Computational Chemistry
Vol.04 No.01(2016), Article ID:63266,15 pages
10.4236/cc.2016.41003
Density Functional Theory Study of Exohedral Carbon Atoms Effect on Electrophilicity of Nicotine: Comparative Analysis
S. Dheivamalar1*, L. Sugi1, K. Ambigai2
1Department of Physics, Government Arts College for Women, (Autonomous) Pudukkottai, India
2Department of Physics, Bharath College of Science & Management, Thanjavur, India
![]()
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 2 December 2015; accepted 26 January 2016; published 29 January 2016
ABSTRACT
In recent years, many studies have been done on the structure of fullerene as medicine nano carrier compounds. On this basis, Quantum mechanical calculations have been done and the effect of the nicotine compound in structure of Nanofullerene C12 was studied. Density Functional Theory (DFT) can be used to calculate an accurate electronic structure, HOMO and LUMO energies, Mulliken charge of atoms, energetic orbital levels, global hardness, chemical potential and electrophilicity of systems, and finally chemical, physical properties of fullerene and fullerene derivatives. Theoretical calculations such as Natural Bond Orbital (NBO) are very important to understand the pathways of electron transfer in assemblies. Consequently, the obtained results showed that energy orbital levels decreased considerably by linking structure of Nanofullerene to the structure of Nicotine. The intramolecular interaction is formed by the orbital overlap between C-C, C-N, C-H anti bonding orbital which results an intermolecular charge transfer (ICT) from a Lewis valence orbital (donor), with a decreasing of its occupancy, to a non-Lewis orbital (acceptor). The interacting effect is also discussed in terms of the change in the C-C bond lengths, net atomic charge distribution, total dipole moment. The obtained results indicate that the C-C distances are enlarged interaction. Furthermore, there is a complete change in the net atomic charge distribution, as well as a corresponding increase in the value of the total dipole moment. On the basis of fully optimized ground-state structure, TDDFT//B3LYP/3-21G* calculations have been performed to determine the low-lying excited states of nanofullerene interacting with nicotine (NFN).
Keywords:
DFT, Electrophilicity, Chemical Hardness, Chemical Potential, Nano Fullerene, Nicotine

