Computational Chemistry
Vol.04 No.04(2016), Article ID:71458,18 pages
10.4236/cc.2016.44011
Structural, Spectral (IR and UV/Visible) and Thermodynamic Properties of Some 3d Transition Metal(II) Chloride Complexes of Glyoxime and Its Derivatives: A DFT and TD-DFT Study
Ludovid Ngouo Nogheu1, Julius Numbonui Ghogomu1*, Desire Bikele Mama2, Nyiang Kennet Nkungli1, Elie Younang3, Shridhar Ramachandra Gadre4
1Department of Chemistry, Faculty of Science, University of Dschang, Dschang, Cameroon
2Department of Chemistry, Faculty of Science, University of Douala, Douala, Cameroon
3Department of Inorganic Chemistry, Faculty of Science, University of Yaounde I, Yaounde, Cameroon
4Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur, India

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 5, 2016; Accepted: October 22, 2016; Published: October 25, 2016
ABSTRACT
Metal complexes bearing vic-dioxime ligands have been extensively used as analytical and biochemical reagents, and are well-known antimicrobial agents. Herein is reported a DFT study on the molecular structures, thermodynamic properties, chemical reactivity and spectral properties of some 3d metal(II) chloride complexes of glyoxime. The functionals B3LYP and CAM-B3LYP have each been used in conjunction with LANL2DZ for the metal(II) ions (Fe2+, Co2+, Ni2+ and Cu2+) and the Pople- style basis set 6-31G+(d,p) for the rest of the elements, to perform theoretical calculations. The metal complexation abilities of the glyoxime ligands studied in this work have been evaluated on the basis of metal-ligand binding energies. These ligands were found to have high affinities towards Ni(II) and Fe(II) ions, and all complexation reactions were found to be thermodynamically feasible. Ligand-to-metal electron donations in the complexes studied have been revealed by natural population analysis. The fully optimized geometries of the complexes have adopted square planar structures around the central metal ions. On the basis of orbital composition analysis, the UV-Vis electronic absorption bands of these molecules have been attributed mainly to MLCT, LMCT and d-d electronic transitions involving metal-based orbitals.
Keywords:
Glyoxime Complexes, DFT, Binding Energy, Electronic Absorption

1. Introduction
The interaction of a central metal with surrounding ligands (atoms, ions or molecules) has been of significant interest in coordination chemistry. In the past decades, the coordination chemistry of transition metal complexes bearing oxime ligands has been a subject of intense studies owing to their applications in many scientific domains like biomedical and electrochemistry. Synthesis of Co(III), Ni(II) and Cu(II) complexes based on two glyoxime derivatives namely: dianiline glyoxime and disulfanilamide glyoxime were envisaged and were found to be effective as stimulators in biosynthetic processes of enzymes in some fungi strains [1] . In one review, the synthesis of phenolic oximes and their complexes were reported to be more effective in commercial metal recovery processes based on solvent extraction as a result of their remarkable stability [2] . These studies have been motivated by the fact that oximes are important analytical, biochemical, and antimicrobial agents. They have equally attracted much attention due to their structural features and their uses as liquid crystals and dyes [3] - [6] .
Vicinal dioxime derivatives act as amphoteric ligands due to the presence of weak acidic -OH groups and basic -C = N groups. As such, they are capable of forming highly stable complexes with most transition metals in the periodic table. The exceptional stability and electronic properties displayed by these complexes are attributable to their hydrogen bond-stabilized planar structures [7] - [9] . These complexes have been extensively used for various purposes owing to their high stability, suitability as model compounds for vitamin B12 and as chelators in trace metal analysis [10] [11] . These compounds also play a significant role in domains such as stereochemistry, structure isomerism, spectroscopy, model systems of biochemical interest, cation exchange and ligand exchange chromatography, analytical chemistry, catalysis, stabilizers, polymers, photography, technological improvement, pigments, and dyes [12] - [16] . Furthermore, they have been identified as compounds capable of columnar stacking, which may enhance semiconducting properties [17] . It is well established that different ligand types can affect the stabilities and properties of complexes in various ways. As such, the substitution of some dioxime groups or fragments can significantly modify the structures and stability of its complexes. The stability resulting from metal ion chelation by glyoxime ligands in the complexes currently studied is also dependent on the type of central metal ion present. This research is geared toward facilitating the construction of new vicinal glyoxime derivatives capable of forming very stable 3d metal(II) complexes, and which can be used in trace metal analysis.
