Computational Chemistry
Vol.02 No.02(2014), Article ID:45439,10 pages
10.4236/cc.2014.22005
On the Accuracy of the Complete Basis Set Extrapolation for Anionic Systems: A Case Study of the Electron Affinity of Methane
Alejandro Ramírez-Solís*
Laboratoire de Physico-Chimie des Nano-Objets, INSA-Université de Toulouse. 135, Av. de Rangueil, Toulouse, F-31075, France
Email: alex@uaem.mx
Copyright © 2014 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 2 March 2014; revised 7 April 2014; accepted 15 April 2014
ABSTRACT
Recent experimental evidence suggests again the existence of the metastable methane anion in plasma swarms. In order to test the reliability of the complete basis set (CBS) extrapolation scheme with augmented correlation-consistent basis sets for anionic molecules, we study the evolution of the electron affinity of methane with benchmark ab initio calculations with aug-cc basis sets up to aug-cc-pV6Z + diffuse. Geometry optimizations and vibrational analysis were done at the MP2 level. The electron affinity (EA) was calculated at the MP2 and CCSD(T) levels with and without frozen core and including the extrapolations to the CBS limit. Using the aug-cc-pVnZ basis sets it is found that two non-decreasing CCSD(T) CBS limits exist for the EA (0.29 and 0.53 eV) obtained with the n = 3, 4, 5 and n = 4, 5, 6 series, respectively. A new scheme is proposed which can be generalized for very accurate quantum chemical description of molecular anions: the standard aug-cc-pVnZ basis sets can be supplemented with extra-diffuse orbitals using a simple even-tempered scheme. This yields a reliable CBS extrapolation method to develop a (discrete approximation of a) continuum anionic state near ionization, viz., one that closely matches the energy of the corresponding neutral state. These results show that CH4 has no stable anions of 2A1 symmetry, implying that plasma swarms with anionic methane consist of metastable rather than stable methane anions.
Keywords:
Methane Electron Affinity, Anion Correlated Calculations, Complete Basis Set Limit

*On sabbatical leave from Facultad de Ciencias, Universidad Autonoma del Estado de Morelos, Cuernavaca, Morelos, México.

1. Introduction
It is known that methane is a very stable molecule. It is characterized by five closed shells and its electronic configuration resembles than of the neon atom. Resembling a noble gas atom, it is natural to expect that the methane anion is unstable with respect to the neutral molecule. However, clear experimental evidence of the existence of the methane anion as a metastable species has been provided by several groups [1] -[6] over a period of 50 years. The first observations of electron attachment in methane were reported by Trepka and Neurt [1] and by Sharp and Dowell [2] , who used a beam technique and found that dissociative attachment in CH4 took place over the range of electron energies 8 - 13 eV, with 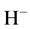 and
and 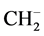 as dissociative product ions. Later, using a pulsed Townsend technique, Hunter et al. [3] measured the density-normalized electron-attachment coefficient over the density-normalized electric field strength, E/N, in the range 52.5 - 250 Td (1 Townsend = 10−17 V cm2). Electron-impact ionization processes have been thoroughly studied for gaseous methane over a very wide range of the density-normalized electric field strength up to 50,000 Td, also suggesting the presence of the methane anion [4] . More than a decade ago de Urquijo et al. [5] studied electron-ionization and electron-attachment processes in methane over the density-normalized electric field strength in the range 120 - 3000 Td through a pulsed Townsend swarm technique. The time-resolved ionic avalanches were obtained under conditions such that the occurrence of electron-attachment processes was detected unambiguously, thus providing irrefutable evidence of the presence of the methane anion. More recently, very accurate swarm measurements of the effective ionization coefficient of CH4/N2 mixtures at high pressures (100 - 600 Torr) reveal again the presence of negative methane ions in the avalanche, formed by a three-body collision process [6] . However, in spite of all these studies showing the formation of the methane anion, to the best of our knowledge, only a single reference reports a direct measurement of the electron affinity (EA) of methane, where it has been determined to be +1.2 eV, i.e., the neutral molecule being slightly more stable than the anion [7] . Of course, the positive value of the EA implies that the anionic methane eventually disintegrates, following at least the two known dissociative pathways [2] .