1. Introduction
Nicotine is a nitrogen organic compound which is mostly found in plants such as tobacco and rarely found in tomato, potato, eggplant and green pepper. The 0.3% to 5% of the tobacco, dried plant is made by nicotine and is effectively on neural system which is used in many insecticides. Nicotine was obtained from tobacco for the first time in 1828 by a German chemist [1] [2] . Melsen obtained its chemical formula experimentally in 1843 [3] and nicotine was processed for the first time in the laboratory in 1893 [4] . Nicotine is a biological stimulus in smaller sizes and causes addiction and many mental characteristics of tobacco smoke [5] [6] . This chemical material has a molecular formula of C10H14N2 and its chemical name is 3-[(2S)-1-methylpyrrolidin-2-yl] pyridine. This compound had different chemical and medical effects such as increased sobriety, memory, and activity, but it results in heartbeat, blood pressure and decreased appetite in larger sizes. This action causes interesting medicinal properties which increase rate of such characters by adding nano properties of this structure [7] -[9] . Extensive HF and DFT calculations on fullerene isomers of C12 and their derivatives have been performed. Relative stabilities of possible isomers of fullerene and the reaction reactivity of the most stable fullerene toward the addition of nicotine have been explored. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry. During this study we report the optimized geometries, and electronic structure calculations for the compound. The structure of the compound has been optimized by using the DFT (B3LYP) method with the 3-21G* basis sets, using the Gaussian 09 program. The electrophilicity index, which measures the stabilization in energy. The interacting effect is also discussed in terms of the change in the C-C bond lengths, net atomic charge distribution and total dipole moment.
The aim of the present work is to investigate the interaction of fullerene with nicotine by using the hybrid DFT-B3LYP functional in conjugation with 3-21G* basis set. These interactions show the stability of the structure. Density Functional Theory is used for calculating the electronic structure, HOMO and LUMO energies, Mulliken charge of atoms, Molecular orbital analyses of the title compound. Electronic properties increase the surface modification which is leading to the novel medical application. By investigating HOMO-LUMO energy gap, the chemical stability against electronic excitation also has been studied. This was done by discussing quantum chemical parameters, local reactivity indices such as a Fukui function in nanofullerene interacting with nicotine by natural bond orbital (NBO) analysis. Thus, it would also possible to produce novel species for biomedical application; by attaching the nitrogen atom of nicotine with the carbon atom of fullerene.
2. Computational Details
All structures relating to the structure of Nicotine and Nano fullerene nicotine (NFN) were designed primarily with the use of Gabedit 2.3.8 software. The computationally predicted various possible conformers are shown in Figure 1. The optimized molecular structure with the numbering of atoms of the title compound is shown in Figure 2. The most optimized structural parameters were also calculated by HF/ B3LYP have depicted in Table 1. Quantum chemical calculation was used for NFN to carry out the optimized geometry with the Gaussian 09W program [10] using the B3LYP and HF functional [11] [12] supplemented with standard 3-21G* basis set. Density Functional Theory (DFT) can be used to calculate an accurate electronic structure, HOMO and LUMO energies, Mulliken charge of atoms, energetic orbital levels, global hardness, chemical potential and electrophilicity of systems, and finally chemical, physical properties of fullerene and fullerene derivatives. Delocalization of electron density between the filled (bond or lone pair) Lewis type NBOs and empty anti-bonding non-Lewis NBOs calculated by NBO (Natural Bond Orbital) analyzing by B3LYP/3-21G* level. The HF and B3LYP methods have also been used to calculate the thermodynamic and electronic parameters like zero point vibrational energy (ZPVE) (kcalmol−1), rotational constants (GHz), heat capacity at constant volume (Cv) (cal・mol−1・K−1), entropy (S) (cal・mol−1・K−1), dipole moment µ (Debye), HOMO-LUMO energies (eV), frontier molecular orbital energy gaps (eV) and atomic charges (a.u) by natural bond orbital analysis.


![]()

![]()
![]()
![]()
![]()
Figure 1. Various possible conformers of Nano fullerene interacting with nicotine (NFN).
![]()
Figure 2. Optimized structure of Nanofullerene interacting with nicotine (NFN).

Table 1. Optimized geometrical parameters for Nanofullerene interacting with Nicotine (NFN) compared at HF, B3LYP levels with 3-21G* basis sets.
3. Results and Discussion
3.1. Global Reactivity Descriptors
The energies of frontier molecular orbital (εHOMO, εLUMO), energy band gap (εLUMO-εHOMO), electro negativity (χ), chemical potential (µ), global hardness (η), global softness (S), and global electrophilicity index (ω) [13] - [16] of Nicotine and NFN. The vertical ionization potential and vertical electron affinity of nicotine and nanofullerene with nicotine (NFN) have been computed at the HF and B3LYP/3-21G* level of theory. The total energies of cation and anion have been acquired on the same optimized geometry of the neutral molecule. Total energy gain upon saturation with electron as the electrophilicity index, atomic orbital contains a higher maximum value of NFN compared to Nicotine have been listed in Table 2. On the basis of εHOMO and εLUMO, these parameters are calculated using the equation (1) as given below
![]()
![]() (1)
(1)
![]()
![]()
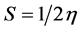
3.2. Dipole Moment, Polarizability, Hyperpolarizability, and Thermodynamic Properties
Dipole moment (µ), polarizability <α>, and total first static hyperpolarizability [17] [18] are also calculated by using density functional theory. These can be expressed in terms of x, y, and z components and are given in equation (2). The first hyperpolarizabilities (β) of this novel molecular system and related properties (β, α) of NFN were calculated using B3LYP/3-21G* basis set, based on the finite field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. Polarizabilities and hyperpolarizabilities characterize the response of a system in an applied electric field [19] .