After a detailed literature survey, we have found to the best of our knowledge that very little theoretical studies have been carried out on glyoxime complexes till date [18] [19] . Moreover, spectroscopic and X-ray crystallographic studies on these molecules have not been performed. In the present paper, density functional theory (DFT) studies have been undertaken on some 3d metal(II) chloride glyoxime complexes. Specifically, the structural, thermodynamic and spectral properties as well as the chemical reactivity of these compounds have been studied. Quantum chemical computations have been used in this research endeavor because they have become an established method for the prediction of novel structures and properties in the last decades. Interestingly, they constitute a powerful tool for analyzing the properties of coordination compounds, and are widely used nowadays to complement experimental works.
2. Computational Procedure
All theoretical calculations were performed using the Gaussian 09 program package [20] . Molecular geometries were optimized in vacuum using the B3LYP exchange-cor- relation hybrid functional in conjunction with the LANL2DZ basis set for the central metal ions and the Pople-style basis set 6-31+G(d,p) for the elements: C, H, O, Cl and N. All optimized geometries were characterized by vibrational frequency calculations at the same level of theory as that used for geometry optimization, and only real frequencies were found confirming the optimized geometries as minima on their potential energy surfaces. For all theoretical calculations, we adopted the restricted closed-shell model for the closed-shell Ni2+ and Fe2+ complexes and the unrestricted model for the open-shell Co2+ and Cu2+ complexes. All complexes were treated as low spin species. Natural population analysis (NPA) calculations were performed using the NBO 3.1 program as implemented in the Gaussian 09 package [20] at the same theoretical level as that used for geometry optimization calculations. TD-DFT calculations were performed on the optimized ground state geometries of the molecules under investigation, in order to simulate their absorption spectra [21] [22] . In these calculations, excitations to the first 20 singlet excited states were considered, and the CAM-B3LYP functional was used together with the same basis set as that used for geometry optimization.
3. Results and Discussion
3.1. Molecular Geometry
The square planar schematic structures of the investigated 3d metal glyoxime complexes, showing atomic numbering are presented in Figure 1. The different structures have been obtained by changing the central metal (M = Fe2+, Co2+, Ni2+ and Cu2+) and the alkyl radical (R = H, -CH3 and -C6H5). The geometric parameters (bond lengths, bond

Figure 1. Schematic structures of investigated complexes.
angles, and dihedral angles) of the ground state geometries of the molecule studied optimized at the DFT/6-31+G(p,d)/(LANL2DZ for metal ions) level, are listed in Table 1.

Table 1. Main optimized bond lengths (Å), bond angles (˚) and dihedral angles (˚) of metal 3d glyoxime complexes at B3LYP/ 6-31+G(d,p) with LANL2DZ for metal(II).
These values ought to be compared with those obtained experimentally, but X-ray crystallographic data is not available in the literature for these complexes. It is clear from the values in Table 1 that all complexes studied are nearly symmetric, and possess two intra-molecular hydrogen bonds (O-H … Cl) each, between the hydrogens of the oxime group and the chloride ligands. These hydrogen bonds contribute in stabilizing the complexes and in maintaining a square planar geometry around their central metal ions.
It is evident from Table 1 that the M-Cl and M-N bond lengths in Fe(II) and Ni(II) complexes are shorter than corresponding bond lengths in Cu(II) and Co(II) complexes the greatest discrepancies being 0.093 Å for M-Cl between 3d and 3c, and 0.2 Å for M-N between 1d and 1a. This indicates that the ligands (glyoxime and chloride ions) interact more strongly with Fe(II) and Ni(II) ions than with Cu(II) and Co(II) ions. In almost all complexes studied, the greatest metal-ligands bond lengths are present in the Cu(II) complexes. It can be seen that the azomethine (C = N) bond lengths decrease with an increase in atomic number of the central metal ions within the 3d period, with calculated differences of about: 0.031 Å for 1a and 1d, 0.033 Å for 2a and 2d, 0.032 Å for 3a and 3d and 0.065 Å for 4a and 4d. The values of the bond angle Cl-M-Cl range from 95.05˚ to 105.963˚ in all complexes studied, and the largest values: 105.963˚, 104.809˚, 100.022˚ and 105.004˚ for 1d, 2d, 3d and 4d respectively, correspond to copper complexes while the smallest values: 97.271˚, 96.768˚, 95.941˚ and 96.654˚ for 1c, 2c, 3c and 4c respectively correspond to nickel complexes. Values of the dihedral angle Cl1-M-N1-C1 are around −180˚ for all the complexes, except 3d for which the value is −175.927˚. This shows that the optimized geometries of all complexes investigated in this research endeavor are nearly square planar around their respective central metal ions.