as dissociative product ions. Later, using a pulsed Townsend technique, Hunter et al. [3] measured the density-normalized electron-attachment coefficient over the density-normalized electric field strength, E/N, in the range 52.5 - 250 Td (1 Townsend = 10−17 V cm2). Electron-impact ionization processes have been thoroughly studied for gaseous methane over a very wide range of the density-normalized electric field strength up to 50,000 Td, also suggesting the presence of the methane anion [4] . More than a decade ago de Urquijo et al. [5] studied electron-ionization and electron-attachment processes in methane over the density-normalized electric field strength in the range 120 - 3000 Td through a pulsed Townsend swarm technique. The time-resolved ionic avalanches were obtained under conditions such that the occurrence of electron-attachment processes was detected unambiguously, thus providing irrefutable evidence of the presence of the methane anion. More recently, very accurate swarm measurements of the effective ionization coefficient of CH4/N2 mixtures at high pressures (100 - 600 Torr) reveal again the presence of negative methane ions in the avalanche, formed by a three-body collision process [6] . However, in spite of all these studies showing the formation of the methane anion, to the best of our knowledge, only a single reference reports a direct measurement of the electron affinity (EA) of methane, where it has been determined to be +1.2 eV, i.e., the neutral molecule being slightly more stable than the anion [7] . Of course, the positive value of the EA implies that the anionic methane eventually disintegrates, following at least the two known dissociative pathways [2] .
From the experimental side work is currently in progress through a hybrid swarm/mass spectrometry technique to directly measure the electron affinity of metastable methane [8] with very high precision (c.a. 10−2 eV). However, from the quantum chemical point of view, the fact that anionic methane is metastable (i.e. that the charged 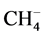 species will eventually produce the CH4 plus a free electron), has a strict logical implication: for a given level of theory, the energy of the anion should approach that of methane as the quality of the atomic basis sets improves. This stems from the fact that larger and more diffuse basis sets will allow the extra electron to move away from the neutral molecule thus lowering the total energy of the (CH4, e-) pair. In this direction we note that that previous electronic structure studies have placed the EA of methane between 6.1 and 1.2 eV [9] using MP2, G2MP2, MP4 and QCISD methods with a variety of gaussian basis sets, ranging from 6-31G(d, p) to 6-3111++G(3df, 2p). However, when the complete basis set (CBS) extrapolation of Peterson et al. [10] was applied, the EA was estimated to be 0.5 eV, less than half the smallest value (1.2 eV) obtained with the G1 [11] , G2 [12] and G2MP2 [13] methods. While in [9] the decrease of the EA with basis set quality is clear, we note that none of the electronic structure methods used the Dunning correlation-consistent (cc-pVnZ) basis sets [14] . These basis sets were specifically developed to smoothly converge toward the complete (infinite) basis set limit of each atom using the Configuration Interaction Singles and Doubles (CISD) method. We note that diffuse basis have also been developed building on the cc-pVnZ basis for each atom and these are known to be particularly important to deal with anions. A prefix aug means one set of diffuse functions is added for every angular momentum-component present in the basis; for instance, the aug-cc-pVTZ basis for carbon has diffuse s, p, d and f orbitals.
species will eventually produce the CH4 plus a free electron), has a strict logical implication: for a given level of theory, the energy of the anion should approach that of methane as the quality of the atomic basis sets improves. This stems from the fact that larger and more diffuse basis sets will allow the extra electron to move away from the neutral molecule thus lowering the total energy of the (CH4, e-) pair. In this direction we note that that previous electronic structure studies have placed the EA of methane between 6.1 and 1.2 eV [9] using MP2, G2MP2, MP4 and QCISD methods with a variety of gaussian basis sets, ranging from 6-31G(d, p) to 6-3111++G(3df, 2p). However, when the complete basis set (CBS) extrapolation of Peterson et al. [10] was applied, the EA was estimated to be 0.5 eV, less than half the smallest value (1.2 eV) obtained with the G1 [11] , G2 [12] and G2MP2 [13] methods. While in [9] the decrease of the EA with basis set quality is clear, we note that none of the electronic structure methods used the Dunning correlation-consistent (cc-pVnZ) basis sets [14] . These basis sets were specifically developed to smoothly converge toward the complete (infinite) basis set limit of each atom using the Configuration Interaction Singles and Doubles (CISD) method. We note that diffuse basis have also been developed building on the cc-pVnZ basis for each atom and these are known to be particularly important to deal with anions. A prefix aug means one set of diffuse functions is added for every angular momentum-component present in the basis; for instance, the aug-cc-pVTZ basis for carbon has diffuse s, p, d and f orbitals.
