portion minimize the polymerization shrinkage and increases the modulus of elasticity of the adhesive layer [11,25].
The lower bond strength values found in G2E and G2D in comparison with G3E and G3D, respectively, may be associated with the low conversion of monomers during the light activation process; that is to say, it is determined by the percentage of double carbon bonds (C=O) which were broken and transformed into simple bonds [26]. The result of this process is directly connected to the behavior of adhesive bonds to dental substrate [26]. The incorporation of HAP nanoparticles into the hydrophobic monomer may have made the interac-

Figure 4. Diffractograms with peaks of greater intensity of HAP, of the hydrophobic monomer of the conventional adhesive system (G1) and of the hydrophobic monomer modified with HAP nanoparticles (G2) (a); diffractograms with peaks of greater intensity of HAP, of the hydrophilic monomer of the conventional adhesive system (G1) and of the hydrophilic monomer modified with HAP nanoparticles (G3) (b), showing peaks characteristic of HAP (a).

Figure 5. Functional Groups of HEMA and Bis-GMA molecules and of the phosphate group of HAP (a); FTIR spectra with the absorption bands in the infrared with reference to the chemical bonds of the functional groups of the composition of HAP, of the hydrophobic monomer of the conventional adhesive (G1), of the hydrophobic monomer modified with HAP nanoparticles (G2), of the hydrophilic monomer of the conventional adhesive system (G1) and of the hydrophilic monomer modified with HAP nanoparticles (G3) (b); FTIR spectra with the absorption bands in the infrared with references to the chemical bonds of the functional groups of the composition of HAP, of the hydrophobic monomer of the adhesive (G1) and of the hydrophobic monomer of the adhesive modified with HAP nanoparticles (G2) (c); FTIR spectra with absorption bands in the infrared with reference to chemical bonds of the functional groups of the composition of HAP, of the hydrophilic monomer of the adhesive (G1) and of the hydrophilic monomer of the adhesive modified with HAP nanoparticles (G3) (d).
tion of these particles difficult by means of dipole-dipole type bonds to the dental structure, and also enabled a greater formation of HAP agglomerates. These, in turn, may have altered the rate of refraction of the wave bands of the light activator appliances to the adjacent layers, altering the degree of polymerization and diminishing the formation of cross links of these polymer systems, generating polymers with a larger quantity of free monomers, and being more susceptible to degradation. Other significant factors were the possibility of agglomerates of HAP nanoparticles having obliterated the enamel prisms and dentinal tubules, difficulty of flowing of the adhesive modified with HAP, and the reduction in the formation of resin tags, responsible for mechanical retention.
The hydrophilic monomer may have acted as a lining of the inorganic load, enabling an improvement in the bond at the HAP nanoparticle interface with the resin matrix of the hydrophobic monomer [27]. The sealing of the HAP surface increases its dispersion and minimizes the formation of inorganic aggregates [27], also making it possible for greater cohesion of these particles with the dental structure to occur.
The lowest bond strength value was found in G4D in comparison with G1D, G2D and G3D, while G4E presented the second highest bond strength value. One of the explanations for this occurrence is the greater complexity of the adhesive bond to dentin [28]. Another probable explanation is related to demineralized dentin drying. The collapse of collagen fibrils and establishment of hydrogen bridges between them may occur, contracting and tensing the collagen network, and leading to the loss of the tridimensional spatial configuration. Thus, thus the permeability of this network is diminished, which hinders the infiltration of the adhesive monomers, leading to the formation of a thinner hybrid layer with deficient sealing of the dentinal tubules [29,30].
The monomer/solvent combination in adhesive systems with their application performed in a larger number of steps must be capable of breaking the interpeptide hydrogen bridges, and thus re-expand the dentin matrix, re-creating the interfibrillar spaces of the collapsed collagen network to allow infiltration of the hydrophobic monomer [29,30].
The bond strength, measured by means of the microshear test, varied with the treatment of the exposed tooth surface. Inferior behavior was observed for Groups G5E and G5D, in which the self-etching adhesive system was used, and which removes a micrometric layer of dental surface [31-33]. Greater unobstruction of enamel and dentinal tubule prisms was verified with the phosphoric acid etching process, [34] as was used in the other experimental groups, allowing better flow of the hydrophobic monomer, with more voluminous and deeper tag formation [32-34].