3.2. Thermodynamic Properties
In order to investigate thermodynamic properties of complexation reactions, statistical thermodynamic calculations were carried out at 1 atm and 298.15 K. To investigate the stability of the complexes studied, the thermodynamic parameters: binding energy (∆Eint), Gibbs free energy (∆G˚) and enthalpy (∆H˚) of formation were calculated.
3.2.1. Complexation Energy
The complexation ability of the ligands with different metal cations is characterized by their binding energy as defined in Equation (1).
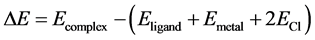 (1)
(1)
where EComplex, ELigand, EMetal(II) and ECl are respectively the thermal energy of complex, free glyoxime ligand, free metal ions and free chloride ligands.
A large variation in the complexation energies is clear from Table 2. The binding energies are negative in all cases, implying that the complexation reactions are highly favorable. For all the ligands studied, the most negative complexation energy values have been obtained for the formation of the Ni(II) complexes whereas least negative
Table 2. Main energetical parameters of metal 3d glyoximes complexes in Kcal/mol at B3LYP/ 6-31+G(d,p) with LANL2DZ for metal(II).
values have been obtained for the formation of the Cu(II) complexes. Hence, the ligands exhibit their greatest affinity toward the Ni(II) ion and the least toward the Cu(II) ion. For instance, the complexation energy for the formation of 1d is higher than that of formation of 1c by 49 kcal∙mol−1. Generally, more negative complexation energies are more thermodynamically favored. Based on this fact, it can be concluded that the formation of the Ni(II) complexes is most favorable of all the complexes investigated. It is worthy of note that the formation processes for the complexes bearing ligand 4 are most favored.
3.2.2. Gibbs Free Energy and Enthalpy Change
Besides binding energies, the standard complexation Gibbs free energy (∆G˚) and enthalpy change (∆H˚) were also calculated using Equations (2) and (3) and the results are presented in Table 2.
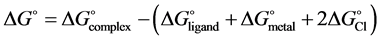 (2)
(2)
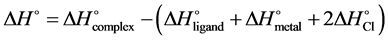 (3)
(3)
It is clear from Table 2 that the complexation Gibbs free energy change for the formation of all complexes investigated is negative, showing that the process is thermodynamically feasible at ambient temperature for each complex. The formation processes for the nickel complexes (1c, 2c, 3c, 4c) have been found to be the most exergonic and, therefore, the most favored compared to the formation of the other complexes. Similarly, the enthalpies of the complexation reactions that yield the complexes studied have been found to be negative which is a clear indication that these reactions are highly exothermic.
3.3. Electronic Properties
3.3.1. Natural Population Analysis (NPA)
The NPA charges on the metal(II) ions in the complexes investigated are listed in Table 3. The positive charge on each metal ion in the complexes is less than +2, the charge on the free state metal ion, whereas the magnitudes of the negative charges on the chloride ions in the complexes is lower than −1, the charge on the free state chloride ion. This result indicates that electron transfer from the ligands to the metal ions occurred during the formation of the coordination bonds. This charge transfer is greatest for the Fe(II), Ni(II) and Co(II) complexes and least for the Cu(II) complexes. From this observation, it can be concluded that the metal-ligand bonds in the Fe(II) Ni(II) and Co(II) complexes are stronger than those in the Cu(II) complexes. This is corroborated
Table 3. Variation of natural population atomic charges (e) at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).
by the complexation energies (Table 2) which have shown that the glyoxime and chloride ligands have a higher affinity for Fe(II) Ni(II) and Co(II) ions than the Cu(II) ion. From the foregoing results, it is clear that coordinate bonding is responsible for complex formation, and ligand to metal ion charge transfer is the underlying mechanism.