The aug-cc-pVnZ basis sets were developed applying stringent conditions on the atoms, but their combination yields molecular basis sets that, in general, provide similar asymptotic behavior of the most important molecular quantities to their CBS limit values, such as energy, equilibrium geometries and harmonic vibrational frequencies [15] . In this study we aim at finding out if the expected convergence of the energy of the methane anion with correlated ab initio methods follows the expected behavior, i.e., that the calculated electron affinity of methane goes to zero with increasing basis set quality using the aug-cc-pVnZ series. As we shall see, even with the largest augmented correlation-consistent basis sets, the most widely used CBS extrapolation scheme fails, leading in this case to unreliable predictions concerning the energy difference of the anion with respect to neutral methane. Section II presents the methodology and computational details. Section III presents a discussion of the applicability of the CBS extrapolation for the EA and we show how the extended aug-cc basis sets can be modified to yield the correct CBS electron affinity. We conclude with some comments in the more general context of methane-anion production and detection in plasma swarms.
2. Computational Methods
Atomic Basis Sets and Electronic Correlation Treatments
From the work of Jursic [9] it is clear that both, the quality of the atomic basis sets and the electronic correlation effects are crucial for an accurate determination of the EA of methane. More than 14 years have passed since that work was published and computer resources have vastly improved since then. Although the correlation- consistent atomic basis sets and their augmented counterparts (aug-cc) were already available at the time of that investigation, the large computational cost of using such extended basis sets was such that no MP2 or Coupled Cluster calculations were performed for the neutral and ionic species of methane with these correlation-consis- tent basis sets.
With present day computational resources, electronic structure calculations at the Coupled Cluster with singles, doubles and perturbative triple excitations -CCSD(T)- level can be done in a few days or weeks even with the largest aug-cc basis sets in parallel computers with large core memories. In this work we shall use the aug-cc-pVnZ basis sets with n = 2, 3, 4, 5, 6 and 6 + diffuse [16] , which will be abbreviated as AVDZ, AVTZ, AVQZ, AV5Z, AV6Z and AV6Z + diff, respectively. Note that these imply benchmark-type calculations since the largest basis sets involve nearly 900 molecular orbitals for 10 (methane) and 11 (the anion) electrons.
Table 1 presents the total number of basis functions and their orbital decomposition on each atom. Since the electronic structure of methane and its anion are strongly monoreferential, their geometries have been optimized at the MP2 (UMP2 for the anion) level with every basis set up to aug-cc-pV6Z. In order to assess the role of the core-valence correlation effects on the geometries and the EA, we have performed both frozen-core (FC) and full MP2 calculations. For the MP2 optimizations tighter convergence criteria were used to obtain geometries within 2 × 10−4 angstroms. MP2 vibrational analysis of the optimized structures was done for each basis set up to aug-cc-pV5Z. The CCSD(T) (and the unrestricted counterpart for the anion) single-point calculations were performed using the MP2 optimized geometries obtained with and without the frozen core for each basis set, thus the CCSD(T) calculations were also done with and without the frozen core. To give an idea of the computational resources needed, the (U)MP2(full) optimizations and the (U)CCSD(T)-full single point calculations with the aug-cc-pV6Z + diffuse basis sets required 40(17 + 23) and 133(56 + 77) CPU days, respectively. The Gaussian03 parallel code [17] was used throughout. Complete basis set limits have been obtained for the geometric parameters and for the EA with the mixed exponential/gaussian extrapolation approach of [18] , which has been shown to yield better convergence properties than the simple exponential form [19] .

Table 1. Basis set composition and total number of basis functions. The atomic decomposition shows the number of contracted basis function on each atom.
3. Results and Discussion
3.1. The Electronic Structure of 
The methane anion has a 2A1 ground state, the extra electron occupying a molecular orbital which is basically derived from the 3 s virtual orbital of carbon, with small equal contributions from all four hydrogen 2 s atomic orbitals. Thus the anion can be described as arising from the first Rydberg state on the molecule. Figure 1 shows the singly occupied HOMO of the methane anion with the aug-cc-pV6Z basis set.