Analysis of the fracture surfaces of the tsps by LM identified a larger number of adhesive failures, which may be justified by the better stress distribution at the bond interface of small specimens demanded in the microshear test [34]. In Groups G2E, G3E and G4E a larger number of cohesive fractures in the dental structure were observed. This is justified by the higher bond strength values in the mentioned groups, with emphasis on G2E and G3E, in which the HAP nanoparticles may have interacted with the hydroxyapatite of enamel tissue, strengthening the bond even further.
In Groups G3E and G4E there were a significant number of specimens whose bond strength value was higher than the rupture strength of the orthodontic wire used in the microshear test, preventing their fracture. This may be associated with the presence of HAP nanoparticles in G3E and silicon in G4E, promoting greater bond strength.
The capacity of HAP nanoparticles to bond to the dentinal structure is potentiated when mixed with the hydrophilic monomer, and this may justify the occurrence of cohesive fractures in dentin in Group G3D. The relationship between carbonate and apatite is of great importance, since carbonate increases the chemical reactivity of apatite, particularly by the increase in the product of solubility and dissolution rate [35].
The clinical consequences of failures that may occur at the tooth/adhesive system interface depend on where they occur at the bond interface. Only if the failure occurs between the base of the hybrid layer and the subjacent dentin can it enable the demineralization of the sealed dentin, leading to bacterial invasion, dentinal sensitivity and pulp irritation [10].
The microshear test was used in this study in accordance with the specifications of the ISO 527-1 and 527-2 standards for determining the properties of materials [36] Imperfections or defects at the bond interfaces, such as air bubbles, water or solvent may allow the development of local stresses during the bond strength test in larger areas. The gaps formed may propagate rapidly and cause failures. In smaller areas, there are fewer imperfections, bearing higher pressures before fracturing [37]. The speed of 0.5 mm per minute used in the test was also an important factor for a uniform distribution of the force. The ISO standard recommends a speed of at least 0.45 mm/min to a maximum of 1.05 mm/min [38].
The bonds between dentin and resin are friable, and tend to fail when subjected to various types of stresses, such as those that typically occur in the mouth. It is common for standard deviations of 30% to 40% to occur around the mean values, as mentioned in TC106 ISO (TR 11405) for the measurement of bond strength to the dental substrate [38].
Element analysis by EDX of the adhesive fracture areas on the dental remainder identified not only the Si in the organic matrix of the hydrophobic monomer, but also Ca and P, probably from the tooth structure located below this thin layer. A large quantity of Ca and P was observed at the interface of fractures of the cohesive type in dental structure, chemical elements of dental tissue, in addition to peaks of Si, characteristic remnants of the organic matrix of the hydrophobic monomers used. In the cohesive fractures in resin, in addition to Ca and P, peaks of Zr and Si - components of the restorative resin composite were found. Mixed fractures are well characterized by the presence of spectra with peaks of Ca and P and of Zr and Si, characteristic elements of the hydrophobic monomer and restorative resin composite. In the cohesive fracture areas in enamel and dentin of Group G3, we observed spectra with higher peaks of Ca and P coming from the dental structure and HAP nanoparticles incorporated into the hydrophilic monomer of the adhesive system.
Qualitative analysis by X-ray diffraction is a powerful method of chemical analysis, considering the entire crystalline structure has a diffraction pattern and that the same substance always presents the same model [39]. Thus, in the analysis of the diffractograms of the studied materials, it was observed that HAP presented standardized peaks, in agreement with reports in the literature [20,40,41]. The presence of a large quantity of amorphous phase in the hydrophobic monomer of Group G1 is owing to the presence of HEMA and Bis-GMA as constituents of its organic matrix. The presence of a large halo of the amorphous phase in the diffractograms, probably supplied the standard peaks in the DRX of HAP, which was added to the material as inorganic load in the proportion of only 10% of its total load. This small quantity of HAP was already sufficient to increase the viscosity of the hydrophobic monomer even further, which contains Bis-GMA, a component considered viscous due to the intermolecular union of hydrogen, by the presence of two methacrylate groups in its molecule, as well as aromatic rings and hydroxyl groups [8]. The formation of agglomerates of HAP nanoparticles in the hydrophobic monomer diminished its flowability and penetration into the etched enamel and dentin surfaces, and consequently, made it difficult for them to bond.