3.3.2. Reactivity Descriptors
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are very important parameters for quantum chemistry. The frontier orbital gap helps to characterize the chemical reactivity and kinetic stability of molecules. A molecule with a small frontier orbital gap is more polarizable and is generally associated with a high chemical reactivity, low kinetic stability and is also termed soft molecule. The eigenvalues of the HOMO and LUMO as well as their energy gap reflect the chemical reactivity of a molecule. A large HOMO-LUMO energy gap has been associated with high stability of molecules [23] . The chemical potential (μ), global hardness (η) and electrophilicity (w) are global descriptors which indicate the overall stability of a chemical system [24] - [27] .
The values of some global reactivity descriptors investigated in this paper are presented in Figure 2 (Table 4). It can be seen from this table that the largest HOMO- LUMO energy gaps (∆EH-L) and chemical hardness (η) values correspond to the Cu(II) complexes (1d, 2d, 3d and 4d) and the Co(II) complexes (1b, 2b, 3b and 4b), indicating that they are kinetically more stable and less reactive than the rest of the complexes. It is also observed in Figure 3 that Fe2+ complexes are the most reactive complexes in each series with the lower value of chemical hardness. The other reactivity descriptors presented in this figure have shown the same reactivity trend for these complexes (Table 4).

Figure 2. Superposition of the global reactivity descriptors of the systems in eV except those of global softness which are in eV−1, calculated at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).
Table 4. Values of the global reactivity descriptors of the systems in eV except those of global softness which are in eV−1, calculated at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).

Figure 3. Picture of the HOMO-LUMO orbitals for Fe2+ complexes.
3.4. Infra-Red Spectroscopy
To the best of our knowledge, no IR spectral data is available in the literature for the complexes investigated in this research endeavor. The DFT/B3LYP method in conjunction with a doubly split valence basis set along with diffuse and polarization functions, 6-31+G(d,p) and LANL2DZ for the central metal(II) ions was used to calculate harmonic vibrational frequencies of the glyoximes complexes studied. IR frequencies of two complexes (1a’ and 1b’) were also calculated using different basis set (B3LYP/6-31+ G(d,p)/SDD for metal ion) with same method in order to study the effect of basis set in the frequency calculation. The Gauss View 5.0.8 molecular visualization program was used to perform the IR vibrational band assignments for the molecules. Although experimentally determined IR vibrational frequencies were not available for these molecules, the calculated values were still scaled down because the DFT/B3LYP method tends to overestimate normal mode frequencies due to a combination of electron correlation effects and basis set deficiencies [28] . Scaling factor for B3LYP/LANL2DZ level is 0.9978 [29] . Given the fact that this factor is very close to unity, coupled with the fact that LANL2DZ has only been used for the central metal ions, the scale factor for B3LYP/6-31+G(d,p) with numerical value 0.9614 was used for same reason [29] - [31] to scale down the computed IR frequencies. Also, it is worth noting that in a preliminary study, the IR frequencies were calculated at B3LYP/6-31+G(d,p)/(SDD for metal ions) for some of the complexes in order to compared with those calculated at B3LYP/6-31+ G(d,p)/(LANL2DZ for metal ions) and very little discrepancies were observed with the calculated frequencies.
IR vibrations of the OH group are generally found to be most sensitive to the environment. As such, they show pronounced shifts in the spectra of the hydrogen-bonded species. The optimum absorption region of non-hydrogen bonded or a free hydroxyl group is 3550 - 3700 cm−1 [32] . In the case of their presence in a molecule, intra and intermolecular hydrogen bonding reduces the O-H stretching band to 3000 - 3550 cm−1 region [33] . In the complexes studied the O-H stretching band are found between 3082 - 3303 cm−1, signifying that all O-H groups in the complexes studied are engaged in intramolecular hydrogen bonding with the chloride ligands. The simulated IR spectra have also shown an intense and relatively sharp band in the range 1426 - 1554 cm−1 assignable to the stretching C-C vibrations of glyoxime moiety of the complexes. Compared to the C-C stretching vibrations in the free glyoxime ligands (1234 - 1355 cm−1), these vibrations have shifted toward regions of very large wavelengths upon complexation, showing that the electron density in C-C bond is increased by transition metal complexation by the stated ligands.