3.2. Geometry and Vibrational Spectrum of 
As mentioned in the previous section, in order to calculate the EA with the CCSD and CCSD(T) methods, we have used the MP2 optimized geometries of methane and its anion corresponding to a given basis set with and without the frozen core orbital. Both species are tetrahedral (Td point group) so that the C-H distances completely describe their geometries. The evolution of these distances with basis set quality is presented in Table 2 along with the corresponding CBS limits. Note that while the MP2(FC) C-H distance for methane is already converged at 1.0844 Å with the AVQZ basis set, for the anion it continues to decrease slightly (less than 0.001 Å) with the AV5Z basis set. However, C-H distances are already converged to less than 0.004 Å with the AVTZ basis set, both for the neutral molecule and for the anion (see Table 2). The C-H distances for the neutral molecule and for the anion are shortened by 0.003 - 0.004 Å when the MP2 optimizations are done without the frozen core and the larger basis sets. It is interesting to note that the anion C-H distance is only 0.0023 Å longer than for the neutral molecule at the MP2(full)/AV6Z + diffuse level, in agreement with the fact that the extra electron in the HOMO of the anion lies mostly outside the nuclear framework. For comparison, Table 2 also shows the previous optimized C-H distances of methane and the anion at the MP2 level with other basis sets from [9] .
The vibrational spectra of methane and its anion have been obtained at the MP2(full) level for the aug-cc-pVnZ (n = 2, 3, 4, 5) basis sets. We found that the vibrational frequencies of both species are already converged to a few (<5) wavenumbers at the MP2/AV5Z level, thus Table 3 shows the corresponding spectra at this level of theory. It is important to note that while the vibrational frequencies of both species are very similar they are still not identical; for the anion all of them are slightly (13 - 35 cm−1) lower than those of the neutral molecule, even with the largest aug-cc basis sets. A discussion on the reliability of these results is given in the following sections.
3.3. Evolution of Electron Affinity with Basis Set Quality
Table 4 shows the total energies of methane and its anion along with the EA at the MP2(FC), MP2(full), CCSD(T)-FC and CCSD(T)-full levels with the various aug-cc-pVnZ basis sets. CCSD values have been omitted from that table since their derived EAs are very similar to those obtained at the MP2 level with all the basis

Figure 1. The singly occupied molecular orbital of the methane anion (isosurface = 0.002). While in line with the Td symmetry of the nuclear framework, note the nearly spherical shape of the HOMO.
Table 2. Evolution of the MP2 optimized C-H distances (Å) for methane and its anion. First column with frozen core, second column without frozen core orbitals.
aPrevious values from [9] . bAfter 48 steps: Max displacement = 0.000306 a.u., RMS displacement = 0.00016 a.u. cAfter 27 steps: Max displacement = 0.000287 a.u., RMS displacement = 0.00017 a.u. dAfter 64 steps: Max displacement = 0.000313 a.u., RMS displacement = 0.00014 a.u.
Table 3. MP2/AV5Z vibrational frequencies (cm−1) for methane and its anion.
sets used here. For comparison we have also included the previous estimations obtained at different levels of theory.
A careful analysis of this table reveals interesting facts. Jursic [9] showed that basis set quality is pivotal for an accurate determination of the electron affinity; for instance, at the MP2(FC) level, the EA is 6.1 eV with the 6 - 31 G basis sets while the value drops down to 0.9 with the 6-311++G(3df, 3pd) basis sets. Previous extrapolations such as G1, G2 and G2MP2 all lead to a value of 1.2 eV for the EA [9] . Here, at all levels of theory, the EA diminishes monotonically with basis set quality all the way to the aug-cc-pV5Z basis set. At the CCSD(T)-FC level the EA goes from 6.05 eV with the 6 - 31 G** basis set to 0.41 eV with the latter basis. Both the MP2 (frozen core and full) electron affinities are marginally larger (c.a. 0.04 eV) than the CCSD(T) values with all basis sets. The CCSD EAs obtained are very similar to the MP2 values with all basis sets used here. This shows that the triple excitations, which are absent in the MP2 scheme but are taken into account perturbatively in the CCSD(T) approach, systematically favor the stabilization of the anion with respect to the neutral molecule by around 0.03 eV. However, quite unexpectedly, when the aug-cc-pV6Z basis sets are used the EA becomes larger (0.08 eV with CCSD(T) and 0.07 eV with MP2) than the values obtained with the aug-cc-pV5Z basis sets. With the frozen-core approximation this surprising result arises since, while for the neutral molecule the MP2 and CCSD(T) energies are lower with the AV6Z than with the AV5Z basis sets, for the anion the total frozen core MP2 and CCSD(T) energies are c.a. 1.2 mH higher with the AV6Z basis sets than with the AV5Z ones.
We note that the aug-cc-pV6Z + diffuse basis sets yield the same optimized MP2 distances and the same electron affinity values as the ones obtained with the aug-cc-pV6Z basis sets, at all levels of theory. Figure 2 shows the evolution of the electron affinity of methane (eV) as function of the cardinal number n in the aug-cc-pVnZ sequence at the CCSD(T)-full level. With this puzzling result we pass now to the CBS extrapolation.