The isotropic polarizability is
 (2)
(2)
and the average hyperpolarizability is

The total molecular dipole moment (μ), mean polarizability (α) and total static hyperpolarizability (β) of NFN molecule have been collected in Table 3. For Nicotine, the calculated dipole moment value is 3.86 Debye. The dipole moment of Nicotine is higher than the dipole moment of NFN. The dipole moment of NFN is calculated 13.1904 Debye. In the above compound αxx have given a greater contribution in the molecule which shows that the molecule is elongated more towards X direction and is more contracted to Y direction. βxxx and βxyy contribute with a larger part of hyperpolarizability in the molecule. These show that XX plane X-axis is more optically active in these directions. Standard thermodynamic functions such as free energy, constant volume heat capacity CV, and entropy S have also been calculated for NFN. These functions can provide helpful information for further study of the title compounds.
Table 2. Calculated (εHOMO, εLUMO), energy band gap (εLUMO-εHOMO), electronegativity (χ), chemical potential (µ), global hardness (η), global softness (S), and global electrophilicity index (ω) for the Nicotine and Nanofullerene interacting with Nicotine (NFN) by B3LYP/3-21G* levels of theory.
Table 3. Calculated values of polarizability and hyperpolarizability using HF/3-21G* and DFT/3-21G* for Nanofullerene interacting with Nicotine (NFN).
3.3. Frontier Molecular Orbital
The HOMO and LUMO energy were calculated by B3LYP/3-21G* method. The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron. The difference between HOMO and LUMO orbital is called as energy gap that is an important stability for structures. This electronic absorption corresponds to the transition from the ground to the first excited state and is mainly described by one electron excitation from the highest occupied molecular or orbital (LUMO) both the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbital take part in chemical stability. Therefore, while the energy of the HOMO is directly related to the ionization potential, LUMO energy is directly related to the electron affinity is shown in Figure 3. In principle, there are several ways to calculate the excitation energies. The frozen orbital approximation and the ground state properties are used to calculate the excitation values. This method is very practical, particularly in calculating large system [20] . Rigorously, the density functional methods which are based on Hohenberg and Kohn theorem [21] are designed to yield total energies. The HOMO energies, the LUMO energies and the energy gap for NFN molecules have been calculated using B3LYP level with 3-21G* basis set. The kinetic stability against electronic excitations is relevant to the LUMO-HOMO energy gap. In NFN the highest occupied molecular orbital is localized mainly on all atoms, the lowest unoccupied molecular orbital is also mainly on the carbon atoms having single bonds only in the ring, which also indicate that, the electron density transfer from n with the p* orbital, so electronic transitions from the HOMO to the LUMO are mainly derived from the electronic transitions of n → p*. From the result the HOMO energy = −5.10625 eV LUMO energy = −4.24310 eV and the Energy Gap = 5.02898 eV. Large HOMO-LUMO gaps are associated with higher kinetic stability, because it is not energetically favorable to add electrons to a high lying LUMO and to extract electrons from a low lying HOMO.
3.4. Electronic Spectra of Nano Fullerene C12 Interacting with Nicotine
On the basis of fully optimized ground-state structure, DFT/B3LYP/3-21G* calculations have been used to determine the low-lying excited states of fullerene. The theoretical results involving the vertical excitation energies, oscillator strength (f) and wavelength are carried out using the Gaussian 09 program. Electronic transition determined from excited-state calculations are listed in Table 4 shows that excited electron energy using HF is more than the value obtained in DFT. The excitation energy is 3.2610 (eV) in HF for the wavelength 380.21 (nm).
3.5. Mulliken Population Analysis
Mulliken atomic charge calculation has an important role in the application of quantum chemical calculation of molecular system because of atomic charge effect dipole moment, molecular polarizability, electronic structure and more a lot of properties of molecular systems. The atomic charge values were obtained by the Mulliken population analysis [22] . The Mulliken atomic charges of NFN are listed in Table 5. The charge changes with basis set presumably occur due to polarization. The charge of N (18) atom is −0.857865e for B3LYP/3-21G* and −1.044565e for HF/3-21G*. The charge distribution of nitrogen atom is increasing trend in HF and B3LYP method. In spite of the enhancement of charge separation, the dipole moments of nicotine and NFN are attributed to higher molecular symmetry. It is well known that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) play a predominant role in chemical reactions. The basis sets used in the atomic charge calculation, the carbon atoms exhibit a substantial negative charge, which are donor atom is shown in Figure 4 hydrogen atom exhibits a positive charge, which is an acceptor atom.