The hydrophilic monomer in Group G1 had only HEMA in its composition. This less viscous monomer with the characteristic of hydrophilicity is considered a wetting agent which, associated with polyalkenoic acid and also to alcohol and water as solvent, facilitates the penetration of the adhesive into the dental structure [8]. These agents would probably allow the dispersion of the HAP nanoparticles, which made it possible to visualize the HAP peaks of low intensity in the diffractograms of G3. No formation of a new phase detectable by the characterization method used was observed. In the diffractograms of the hydrophobic and hydrophilic monomers modified with HAP there were absorption bands characteristic of HAP; a contribution of the HAP nanoparticles to thickening the absorption band of these modified composites was also observed, allowing one to suppose the occurrence of strong interatomic bonds of the functional and specific groups present in the constitution of materials [39].
The molecular structure of HAP and the composites developed presented vibrational excitations resulting from the strong interatomic bonds (covalent bonds) of the specific functional groups present in the materials. The graphs resulting from the energy absorbed by the sample, by reason of the wave number in which this energy was absorbed are represented by the FTIR spectrum [42]. The profile of the FTIR spectrum of HAP showed agreement with the spectra found in the literature [20,40,41]. In this FTIR spectrum of hydrophobic monomers modified with HAP (G2) no evident modifications were observed from the inclusion of HAP as its inorganic constituent, showing it to have the same characteristics as the bands presented by the presence of Bis-GMA [41] in the hydrophobic monomer. To the contrary, the hydrophilic monomer modified with HAP (G3) presented absorption bands of HEMA [44] end more intense absorption bands at 830 cm−1, 1044 cm−1 and 1090 cm−1 belonging to the phosphate groups of HAP. This fact added to the presence of bands that identify free OH groups, probably contributed to the higher bond strength of this composite to both enamel and dentin.
The conventional adhesive system without load is a low cost material, available on the dental market, which modified with HAP nanoparticles processed with accessible reagents and by simple techniques, made it possible for a true bond to the dentin substrate to occur. This study enables a review of scientific knowledge and development of polymer composites with more effective bonds to enamel, and particularly to dentin. In addition, it incites new methodologies to be performed to confirm or refute the results found.
5. Conclusions
Considering the experimental conditions of this study it could be concluded that:
- the addition of HAP nanoparticles to the hydrophilic monomer of a conventional adhesive system increased the bond strength to enamel and dentin;
- the bond strength to the dental structure, of a HAPmodified hydrophobic monomer of the conventional adhesive system presented limited improvement;
- the bond strength to the dental structure, of the hydrophilic monomer with HAP, of the conventional adhesive system was shown to be higher than that of the monocomponent and self-etching adhesive systems;
- the majority of the fractures that occurred at the adhesive system/dental structure interfaces of the composites were of the adhesive type, and the groups with the presence of HAP in their constitution presented a higher percentage of cohesive fractures in enamel;
- the composites developed were shown to be stable, with the presence of crystallographic and amorphous phases, in addition to a similar morphology and chemical structure to those of their precursor materials.
6. Acknowledgements
The authors thank the “Laboratório Multiusuário de Microscopia Avançada—LMMA” (FAPEMIG-Processo CEX 112/10) of the Department of Chemistry/UFVJM for the obtainment of the XRD spectra; the Pathology Laboratory of the Department of Basic Sciences/UFVJM, and thank CAPES, SECTES/FAPEMIG and CNPq for the financial support.