3.4. CBS Extrapolations of Electron Affinity
Following the suggestion of Peterson et al. [18] the property (F) for each correlation consistent basis set with cardinal number n is approximated using a mixed exponential/gaussian function of the form
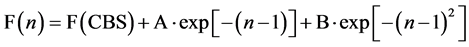 , so that three computed values of F are needed to de-
, so that three computed values of F are needed to de-
Table 4. Evolution of the MP2 and CCSD(T) (frozen core, full) total energies (a.u.) with basis set quality for methane and its anion. Electron affinities (EA) in eV.
aThese CCSD(T) calculations required 133 CPU days and 96 Gb RAM; bFrom [9] .
termine A, B and the asymptotic complete basis set limit value of F, F(CBS). This is usually achieved using the properties obtained with n = 2, 3 and 4. In this case, since we have the larger sequence n = 2, 3, 4, 5, 6, 6 + diffuse, we have chosen to explore the asymptotic EA with two sequences (n = 3, 4, 5) and (n = 4, 5, 6), leading to two estimations labeled EA(CBS-345) and EA(CBS-456), respectively. Table 5 presents these CBS limits at each level of theory.
Focusing on the results obtained with the most accurate correlation treatment, the CCSD(T)-full method, the table shows that in the first case we obtain 0.29 eV while, quite surprisingly, the second extrapolation yields a

Figure 2. Evolution of the electron affinity of methane (eV) with the cardinal number n in the aug-cc-pVnZ sequence at the CCSD(T)-full level.
Table 5. Asymptotic CBS electron affinities (eV) with two correlation-consistent basis set sequences (n = 3, 4, 5) and (n = 4, 5, 6), leading to two estimations labeled EA(CBS-345) and EA(CBS-456), see the text.
larger value of 0.53 eV. Although the 0.24 eV difference between these two asymptotic values is energetically small, the percent relative error between them is very large,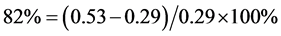 . This fact reveals some of the problems discussed in [20] concerning the robustness of approximate extrapolation schemes when using extremely large correlation-consistent basis sets. Some of the fundamental questions raised are: what are the scaling properties of the basis set superposition error with very large correlation-consistent basis sets? Does the coupling of the BSSE and electron correlation energy affect the CBS convergence? In our case, since the optimized C-H distances show the expected convergence properties for both the neutral and the ionic species, we might add another question: is this coupling more important for energetic properties than for structural ones? Is it reasonable to trust the optimized distances and vibrational frequencies of the anion obtained with the CBS scheme using the aug-cc-pVnZ series when such a flawed description appears for the energy?
. This fact reveals some of the problems discussed in [20] concerning the robustness of approximate extrapolation schemes when using extremely large correlation-consistent basis sets. Some of the fundamental questions raised are: what are the scaling properties of the basis set superposition error with very large correlation-consistent basis sets? Does the coupling of the BSSE and electron correlation energy affect the CBS convergence? In our case, since the optimized C-H distances show the expected convergence properties for both the neutral and the ionic species, we might add another question: is this coupling more important for energetic properties than for structural ones? Is it reasonable to trust the optimized distances and vibrational frequencies of the anion obtained with the CBS scheme using the aug-cc-pVnZ series when such a flawed description appears for the energy?
We know that in this case the physical problem at hand has to do with the way the extra electron interacts with the neutral neon-like core of methane, which can be translated into what is the exact spatial distribution of the HOMO for the anion? In the following section we address this problem by providing extra degrees of freedom to the unpaired electron of the anion.