Figure 3. HOMO-LUMO energy level diagram for Nanofullerene interacting with nicotine (NFN).

Figure 4. Mulliken Atomic Charges the carbon atoms exhibit a sub- stantial negative charge, which are donor atom and Hydrogen atom exhibits a positive charge, which is an acceptor atom for Nanoful- lerene interacting with Nicotine (NFN) at B3LYP/ 3-21G*.
Table 4. Calculated parameters using TDDFT/B3LYP/3-21G*and HF/3-21G* for Nanofullerene interacting with Nicotine (NFN).
3.6. Local Reactivity Descriptors
The Fukui Function (FF) of a molecule provides information on the reactivity. The atom with the highest Fukui function value is highly reactive when compared to the other atoms in the molecule. These values represent the qualitative description of reactivity of different atoms in the molecule. The Fukui Function successfully predicts relative reactivity for most chemical systems and as such it provides a method for understanding and categorizing chemical reactions. The use of the Fukui Function for the selectivity of the nicotine molecule for nucleophilic and electrophilic attracts has been made with special emphasis to the dependence of the Fukui values on the basis to B3LYP/3-21G* level of theory. Ayers and Parr [23] have elucidated that molecules tend to react where the Fukui Function is the largest when attacked by soft reagents and in places where the Fukui Function is found to be smaller when attacked by hard reagents. Using the Mulliken atomic charges of neutral, cation, and anion, state of Nicotine, the Fukui Function ( ,
,  ,
, ), local softness (
), local softness ( ,
,  ,
, ), and local electrophilicity indices (
), and local electrophilicity indices ( ,
,  ,
, ) [15] [24] the Fukui Function are calculated using the equations (3, 4): The N
) [15] [24] the Fukui Function are calculated using the equations (3, 4): The N
Table 5. Calculated Values of Mulliken Atomic Charges (a.u.) for Nanofullerene interacting with Nicotine (NFN) at HF/3-21G* and DFT/3-21G* methods.
corresponds to the number of electrons in the molecule. N + 1 corresponds to an anion, with an electron added to the LUMO of the neutral molecule. N − 1 correspondingly is the cation with an electron removed from the HOMO of the neutral. All calculations are done at the ground-state geometry. These functions can be condensed to the nuclei by using an atomic charge partitioning scheme, such as Mulliken population analysis:

 (3)
(3)

Local softness and electrophilicity indices are calculated using (4)
 (4)
(4)

where +, −, and 0 signs show nucleophilic, electrophilic, and radical attack, respectively. Redistribution of the electrons on the f+ species the atomic charges of each atom are slightly increased (Table 6 and Figure 5). The maximum values of all the three local electrophilic reactivity descriptors ( ,
,  ,
, ) at N6 and N13 indicate
) at N6 and N13 indicate