REFERENCES
- A. B. Matos, C. H. C. Saraceni, M. M. Jacobs and M. Oda, “Estudo de Resistência à Tração de Três Sistemas Adesivos Associados a Resina Composta em Superfícies Dentinárias,” Pesquisa Odontológica Brasileira, Vol. 15, No. 2, 2001, pp. 161-165. http://dx.doi.org/10.1590/S1517-74912001000200014
- K. L. Van Landuyt, J. Snauwaert, J. De Munck, M. Peumans, Y. Yoshida, A. Poitevin, E. Coutinho, K. Suzuki, P. Lambrechts and B. Van Meerbeeka, “Systematic Review of the Chemical Composition of Contemporary Dental Adhesives,” Biomateriais, Vol. 28, No. 26, 2007, pp. 3757- 3785. http://dx.doi.org/10.1016/j.biomaterials.2007.04.044
- G. Inoue, T. Nikaido, M. R. Foxton and J. Tagami, “The Acid-Base Resistant Zone in Three Dentin Bonding Systems,” Dental Materials Journal, Vol. 28, No. 6, 2009, pp. 717-721. http://dx.doi.org/10.4012/dmj.28.717
- M. Sarr, A.W. Kane, J. Vreven, A. Mina, K.L. Van Landuyt, M. Peumans, P. Lambrechts, B. Van Meerbeek and J. De Munck, “Microtensile Bond Strength and Interfacial Characterization of 11 Contemporary Adhesives Bonded to Bur-Cut Dentin,” Operative Dentistry, Vol. 35, No. 1, 2010, pp. 94-104. http://dx.doi.org/10.2341/09-076-L
- T. Yoshikawa, N. Wattanawongpitak, E. Cho and J. Tagami, “Effect of Remaining Dentin Thickness on Bond Strength of Various Adhesive Systems to Dentin,” Dental Materials Journal, Vol. 31, No. 6, 2012, pp. 1033-1038. http://dx.doi.org/10.4012/dmj.2012-143
- N. B. Suryakumari, P. S. Reddy, L. R. Surender and R. Kiran, “In Vitro Evaluation of Influence of Salivary Contamination on the Dentin Bond Strength of One-Bottle Adhesive Systems,” Contemporary Clinical Dentistry, Vol. 2, No. 3, 2011, pp. 160-164. http://dx.doi.org/10.4103/0976-237X.86440
- R. N. Garcia, A. E .G. Alvarez, C. E. Dias, M. A. Mazaro, T. Firmo, H. Stuker and M. Giannini, “Bond Strength of Contemporary Restorative Systems to Enamel and Dentin,” Revista Sul-Brasileira de Odontologia, Vol. 8, 2011, pp. 60-67.
- A. Peutzfeldt, “Resin Composites in Dentistry: The Monomer Systems,” European Journal of Oral Science, Vol. 105, No. 2, 1997, pp. 97-116. http://dx.doi.org/10.1111/j.1600-0722.1997.tb00188.x
- L. M. Cavalcante, L. F. J. Schneide, L. S. Silva, A. K. Bedran-Russo and L. A. F. Pimenta, “Efeito da Ciclagem Térmica na Microinfiltração e Microtração de Restaura- ções de Resina Composta,” Revista da Faculdade de Odontologia-UPF, Vol. 14, 2009, pp. 132-138.
- J. Perdigão, “Dentin Bonding—Variables Related to the Clinical Situation and the Substrate Treatment,” Dental Material, Vol. 26, No. 2, 2010, pp. 24-37. http://dx.doi.org/10.1016/j.dental.2009.11.149
- M. Sadat-shojai, M. Atai, A. Nodehi and L. N. Khanlar, “Hydroxyapatite Nanorods as Novel Fillers for Improving the Properties of Dental Adhesives: Synthesis and Application,” Dental Materials, Vol. 26, No. 5, 2010, pp 471- 482. http://dx.doi.org/10.1016/j.dental.2010.01.005
- U. Lohbauer, A. Wagner, R. Belli, C. Stoetzel, A. Hilpert, H. D. Kurland, J. Grabow and F. A. Müller, “Zirconia Nanoparticles Prepared by Laser Vaporization as Fillers for Dental Adhesives,” Acta Biomaterialia, Vol. 6, No. 12, 2010, pp. 4539-4546. http://dx.doi.org/10.1016/j.actbio.2010.07.002
- M. A. Ribeiro, I. X. Ferreira, R. P. Lima, A. L. A. Mariz, J. G. F. Pompeu and C. H. V. Silva, “The Insertion Technique’s Infl Uence of Composite resin on the Microleakage in Occlusal Esthetic Restorations,” Revista Odontológica Clínico Científica, Vol. 9, 2010, pp. 345-348.