3.5. Addition of Extra-Diffuse Orbitals to the aug-cc-pV6Z Basis Set
The appearance of a non-monotonic behavior of the electron affinity with increasing n in the aug-cc-pVnZ series at all levels of theory casts serious doubts on the reliability of these basis set for a truly accurate description of the anion. In particular, knowing that the electron affinity must decrease with basis set quality, it is completely against the core philosophy of the aug-cc-pVnZ basis sets that larger estimations of the EA are obtained with the n = 4, 5, 6 series than with the n = 3, 4, 5 series. Therefore we have decided to add one to four diffuse orbitals on the carbon atom to study the convergence of the energy for methane and for the anion at the CCSD(T)-full level. These diffuse orbitals were chosen with spherical symmetry for two reasons: 1) the HOMO of the anion closely approaches a spherical distribution with increasing basis set quality and, 2) there is only one orbital per added exponent against three or five harmonics for p and d symmetries so that these very large CCSD(T) calculations are faster (recall the O(n6) behavior of the method). The four extra-diffuse s exponents (diff2 = 0.012, diff3 = 0.004, diff4 = 0.0013, diff5 = 0.0004) have been chosen using an even-tempered scheme with α = 1/3, taking as starting point the exponent (diff1 = 0.0354) of the last s orbital of the aug-cc-pV6Z + diffuse carbon basis set. In this way a sequence of aug(m)-aug-cc-pV6Z basis sets can be defined with a new cardinal number m = 7, 8, 9, 10 and 11, where m is the sum of n = 6 plus the number of diffuse s orbitals added to the aug-cc-pV6Z basis set of carbon; therefore, m = 7 corresponds to the published aug-cc-pV6Z + diffuse basis set. Table 6 shows the energies of methane and its anion as well as the electron affinity calculated with each of these aug(m)-aug-cc- pV6Z basis sets. The molecular geometries are those optimized at the MP2/aug-cc-pV6Z level.
As expected, the CCSD(T) energy of methane shows much smaller changes (ca. 10−6 a.u.) from the aug(7)- AV6Z to the aug(11)-AV6Z basis set, while the energy decrease of the anion is three orders of magnitude larger. With the largest basis set the energy of the anion is only 13 meV higher than that of the neutral molecule thus yielding an EA nearly 40 times smaller than that estimated with the aug-cc-pV6Z + diffuse basis set. Figure 3 shows the evolution of the CCSD(T) with these extra-diffuse basis sets.
Although not strictly zero, using the same three-parameter CBS extrapolation scheme we obtain now −0.003 eV as the limit for the electron affinity of methane with the m = 9, 10, 11 sequence. This is consistent with the expected asymptotic behavior of the EA towards zero with increasing basis set quality. Accordingly, note in Ta- ble 6 that the energy of the HOMO of the anion monotonically decreases towards zero, thus confirming the CH4 + e− asymptotic description of the anion. With this knowledge, we have performed a new MP2 optimization of
Table 6. CCSD(T)-full total energies (a.u.) with diffuse and extra-diffuse s orbitals added to the aug-cc-pV6Z + diffuse basis set for methane and its anion. For details of the exponents, see text. Electron affinities (EA) in eV and HOMO energy of the anion in a.u.

Figure 3. Evolution of the CCSD(T)-full electron affinity of methane (eV) with the new cardinal number m in the aug(m)-aug-cc-pV6Z sequence of carbon basis sets; m = 7 corresponds to the aug-cc-pV6Z + diffuse basis set, see the text. The curve in red shows the extrapolated values for m up to 14.
the anion with the aug(11)-aug-cc-pV6Z basis set and, as expected, the C-H distance decreases to 1.0815 Å, in closer agreement with that obtained for neutral methane with the aug-cc-pV6Z basis set. However, we note that after 16 optimization steps the Max Displacement = 0.000319 a.u. and RMS displacement = 0.00016 a.u parameters are found but the geometry optimization has not been completely achieved due to small oscillations (<0.001 and <0.0008) of the Max-force and RMS-force parameters; this leads to non-compliance of the convergence criteria thus precluding the calculation of the vibrational spectrum of the anion with the aug(11)-aug-cc- pV6Z basis set. These oscillations are due to an unbalanced growth of the l = 0 diffuse basis set and other higher angular momentum orbitals might be needed to stabilize the geometry optimization procedure with such a large basis set.
4. Conclusions
Experimental evidence for the existence of anionic methane has accumulated over the last 50 years in plasma swarm experiments and the best previous theoretical extrapolated estimations have placed the electron affinity of methane in the 1.2 - 0.5 eV range. However, the various extrapolation schemes used were not based on the augmented and correlation-consistent pVnZ series of basis sets, which are known to yield the correct asymptotic behavior with basis set quality for a wide variety of molecular systems.
In order to test the reliability of the electron affinity with the widely used complete basis set extrapolation scheme in this complex case, geometry optimizations and vibrational calculations for methane and its anion were performed at the MP2 level with the aug-cc-pVnZ (n = 2, 3, 4, 5, 6 and 6-diffuse) basis sets. Using these optimized geometries we have performed single-point CCSD(T) calculations in order to obtain the electron affinity of methane with each basis set. The MP2 and CCSD(T) calculations were done with and without the frozen core approximation and, as expected, yield practically identical energetic results.