Figure 5. Nicotine charges on the atomic sites of the fk and its ionic radicals.
Table 6. Fukui Functions ( ,
, ), local softnesses (
), local softnesses ( ,
, ), and local electrophilicity indices (
), and local electrophilicity indices ( ,
, ) for atomic charges of Nicotine, using the Mulliken population analysis at B3LYP/3-21G* level.
) for atomic charges of Nicotine, using the Mulliken population analysis at B3LYP/3-21G* level.
that this atom is nucleophilic attack, while for electrophilic attack, H23 are found to be the most active atoms. The calculated Fukui’s functions for all the inhibitors are presented in Table 6 as well as the corresponding population for the neutral and ionic species.
3.7. Natural Bond Orbital Analysis
Natural Bond Orbital Analysis was originally developed as a way of quantifying resonance structure contributions to molecules. NBO analysis is carried out by examining all possible interactions between “filled” (donor) Lewis-type NBOs and “empty” (acceptor) non-Lewis NBOs, and estimating their energetic importance of 2nd-order perturbation theory. NBO analysis is an essential tool for studying intra and intermolecular bonding and interaction among bonds, and also provides a convenient basis for charge transfer or conjugative interaction in molecular systems. NBO analysis has been performed on NFN molecule using Gaussian 03 package at the B3LYP/3-21G* level in order to understand various second order interactions between electron donors and electron acceptors. In the NBO analysis [25] , the hyper conjugative BD-BD* interactions play a highly important role. Delocalization of the electron density between occupied Lewis type (bond (or) lone pair) NBO orbital’s and formally unoccupied (anti-bond (or) Rydberg) non Lewis NBO orbital’s corresponding to a stabilizing donor-acceptor interaction. The energy of this interaction can be estimated by the second order perturbation theory [26] . The intermolecular interaction is formed by the orbital overlap between C-C, C-N, C-H anti bonding orbital which results an intermolecular charge transfer (ICT) from a Lewis valence orbital (donor), with a decreasing of its occupancy, to a non-Lewis orbital (acceptor). Table 7 shows that the first two columns give the type of
Table 7. NBO results showing the formation of Lewis and non Lewis orbital’s the valence hybrids corresponding to the intramolecular N… C Hydrogen bonds in NFN.
Table 8. The second order perturbation energies E(2) (kcal/mol) corresponding to the most important charge transfer interactions (donor-acceptor) in the compounds studied by B3LYP/ 3-21G* method.
orbital and occupancy between 0.8 and 1.9 electrons. Several other types of valence data, such as directionality, hybridization and partial charges were analyzed in Table 6, the output of NBO analysis electrons. In this table, BD (C27-C28) orbital with 1.99014 has 50.63% C27 character in a sp 3.02 hybrids and has 49.37% C28 character in a sp 2.81 hybrid. The sp3.02 hybrid on C27 has 75.12% p-character. The sp 2.81 hybrid on O has 73.74% p character. The BD (C27-C28) bond then corresponds roughly to the quantitative concept of interacting sp3 hybrids. The two coefficients, 0.7116 and 0.7026 are called polarization coefficients. The sizes of these coefficients show the importance of the two hybrids in the formation of the bond. The oxygen has the larger percentage of this NBO, at 50.63% and gives the larger polarization coefficient of 0.7026 because it has the higher electro negativity. Similarly BD (C5-C10), BD (C4-C9), BD (C13-N18), BD (C7-C8), BD (C6-C11), BD (C1-C2), BD (C13-H20), BD (C16-H22) bonding orbital are also shows hydrogen and nitrogen have the lesser percentage of NBOs and gives the lesser polarization coefficients as compared to BD (C27-C28) bond. This