- H. Zhang and B. W. Darvell, “Mechanical Properties of Hydroxyapatite Whisker-Reinforced Bis-GMA-Based Resin Composites,” Dental materials, Vol. 28, No. 8, 2012, pp. 824-830. http://dx.doi.org/10.1016/j.dental.2012.04.030
- V. C. Leitune, F. M. Collares, R. M. Trommer, D. G. Andrioli, C. P. Bergmann and S. M. Samuel, “The Addition of Nanostructure Hydroxyapatite to an Experimental Adhesive resin,” Journal of Dentistry, Vol. 41, No. 4, 2013, pp. 321-327. http://dx.doi.org/10.1016/j.jdent.2013.01.001
- A. Akhavan, A. Sodagar, F. Motjahedzadeh and K. Sodagar, “Investigating the Effect of Incorporating nanosilver/Nanohydroxyapatite Particles on the Shear Bond Strength of Orthodontic Adhesives,” Acta Odontologica Scandinavica, Vol. 71, No. 5, 2013, pp. 1038-1042. http://dx.doi.org/10.3109/00016357.2012.741699
- J. Vandiver, D. Dean, N. Patel, W. Bonfield and C. Ortiz, “Nanoscale Variation in suRface Charge of Synthetic Hydroxyapatite Detected by Chemically and Spatially Specific High-Resolution Force Spectroscopy,” Biomaterials, Vol. 26, No. 3, 2005, pp. 271-283. http://dx.doi.org/10.1016/j.biomaterials.2004.02.053
- A. S. Khan, K. R. Hassan, S. F. Bukhari, W. Ferranti and S. L. Rehmaniu, “Structural and in Vitro Adhesion Analysis of a Novel Covalently Coupled Bioactive Composite,” Journal of Biomedical Materials Research Part B, Vol. 100, No. 1, 2012, pp. 239-248. http://dx.doi.org/10.1002/jbm.b.31945
- Y. Zhang and Y. Wang, “Hydroxyapatite Effect on Photopolymerization of Self Etching Adhesives with Different Aggressiveness,” Journal of Dentistry, Vol. 40, No. 7, 2012, pp. 564-570. http://dx.doi.org/10.1016/j.jdent.2012.03.005
- J. K. Jekel, D. L. Katz and J. G. Elmore, “Epidemiology, Biostatistics, and Preventive Medicine,” Elsevier, Philadelphia, 2001.
- P. Jacques and J. Hebling, “Influence of Autoclave Sterilization of Human Teeth on Dentin Bonding,” Pesquisa Brasileira em Odontopediatria e Clínica Integrada, Vol. 6, 2006, pp. 9-13.
- M. H. Santos, M. Oliveira, L. F. P. Souza, H. S. Mansur and W. L. Vasconcelos, “Synthesis Control and Characterization of Hydroxyapatite Prepared by Wet Precipitation Process,” Materials Research, Vol. 7, No. 4, 2004, pp. 1- 6. http://dx.doi.org/10.1590/S1516-14392004000400017
- C. S. Fowler, M. L. Swartz, B. K. Moore and B. F. Rhodes, “Influence of Selected Variables on Adhesion Testing,” Dental Materials, Vol. 8, No. 4, 1992, pp. 265-269. http://dx.doi.org/10.1016/0109-5641(92)90097-V
- J. Perdigão, “Effect of Substrate Age and Adhesive Composition on Dentin Bonding,” Operative Dentistry, Vol. 38, 2012.
- Y. Liu, Y. Tan, T. Lei, Q. Xiang, Y. Han and B. Huang, “Effect of Porous Glass-Ceramic Fillers on Mechanical Properties of Light-Cured Dental Resin Composites,” Dental Materials, Vol. 25, No. 6, 2009, pp. 709-715. http://dx.doi.org/10.1016/j.dental.2008.10.013
- T. F. Boing, G. M. Gomes, C. Z. Grande, A. Reis, J. C. Gomes and O. M. M. Gomes, “Evaluation of the Degree of Conversion of a Composite Resin Using Different Surface Treatments before Final Curing,” Revista Dentística on line, Vol. 10, 2011.
- H. Zhang and M. Zhang, “Effect of Surface Treatment of Hydroxyapatite Whiskers on the Mechanical Properties of Bis-GMA-Based Composites,” Biomedical Materials, Vol. 5, 2010.