As reported in previous studies, the methane EA systematically decreases with basis set quality at the MP2, CCSD and CCSD(T) levels of theory. Using the aug-cc-pVnZ (n = 3, 4, 5) series, the CBS limit for the electron affinity is 0.29 eV at the CCSD(T) level. However, with the aug-cc-pVnZ (n = 4, 5, 6) series the CBS limit yields an even larger (0.53 eV) electron affinity, thus revealing the inadequacy of the CBS extrapolation scheme with the aug-cc-pVnZ series of basis sets for the anionic molecule. However, when even-tempered diffuse and extra-diffuse orbitals are added to the aug-cc-pV6Z + diffuse basis set this leads to further lowering of the CCSD(T) energy of the anion, so that the new CBS energy limit correctly converges to that of CH4 + e−. MP2 optimizations for the anion using these aug(m)-aug-cc-pV6Z correctly leads to a shorter C-H distance, in closer agreement with that estimated as the CBS limit for the neutral molecule. Thus the G1, G2, G2MP2 and CBS extrapolations for the energy and geometry of the methane anion previously reported [9] , as well as the CBS limits reported here with the aug-cc-pVnZ sequences are wrong, even with the largest available (n = 4. 5, 6) augmented correlation-consistent basis sets.
A practical scheme for very accurate quantum chemical descriptions of molecular anions has been proposed here: the standard aug-cc-pVnZ basis sets can be supplemented with extra-diffuse orbitals using a simple even- tempered scheme. With this scheme the EA of methane has been shown to follow the asymptotic behavior towards zero.
From the more general perspective concerning the experimental detection of the methane anion in plasma swarms, these calculations confirm that if true electronic capture occurs, the negatively charged methane molecule will rather swiftly relax towards methane plus a free electron. However, given the experimental evidence [1] - [7] of anionic methane that has accumulated over the last 50 years, it cannot be excluded that if a density of negatively charged plasma exists above a certain threshold in a sufficiently large spatial region, a small ensemble of anionic methane molecules could actually be formed, where the extra electron is perhaps in resonance with some C− anionic Rydberg states, and these could briefly exist before the anionic methane decomposition takes place. Further exploration of the right experimental conditions (pressure, density-normalized electric field strength and perhaps even the geometry/dimensions of the Townsend chamber) of the ionic avalanche might lead to relaxation times that could give experimentalists the opportunity to obtain refined data of a yet undiscovered meta-stable anionic structure of mehane in those special conditions. This work yields a reliable CBS extrapolation method to develop a (discrete approximation of a) continuum anionic state near ionization, viz., one that closely matches the energy of the corresponding neutral state.
Finally, since the ground state of the neutral methane is 1A1, the metastable anion (long-lived to be detectable in experiments) is unlikely to be 2A1, as this spatial symmetry is identical with that one of the neutral molecule and one free electron, giving rise to a shape resonance [21] that would decay through the Coulomb electron-electron repulsion operator in 10−10 - 10−15 s, too soon to be detected. This work shows that CH4 has no stable anions of 2A1 symmetry, implying that plasma swarms with anionic methane consist of metastable rather than stable anions.
Acknowledgements
The author is indebted to Prof. Jaime de Urquijo for interesting discussions on methane electron attachment and for pointing out the previous and new experimental results showing the presence of the metastable methane anion in Townsend plasma swarms. Support from CONACYT Sabbatical Program is acknowledged. Financial support from the CONACYT-México Basic Science project No. 130931 is also acknowledged.
References
- Trepka, L.V. and Neurt, H. (1963) Uberdie Entstehung von Negativen Ionen. Zeitschrift für Naturforschung A, 18, 1295.
- Sharp, T.E. and Dowell, J.T. (1967) Isotope Effects in Dissociative Attachment of Electrons in Methane. The Journal of Chemical Physics, 46, 1530. http://dx.doi.org/10.1063/1.1840885
- Hunter, S.R., Carter, J.G. and Christophorou, L.G. (1986) Electron Transport Measurements in Methane Using an Improved Pulsed Townsend Technique. Journal of Applied Physics, 60, 24. http://dx.doi.org/10.1063/1.337690
- Bordage, M.C. (1995) Thèse de Doctorat. Université Paul Sabatier de Toulouse, France.