shows that nitrogen and hydrogen in above bonding orbital have less electronegative as compare to BD (C27-C28). Table 8 lists the calculated second order interaction energies (E (2)) between the donors-acceptor orbital’s in nano fullerene interacting with Nicotine (NFN). The most important interaction energies, related to the electron donating from the LP (1) C8, LP (1) N25, BD (C16-H22) to the anti-bonding acceptor BD* (C3-C8), BD* (C29-H36), BD* (C17-N18), BD* orbital’s and their corresponding energies are 7.60, 6.67, 4.36 Kcal/Mol, respectively. These interactions lead to the stability of these compounds.
3.8. Thermodynamic Properties
The values of some thermodynamic parameters such as zero-point vibrational energy, thermal energy, specific heat capacity, rotational constants, and entropy of nano fullerene with nicotine (NFN) at 298.15 K in ground state are listed in Table 9. The variation in zero-point vibrational energies (ZPVEs) seems to be significant. The HF/B3LYP level of ZPVE is much lower by the DFT/B3LYP. The biggest values of ZPVE of HF (NFN) are
Table 9. The thermodynamic parameters of Nanofullerene interacting with Nicotine (NFN) at B3LYP/ 3-21G* and HF/3-21G*.
Table 10. Thermodynamic parameters for Nanofullerene interacting with Nicotine (NFN) at B3LYP/3-21G* and HF/3-21G*.
195.957 a.u. Whereas the smallest values of ZPVE of B3LYP (NFN) are 181.9345 a.u obtained at B3LYP/ 3-21G*. Dipole moment reflects the molecular charge distribution and is given as a vector in three dimensions. Therefore, it can be used as a descriptor to depict the charge movement across the molecule. Direction of the dipole moment vector in a molecule depends on the centers of positive and negative charges. Dipole moments are strictly determined for neutral molecules. For charged systems, its value depends on the choice of origin and molecular orientation. On the basis of vibration analysis, the statically thermodynamic functions: heat capacity (Cp), entropy (S), and enthalpy changes (H), Gibbs free energy for the title molecule were obtained from the theoretical thermodynamic parameters are listed in Table 10.
4. Conclusion
In this work, the optimized geometry of the “nano fullerene interacting with nicotine” molecule has been determined by the method of density functional theory (DFT). On the basis of fully optimized ground-state structure, TDDFT//B3LYP/3-21G* calculations have been used to determine the low-lying excited states of “Nanofullerene interacting with Nicotine.” The hyperpolarizabilities indicate a possible use of these compounds in electro optical applications. The dipole moment of Nicotine is higher than the dipole moment of NFN. We have also discussed global and local reactivity descriptors sites for Nicotine molecules during electrophilic, nucleophilic and radical attack. The charge distribution of nitrogen atom is increasing trend in HF and B3LYP method. These values represent the qualitative description of reactivity of different atoms in the molecule. NBO analysis indicated that the higher second-order perturbation interaction (E2) and the electronic, chemical potential energy (µ) and the HOMO-LUMO gap in Nanofullerene interacting with Nicotine. This compound shows that the stability of the molecules increases because of interaction of nicotine.
Cite this paper
S.Dheivamalar,L.Sugi,K.Ambigai, (2016) Density Functional Theory Study of Exohedral Carbon Atoms Effect on Electrophilicity of Nicotine: Comparative Analysis. Computational Chemistry,04,17-31. doi: 10.4236/cc.2016.41003
References
- 1. Posselt, W. and Reimann, L. (1828) Geiger’s Magazinfür Pharmacies, 6, 138.
- 2. Henning Field, J.E. and Zeller, M. (2006) Psychopharmacology, 184, 286.
- 3. Melsens (1844) Journal für Praktische Chemie, 32, 372.