As a general observation in Table 5, significant shifts in IR vibrations occurred for the C = N bonds, implying that their vibrations are the most affected by complexation since their vibrational frequencies in the complexes significantly differ from those in the free ligands. The coordination of the vic-dioxime ligands to the metal center via the two nitrogen atoms is expected to reduce the electron density in the azomethine link and thus lowers the C = N absorption frequency. The C = N peaks of the complexes were indeed shifted to lower frequencies indicating the coordination of the azomethine nitrogen to the metal ions.
3.5. Absorption Spectra Analysis
The absorption spectral simulation of 3d metal(II) glyoximes complexes have been performed using Time-Dependent DFT calculations at the Cam-B3LYP/6-31G+(d,p)/ (LanL2DZ for metal(II) ions) levels of theory in gas phase. The calculated absorption energy, corresponding oscillator strength and orbital coefficients are summarized in
Table 5. Calculated vibrational frequencies (cm−1) at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).
1a’: Calculated frequencies of Fe(II) complex with ligand 1 at B3LYP/6-31+G(d,p)/(SDD for metal ion). 1b’: Calculated frequencies of Co(II) complex with ligand 1 at B3LYP/6-31+G(d,p)/(SDD for metal ion).
Table 6. To obtain the nature and the energies of the electronic transitions within the investigated complexes, the first twenty low-lying excited states have been calculated. The absorption energies with first two higher oscillator strengths are considered throughout the discussion in this paper. The TD-DFT results in Table 7 have shown that the lowest energy transition in the complexes correspond to electronic excitations from HOMO to LUMO. The absorption intensity is directly related to the dimensionless oscillator strength value and the dominant absorption bands are the transitions with highest oscillator strength.
For the first series of complexes, the most intense band was observed at: 295.02, 326.80, 216.88 and 264.83 nm for the 1a, 1b, 1c and 1d complexes respectively. As evidenced in Figure 4(a), the absorption band for complex 1c has been found to be the most intense, with high oscillator strength, 0.4923. The intensities of the absorption
Table 6. Computed UV-Vis absorption bands, dominant transitions, and oscillator strengths (f) of metal 3d glyoximes complexes at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).
bands of this series of complexes are in the decreasing order: 1c > 1a > 1d > 1b.
The transition energy is inversely proportional to the absorption wavelength in each case, for instance, complex 1c has a high absorption energy (5.7167 ev) corresponding to the HOMO − 7 → LUMO + 1 electronic transition, which is observed at the wavelength 216.88 nm. The molecular orbital compositions in Table 7 have revealed that 64.44% of the electron density in HOMO − 7 of 1c is localized mainly on the two chloride ligands and 61.81% of the electron density in LUMO + 1 is localized on the metal ions, hence the character for this transition is mainly ligand-to-metal charge transfer (LMCT).
For the second series of complexes containing dimethylglyoxime as one of the ligands, the maximum absorption bands appeared at: 356.69, 316.65, 214.29 and 272.61 nm for the 2a, 2b, 2c and 2d complexes respectively. It is clear from Figure 4(b) that the absorption band for complex 2c with high oscillator strength, 0.4516 is the most intense. For this series, absorption intensities decrease in the order: 2c > 2a > 2d > 2b. As such, the most intense band in 2c is due mainly to the transition HOMO − 8 → LUMO + 1. On the basis of the molecular orbital compositions in Table 7, the electron density in HOMO − 8 is localized mainly on the two chloride ligands (65.56%), and that of LUMO + 1 is localized mainly on the metal ions (62.15%), hence, the HOMO − 8 is essentially the p-type orbitals p(Cl) on the chloride ligands (donors) whereas LUMO + 1 is mainly the d-type orbital, d(Ni) on nickel(II) ion (acceptor). Therefore, the transition
Table 7. Frontier molecular orbital compositions for the ground states of metal 3d glyoximes complexes at B3LYP/6-31+G(d,p) with LANL2DZ for metal(II).
character of the most intense band in 2c is principally ligand-to-metal charge transfer (LMCT).