- de Urquijo, J., Arriaga, C.A., Cisneros, C. and Alvarez, I. (1999) A Time-Resolved Study of Ionization, Electron Attachment and Positive-Ion Drift in Methane. Journal of Physics D: Applied Physics, 32, 41. http://dx.doi.org/10.1088/0022-3727/32/1/008
- de Urquijo, J., Herrera, J. and Private Communication (2014)
- CRC Handbook of Chemistry and Physics 1986-1987, Vol. 47. CRC Press, Boca Raton.
- Hernández, E.M., Hernández, L., Martíınez-Flores, C., Trujillo, N., Salazar, M., Chávez, A. and Hinojosa, G. (2014) Electron Detachment cross Sections of CH4− Colliding with O2 and N2 below 10 kEV. Plasma Sources Science and Technology, 23, Article ID: 015018. http://dx.doi.org/10.1088/0963-0252/23/1/015018
- Jursic, B.S. (2000) Theoretical Investigation of Structures and Energies of the Protonated Methane Radical Cation and Ethane. Journal of Molecular Structure: THEOCHEM, 498, 149. http://dx.doi.org/10.1016/S0166-1280(99)00254-7
- Montgomery, J.A., Ochterski, J.W. and Petersson, G.A. (1994) A Complete Basis Set Model Chemistry. IV. An Improved Atomic Pair Natural Orbital Method. The Journal of Chemical Physics, 101, 5900. http://dx.doi.org/10.1063/1.467306
- Pople, J.A., Head-Gordon, M., Fox, D.J., Raghavachari, K., Curtiss, L.A. (1989) Gaussian-1 Theory: A General Procedure for Prediction of Molecular Energies. The Journal of Chemical Physics, 90, 5622. http://dx.doi.org/10.1063/1.456415
- Raghavachari, K., Trucks, G.W. and Pople, J.A. (1991) Electronic Structures of the Negative Ions Si−2 –Si− 10: Electron Affinities of Small Silicon Clusters. The Journal of Chemical Physics, 94, 7221. http://dx.doi.org/10.1063/1.459738
- Curtiss, L.A., Raghavachari, K. and Pople, J.A. (1993) Gaussian-2 Theory Using Reduced Möller-Plesset Orders. The Journal of Chemical Physics, 98, 1293. http://dx.doi.org/10.1063/1.464297
- Dunning, T.H. (1989) Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. The Journal of Chemical Physics, 90, 1007. http://dx.doi.org/10.1063/1.456153
- Temelso, B., Valeev, E.F. and Sherrill, C.D. (2004) A Comparison of One-Particle Basis Set Completeness, Higher-Order Electron Correlation, Relativistic Effects, and Adiabatic Corrections for Spectroscopic Constants of BH, CH+, and NH+. The Journal of Chemical Physics A, 108, 3068. http://dx.doi.org/10.1021/jp036933+
- Wilson, A.K., van Mourik, T. and Dunning Jr., T.H. (1997) Gaussian Basis Sets for Use in Correlated Molecular Calculations. VI. Sextuple Zeta Correlation Consistent Basis Sets for Boron through Neon. Journal of Molecular Structure: THEOCHEM, 388, 339; aug-cc-pVnZ basis sets from https://bse.pnl.gov/bse/portal http://dx.doi.org/10.1016/S0166-1280(96)80048-0
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al. (2009) Gaussian 09, Gaussian Inc., Pittsburgh.
- Peterson, K.A., Woon, D.E. and Dunning Jr., T.H. (1994) Benchmark Calculations with Correlated Molecular Wave Functions. IV. The Classical Barrier Height of the H+H2→H2+H Reaction. The Journal of Chemical Physics, 100, 7410. http://dx.doi.org/10.1063/1.466884
- Feller, D. and Sordo, J.A. (2000) A CCSDT Study of the Effects of Higher Order Correlation on Spectroscopic Constants. I. First Row Diatomic Hydrides. The Journal of Chemical Physics, 112, 5604. http://dx.doi.org/10.1063/1.481135
- de Lara-Castells, M.P., Krems, R.V., Buchachenko, A.A., Delgado-Barrio, G. and Villarreal, P. (2001) Complete Basis Set Extrapolation Limit for Electronic Structure Calculations: Energetic and Nonenergetic Properties of HeBr and HeBr2 (with 2 as Subscript) van der Waals Dimers. The Journal of Chemical Physics, 115, 10438. http://dx.doi.org/10.1063/1.481135
- Schulz, G.J. (1973) Resonances in Electron Impact on Diatomic Molecules. Reviews of Modern Physics, 45, 423. http://dx.doi.org/10.1103/RevModPhys.45.423