- 4. Pictet, A. and Crepieux, P. (1903) Comptesrendus, 137, 860.
- 5. Hoffmann, D. and Hoffmann, I. Smoking and Tobacco Control Monograph, 9, 55.
- 6. Siegmund, B., Leitner, E. and Pfannhauser, W. (1999) Determination of the Nicotine Content of Various Edible Nightshades (Solanaceae) and Their Products and Estimation of the Associated Dietary Nicotine Intake. Journal of Agriculture and Food Chemistry, 47, 3113-3120. http://dx.doi.org/10.1021/jf990089w
- 7. Hirsch, A. and Brettreich, M. (2005) Fullerenes: Chemistry and Reactions. Willey VCH Verlag GmbH.
- 8. Holister, P., Roman, C. and Harper, T. (2003) Fullerenes. Cientifica Ltd.
- 9. Panina, L.K., Kurochkin, V.E., Bogomolova, E.V., Evstrapov, A.A. and Spitsyna, N.G. (1997) Doklady Biological Science, 357, 530.
- 10. Frisch, J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., et al. (2009) Gauss 09, Rev A. Vol. 11.4 Gaussian Inc., Pitts Burgh.
- 11. Becker, A.D. (1993) Density-Functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics, 98, 5648-5652. http://dx.doi.org/10.1063/1.464913
- 12. Lee, C., Yang, W. and Parr, R.G. (1998) Physical Review, 1377, 785-789.
- 13. Pearson, R.G. (1989) Absolute Electro Negativity and Hardness: Applications to Organic Chemistry. Journal of Organic Chemistry, 54, 1423-1430. http://dx.doi.org/10.1021/jo00267a034
- 14. Parr, R.G., Szentpaly, L.V. and Liu, S. (1999) Electrophilicity Index. Journal of the American Chemical Society, 121, 1922-1924. http://dx.doi.org/10.1021/ja983494x
- 15. Chattaraj, P.K. and Giri, S. (2007) Stability, Reactivity, and Aromaticity of Compounds of a Multivalent Super Atom. Journal of Physical Chemistry A, 111, 11116-11121. http://dx.doi.org/10.1021/jp0760758
- 16. Padmanabhan, J., Parthasarathi, R., Subramanian, V. and Chattaraj, P.K. (2007) Electrophilicity-Based Charge Transfer Descriptor. Journal of Physical Chemistry A, 111, 1358-1361. http://dx.doi.org/10.1021/jp0649549
- 17. Kleinman, D.A. (1962) Nonlinear Dielectric Polarization in Optical Media. Physical Review B, 126, 1977-1979. http://dx.doi.org/10.1103/PhysRev.126.1977
- 18. Pipek, J. and Mezey, P.G. (1989) A Fast Intrinsic Localization Procedure Applicable for ab Initio and Semi Empirical Linear Combination of Atomic Orbital Wave Functions. The Journal of Chemical Physics, 90, 4916-4926. http://dx.doi.org/10.1063/1.456588
- 19. Krishnakumar, V., Keresztury, G., Sundius, T. and Ramasamy, R. (2004) Simulation of IR and Raman Spectra Based on Scaled DFT Force Fields: A Case Study of 2-(Methylthio)benzonitrile, with Emphasis on Band Assignment. Journal of Molecular Structure, 702, 9-21. http://dx.doi.org/10.1016/j.molstruc.2004.06.004
- 20. Curtiss, L.A. and Reghavachari, P.C. (1998) The Journal of Chemical Physics, 42, 117-122.
- 21. Hohenberg, P. and Kohn, W. (1964) Inhomogeneous Electron Gas. Physical Review B, 136, 864-871. http://dx.doi.org/10.1103/PhysRev.136.B864
- 22. Mulliken, R.S. (1955) Electronic Population Analysis on LCAO[Single Bond]MO Molecular Wave Functions. I. The Journal of Chemical Physics, 23, 1833-1840. http://dx.doi.org/10.1063/1.1740588
- 23. Ayers, P.W. and Parr, R.G. (2000) Variational Principles for Describing Chemical Reactions: The Fukui Function and Chemical Hardness Revisited. Journal of the American Chemical Society, 122, 2010-2018. http://dx.doi.org/10.1021/ja9924039
- 24. Parr, R.G., Szentpaly, L.V. and Liu, S. (1999) Electrophilicity Index. Journal of the American Chemical Society, 121, 1922-1924. http://dx.doi.org/10.1021/ja983494x
- 25. Reed, A.E., Curtiss, L.A. and Weinhold, F. (1988) Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chemical Reviews, 88, 899-926. http://dx.doi.org/10.1021/cr00088a005
- 26. Chocholousova, J., Vladimir Spirko, V. and Hobza, P. (2004) Chemical Physics Society, 6, 37.
NOTES

*Corresponding author.