Figure 4. Schematic of calculated electronic transitions of glyoximes complexes.
In the case of the third series, the maximum absorption bands are: 364.96, 330.42, 304.56 and 340.27 nm corresponding to the 3a, 3b; 3c and 3d complexes respectively. Hence, the most intense band with high oscillator strength (0.3154) is exhibited by 3a. The intensities of the dominant transitions in this series can be classified in the increasing order: 3a > 3c > 3d > 3b (Figure 4). The maximum absorption band in 3a corresponds to the transition HOMO - 2 → LUMO. From the molecular orbital compositions in Table 7, it is clear that the electron density in HOMO − 2 is mainly localized on the diphenylglyoxime ligand (88.94%) and that of the LUMO is similarly localized on the central metal (76.66%). As such, the HOMO − 2 is a π-donor and the LUMO is the d-type orbital of Fe. The main transition character of the most intense absorption band in 3a is ligand-to-metal charge transfer (LMCT).
For the last series of complexes studied, the maximum absorption bands are respectively 390.02, 459.55, 349.24 and 326.52 nm for the 4a; 4b; 4c and 4d complexes. Clear from Table 7, the highest absorption energy was observed for the 4a complex (3.1790 eV) corresponding to the HOMO - 4 → LUMO + 1 transition. From the molecular orbital composition analysis result in Table 7, it is obvious that the electron density in HOMO − 4 is mainly localized on the two chloride ligands (72.88%) and that of the LUMO + 1 is localized mainly on the metal ion which has contributed 68.04% of it, and the ligands (L) that have contributed 23.66%. It can be concluded from these results that the most intense electronic band in 4d has a mixed ligand-to-metal charge transfer (LMCT) and ligand-to-ligand charge transfer (LLCT) character (Table 6).
4. Conclusion
DFT and TD-DFT calculations were used to investigate the molecular properties of some 3d metal (II) chloride glyoxime complexes. The optimized geometries of the complexes studied revealed a nearly square planar geometry around the central metal ion in each complex. Our results showed the existence of hydrogen bonds between the two OH groups of glyoxime and the two chloride ligands in all complexes investigated, which significantly contribute toward the stabilization of the complexes. As a general observation for all the metal ions studied, the ligands showed the greatest affinity toward the nickel(II) ion and the least affinity toward copper(II) ion. For the complexes studied, all complexation reactions were found to be thermodynamically feasible and exothermic. Natural population analysis showed the possibility of ligand-to-metal charge transfer. Electronic absorption bands obtained in the UV/Visible region have been attributed mainly to MLCT, LMCT and d-d electronic transitions involving metal-based orbitals.
Acknowledgements
The authors are thankful to the IIT Kanpur, India for the resources made available through a CV Raman International Fellowship award (Grant No. 101F102), offered by the Ministry of External Affairs of India and FICCI (Federation of Indian Chambers of Commerce and Industry).
Cite this paper
Nogheu, L.N., Ghogomu, J.N., Mama, D.B., Nkungli, N.K., Younang, E. and Gadre, S.R. (2016) Structural, Spectral (IR and UV/Visible) and Thermodynamic Properties of Some 3d Transition Metal(II) Chloride Complexes of Glyoxime and Its Derivatives: A DFT and TD-DFT Study. Computational Chemistry, 4, 119-136. http://dx.doi.org/10.4236/cc.2016.44011
References
- 1. Rija, A., Bulhac, I., Coropceanu, E., Gorincioi, E., Calmîc, E. and Barba, O. (2011) Bologasynthesis and Spectroscopic Study of some Coordinative Compounds of Co(III), Ni(II) and Cu(II) with Dianiline- and Disulfanilamideglyoxime. Chemistry Journal of Moldova, 6, 73- 78.