- B. Poptani, K. S. Gohil, J. Ganjiwale and M. Shukla, “Microtensile Dentin Bond Strength of Fifth with Five Seventh-Generation Dentin Bonding Agents after Thermocycling: An in Vitro Study,” Contemporary Clinical Dentistry, Vol. 3, No. 6, 2012, pp. 167-171. http://dx.doi.org/10.4103/0976-237X.101079
- A. O. Spazzin, B. Carlini Júnior, R. R. Moraes and M. F. Mesquita, “Bonding to Wet and Dry Dentin: Microtensile Bond Strength and Marginal Leakage,” Revista de Odontologia da UNESP, Vol. 37, 2008, pp. 91-96.
- T. K. Vaidyanathan and J. Vaidyanathan, “Recent Advances in the Theory and Mechanism of Adhesive Resin Bonding to Dentin: A Critical Review,” Journal of Biomedical Materials Research Part B: Applied Biomaterials, Vol. 88, No. 2, 2009, pp. 558-578. http://dx.doi.org/10.1002/jbm.b.31253
- P. Senawongse, C. Harnirattisai, Y. Shimada and J. Tagami, “Effective Bond Strength of Current Adhesive Systems on Deciduous and Permanent Dentin,” Operative Dentistry, Vol. 29, 2004, pp. 196-202.
- P. Banks and B. Thiruvenkatachari, “Long-Term Clinical Evaluation of Bracket Failure with a Self-Etching Primer: A Randomized Controlled Trial,” Journal of Orthodontics, Vol. 34, No. 4, 2007, pp. 243-251. http://dx.doi.org/10.1179/146531207225022293
- M. A. Montasser, J. L. Drummond, J. R. Roth, L. AlTurki and C. A. Evans, “Rebonding of Orthodontic Brackets. Part II, an XPS and SEM Study,” Angle Orthodontist, Vol. 78, No. 3, 2008, pp. 537-44. http://dx.doi.org/10.2319/022707-102.1
- A. Manabe, M. Kanehira, W. J. Finger, H. Hisamitsu and M. Komatsu, “Effects of Opacity and Oxygen Inhibition of Coating Resin Composites on Bond Strength to Enamel,” Dental Materials Journal, Vol. 28, No. 5, 2009, pp. 552-557. http://dx.doi.org/10.4012/dmj.28.552
- C. Elliot, “Structure and Chemistry of the Apatites and other Calcium Orthophosphates: Studies in Inorganic Chemistry 18,” Elsevier Science, 1994.
- T. Arao and N. Nakabayashi, “Effect of Miniaturized Dumbbell-Shaped Specimen to Identify Bonding of Resin to Bovine Dentin,” Journal Japanese of Dental Materials, Vol. 16, 1997, pp. 175-181.
- H. Sano, T. Shono, H. Sonoda, T. Takatsu, B. Ciucchi and R. M. Carvalho, “Relationship between Surface Area for Adhesion and Tensile Bond Strength-Evaluation of a Microtensile Bond Test,” Dental Materials, Vol. 10, 1994, pp. 236-240. http://dx.doi.org/10.1016/0109-5641(94)90067-1
- International Organization for Standardization. ISO/ TC106 (TR 11405): Dental Materials—Testing of Adhesion to Tooth Structure, Geneva, 2003.
- B. D. Cullity and S. R. Stock, “Elements of X-Ray Diffraction,” Prentice Hall, New Jersey, 2001.
- M. H. Santos and H. S. Mansur, “Low-Cost Processing Technology for the Synthesis of Calcium Phosphates/ Collagen Biocomposites for Potential Bone Tissue Engineering Applications,” Materials Research, Vol. 10, 2007, pp. 431-436. http://dx.doi.org/10.1590/S1516-14392007000400018
- M. H. Santos, L. G. D. Heneine and H. S. Mansur, “Synthesis and Characterization of Calcium Phosphate/Collagen Biocomposites Doped With Zn2+,” Materials Science and Engineering C, Vol. 28, 2008, pp. 563-571. http://dx.doi.org/10.1016/j.msec.2007.07.002
- D. L. Paiva, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, “Introduction to Spectroscopy,” Cengage Learning, Estados Unidos, 2009.
NOTES
*Corresponding author.