- 2. Smith, A.G., Tasker, P.A. and White, D.J. (2003) The Structures of Phenolic Oximes and their Complexes. Coordination Chemistry Reviews, 241, 61-85. http://dx.doi.org/10.1016/S0010-8545(02)00310-7
- 3. Blower, P.J. (1997) Small Coordination Complexes as Radiopharmaceutical for Cancer Targeting. Transition Metal Chemistry, 23, 109-112. http://dx.doi.org/10.1023/A:1006926505754
- 4. Karapcin, F. and Arabali, F. (2006) Synthesis and Characterization of 4-Arylaminobi- Phenylglyoximes and Their Complexes. Journal of the Chilean Chemical Society, 51, 982- 985.
- 5. Chaudhuri, P. (2003) Homo- and Hetero-Polymetallic Exchange Coupled Metal-Oximates. Coordination Chemistry Reviews, 243, 143-190. http://dx.doi.org/10.1016/S0010-8545(03)00081-X
- 6. Kurtoglu, M. and Baydemir, S.A. (2007) Studies on Mononuclear Transition Metal Chelates Derived from a Novel (E,E)-Dioxime: Synthesis, Characterization and Biological Activity. Journal of Coordination Chemistry, 60, 655-665. http://dx.doi.org/10.1080/00958970600896076
- 7. Serin, S. (2001) New Vic-Dioxime Transition Metal Complexes. Transition Metal Chemistry, 26, 300-306. http://dx.doi.org/10.1023/A:1007163418687
- 8. Schrauzer, G.N. (1976) New Developments in the Field of Vitamin B12: Reactions of the Cobalt Atom in Corrins and in Vitamin B12 Model Compounds. Angewandte Chemie International, 15, 417-426. http://dx.doi.org/10.1002/anie.197604171
- 9. Alper, T.O., Gazi, I., Hasene M., Tuncer, H. and Nagihan A. (2009) A Co(III) Complex with a Tridentate Amine-Imine-Oxime Ligand from 1,2,3,4-Tetrahydroquinazoline: Synthesis, Crystal Structure, Spectroscopic and Thermal Characterization. Journal of Coordination Chemistry, 62, 1005-1014. http://dx.doi.org/10.1080/00958970802345831
- 10. Gök, Y. and Kantekin, H. (1997) Synthesis and Characterization of Novel (E,E)-Dioximes and Its Mono- and Heterotrinuclear Complexes. Acta Chemica Scandinavica, 51,664-671. http://dx.doi.org/10.3891/acta.chem.scand.51-0664
- 11. Kuse, S. Motomizu, S. and Toei, K. (1974) Diketonedioxime Compounds as Analytical Reagents for the Spectrophotometric Determination of Nickel. Analytica Chimica Acta, 70, 65-76. http://dx.doi.org/10.1016/S0003-2670(01)82911-1
- 12. Mercimek, B., Ozler, M.A., Irez, G. and Bekaroglu, O. (1999) Synthesis of a Novel Heterocyclic Dioxime and its Mononuclear Complexes with Ni(II), Co(II), Cu(II), Zn(II), Cd(II) and Hg(II). Synthesis and Reactivity in Inorganic and Metal-Organic, 29, 513-524. http://dx.doi.org/10.1080/00945719909349466
- 13. Kurita, K. (1998) Chemistry and Application of Chitin and Chitosan. Polymer Degradation and Stability, 59, 117-120. http://dx.doi.org/10.1016/S0141-3910(97)00160-2
- 14. Soga, S., Sharma, S., Shiotsu, Y., Shimizu, M., Tahara, H., Yamaguchi, K., Ikuina, Y., Murakata, C., Tamaoki, T., Kurebayashi, J., Schulte, T., Neckers L. and Akinaga, S. (2001) Stereospecific Antitumor Activity of Radicicoloxime Derivatives. Cancer Chemotherapy Pharmacology, 48, 435-445. http://dx.doi.org/10.1007/s002800100373
- 15. Underhill, A.E., Watkins D.M. and Petring, R. (1973) Electrical Conduction Properties of Ni(dpg)2I, Ni(dpg)2Br, and Pd(dpg)2I (Where dpg = Diphenylglyoxime). Journal of Inorganic and Nuclear Chemistry, 9, 1269-1273. http://dx.doi.org/10.1016/0020-1650(73)80009-1
- 16. Park, H.J., Lee, K., Park, S.J., Ahn, B., Lee, J.C., Cho, H. and Lee, K.I. (2005) Identification of Antitumor Activity of Pyrazole Oxime Ethers. Bioorganic and Medicinal Chemistry Letters, 13, 3307-3312. http://dx.doi.org/10.1016/j.bmcl.2005.03.082
- 17. Thomas, T.W. and Underhill, A. E. (1972) Metal-metal Interactions in Transition-Metal Complexes Containing Infinite Chains of Metal Atoms. Chemical Society Reviews, 1, 99- 120. http://dx.doi.org/10.1039/CS9720100099
- 18. Hayati, S., Muzaffer, C. and Mustafa, M. (2005) Potentiometric and Theoretical Studies of Stability Constants of Glyoxime Derivatives and Their Nickel, Copper, Cobalt and Zinc Complexes. Acta Chimica Slovenica, 52, 317-322.
- 19. Ulku, S., Omer, D., Ercan, T. and Ayhan, O. (2012) Magnetic Properties of Single Crystal Alpha-Benzoinoxime: An EPR Study. Radiation Physics and Chemistry, 81, 146-151. http://dx.doi.org/10.1016/j.radphyschem.2011.08.013
- 20. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J. and Fox, D.J. (2009) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford.
- 21. Casida, M.E. (1995) In Recent Advances in Density Functional Methods, Part I. World Scientific, Singapore City.
- 22. Gross, E.K.U., Dobson, J.F. and Petersilka, M. (1996) Introduction Density Functional Theory II. Springer, Heidelberg.
- 23. Mariappan, G. and Sundaraganesan, N. (2014) Spectral and Structural Studies of the Anti- Cancer Drug Flutamide by Density Functional Theoretical Method. Spectrochimica Acta Part A, 117, 604-612. http://dx.doi.org/10.1016/j.saa.2013.09.043
- 24. Lee, C., Yang W. and Parr, R.G. (1988) Local Softness and Chemical Reactivity in Molecules CO, SCN- and H2CO. Journal of Molecular Structure, 163, 305-313. http://dx.doi.org/10.1016/0166-1280(88)80397-X
- 25. Parr, R.G. and Pearson, R.G. (1983) Absolute Hardness: Companion Parameter to Absolute Electronegativity. Journal of the American Chemical Society, 105, 7512-7516. http://dx.doi.org/10.1021/ja00364a005
- 26. Parr, R.G., Donnelly, R.A., Levy M. and Palke, W.E. (1978) Electronegativity—The Density Functional Viewpoint. Journal of Chemical Physics, 68, 3801-3807. http://dx.doi.org/10.1063/1.436185
- 27. Parr, R.G. and Yang, W. (1988) Density Functional Theory of Atoms and Molecules. Oxford Science Publications, New York.
- 28. Bayrak, C. and Bayari, S.H. (2010) Vibrational and DFT Studies of Creatinine and Its Metal Complexes. Hacettepe Journal of Biology and Chemistry, 38, 107-118.
- 29. Cramer, C.J. (2004) Essentials of Computational Chemistry: Theories and Models.4th Edition, John Wiley & Sons Ltd., West Sussex.
- 30. Lewars, E.G. (2011) Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics. 2nd Edition, Springer, New York. http://dx.doi.org/10.1007/978-90-481-3862-3
- 31. Foresman, J.B. and Frisch, A. (1996) Exploring Chemistry with Electronic Structure Methods. 2nd Edition, Gaussian Inc., Pittsburgh.
- 32. Panicker, C.Y., Varghese, H.T., Ambujakshan, K.R., Mathew, S., Ganguli, S., Nanda, A.K. and Van Alsenoy, C. (2009) FT-IR and FT-Raman Spectra and ab Initio Calculations of 3-{[(2-Hydroxyphenyl)Methylene]Amino}-2-Phenylquinazolin-4(3H)-One. Journal of Raman Spectroscopy, 40, 1262-1273. http://dx.doi.org/10.1002/jrs.2276
- 33. Teimouri, A.N., Chermahini, A., Taban, K. and Dabbagh, H. (2009) Experimental and CIS, TD-DFT, ab Initio Calculations of Visible Spectra and the Vibrational Frequencies of Sulfonylazide-Azoic Dye. Spectrochimica Acta Part A, 72, 369-377. http://dx.doi.org/10.1016/j.saa.2008.10.006