International Journal of Intelligence Science
Vol.05 No.03(2015), Article ID:55974,14 pages
10.4236/ijis.2015.53014
A Decision Support System Based on Multi-Agent Technology for Gene Expression Analysis
Edna Márquez1, Jesús Savage1, Jaime Berumen2, Christian Lemaitre3, Ana Lilia Laureano-Cruces4, Ana Espinosa2, Ron Leder1, Alfredo Weitzenfeld5
1Facultad de Ingeniería, Universidad Nacional Autónoma de México, México D.F., México
2Unidad de Medicina Genómica, Hospital General de México, México D.F., México
3Departamento de Ciencias de la Comunicación, Universidad Autónoma Metropolitana, México D.F., México
4Departamento de Sistemas, Universidad Autónoma Metropolitana, México D.F., México
5Department of Computer Science and Engineering, University of South Florida, Tampa, FL, USA
Email: cednam@gmail.com, robotssavage@gmail.com, jaimeberumen@hotmail.com, lemaitre@gmail.com, clc@azc.uam.mx, anaesga@hotmail.com, rleder@ieee.org, aweitzenfeld@usf.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 13 March 2015; accepted 21 April 2015; published 27 April 2015
ABSTRACT
The genetic microarrays give to researchers a huge amount of data of many diseases represented by intensities of gene expression. In genomic medicine gene expression analysis is guided to find strategies for prevention and treatment of diseases with high rate of mortality like the different cancers. So, genomic medicine requires the use of complex information technology. The purpose of our paper is to present a multi-agent system developed in order to improve gene expression analysis with the automation of tasks about identification of genes involved in a cancer, and classification of tumors according to molecular biology. Agents that integrate the system, carry out reading files of intensity data of genes from microarrays, pre-processing of this information, and with machine learning methods make groups of genes involved in the process of a disease as well as the classification of samples that could propose new subtypes of tumors difficult to identify based on their morphology. Our results we prove that the multi-agent system requires a minimal intervention of user, and the agents generate knowledge that reduce the time and complexity of the work of prevention and diagnosis, and thus allow a more effective treatment of tumors.
Keywords:
Multi-Agent Systems, Machine Learning, Bioinformatics, Gene Expression Analysis

1. Introduction
In genomic medicine the knowledge derived from gene expression analysis could help to understand the function of normal and neoplastic cells and to identify molecular markers with diagnostic value and medical prognosis in cancers, in order to develop effective prevention, early diagnostic and therapeutic strategies. Two important goals of genomic medicine, in particular in cancer studies, are to search new molecular subtypes of cancer and predict membership to predefined cancer classes [1] . Classification of samples is important because it contributes to the prognosis of cancer.
Our work was designed in order to achieve these objectives, because current medicine requires non-trivial information processing like pre-processing data, filtering data, data mining, characterizing of genes, and visualization of results. Also our system proposed through intelligent agents and machine learning can contribute to these types of tasks that are in the area of Bioinformatics.
The use of agent technology is viewed as an emerging area in Bioinformatics [2] . In Bioinformatics almost all of the applications of agents are dedicated in the integration of resources using different sources that are distributed in different nodes like data bases or web sites. In our multi-agent system for gene expression analysis (MAS-GEN), we are interested in the automation of complete whole process of gene expression analysis through distribution of activities in a group of specialized agents with different abilities.
In the first section we present a background of gene expression in microarrays and the relation between agents and gene expression analysis, and the machine learning methods applied. In the second section we present the architecture implemented in MAS-GEN. In the third section are some example outcomes with study case of cervical cancer and finally the conclusions and the discussion.
1.1. DNA Microarray
Since the deoxyribonucleic acid microarrays appeared in 90’s the studies based in molecular biology can have the expression of thousands of genes in a single matrix. In a typical experiment the study includes dozens of microarrays with the measure of the expression levels of large numbers of genes simultaneously and the comparison of the information extracted from them.
This microarray technology demands to computer science methods and techniques at different levels, like data analysis and statistical information processing, information standardization, and automation of the whole information processes involved in each experiment.
Through the analysis of microarrays is possible for researchers to know which genes are active in a cell at particular situation. The comparison of genes expression patterns (which genes are active) of two cells of the same type, one normal cell and the other belonging to a tumor tissue, can be of great help in understanding what are the genes that might be involved in the tumor formation.
In many cases, for researchers in gene expression analysis, it is not trivial to use several software programs to complete the microarray analyses that we can find in most experiments. The problems include matching of data output and input formats understanding the software performance/limitations, and finding one or more special software programs; this is usually not an easy task given their training. We propose an agent system platform for automating the information analysis process of microarray samples.
1.2. Related Work
Many groups of research have been interested in combining Bioinformatics (where gene expression analysis is included) and intelligent agent software. In [3] we found a Group on Agents in Bioinformatics, BioAgents, which promote the agent work in Bioinformatics fields. In [4] the project Geneweaverthe, agents work with external databases. Another system for gene expression analysis using multi-agent technology is in [5] . They propose three stages of gene expression analysis: pre-processing, statistical analysis and biological inference. With agents they want to get automation and parallel processing. In BioMas [6] the agents work with the sequence and function of genes. A multi-agent system for gene expression classification is presented in [7] . The system searches for significant genes to classify samples of some cancers. The genes are grouped by couples; that are tested with clustering methods if they could classify types of cancer.
In our application we are interested in the automation of complete whole process of gene expression analysis, since the reading of profiles expression data to selection of relevant genes and sample classification, through distribution of activities in a group of specialized agents with different abilities.
At difference with the previous multi-agent systems, MAS-GEN joins in its agents the knowledge of experts with the use of statistical and machine learning methods for supporting decisions in gene expression analysis in samples classification and identification of relevant genes for a not expert user. The intervention of the user could be minimal, he only must give the files with gene expression intensity and MAS-GEN makes all tasks to find relevant genes and create groups of samples.
1.3. Agent Technology
An agent is a computer system that is situated in some environment, and that is capable of autonomous action in this environment in order to meet its design objectives [8] -[10] .
Some characteristics of intelligent agents are:
・ Autonomy, agents can exercise control over their internal state and actions without direct human or other interaction.
・ Reactive, they have to react timely and appropriately to unexpected events.
・ Proactive are goal-oriented and take the initiative where appropriate
・ Planning, the agents can plan their own actions, they have to solve tasks through plans
・ Adaptive, can learn and change their behavior on the basis of their previous experience to adapt to changing environmental conditions
・ Mobile, have the ability to transport themselves from one machine to another
・ Sociability, agents deal with interactions, positive and negative, with other agents.
Agent technology represents a paradigm of software development, which improves the development of systems in solving complex and real world problems [11] .
A multi-agent system is composed of a number of interacting agents. Multi-agent technology offers an alternative to building software in bioinformatics, where the complexity of required systems can be divided into well-defined sub components that can be handled by agents. Decomposition of complex problems into autonomous agents is an effective way of partitioning a problem [11] . In the systems where using different data resources and need an ontology, multi-agent technology has been adopted in several research projects for integration of Bioinformatics resources [12] .
1.4. Machine Learning
The data obtained from digital image files of multiple microarrays with thousands of genes must be transformed and organized in a gene expression matrix, where each row represents the expression of one gene in many samples represented by columns. After the matrix of gene expression profiles has been generated, we can begin the analysis which in non trivial doubt the dimension of data. Machine learning contributes with the supervised and non-supervised algorithms in order to make groups of similar data, genes or samples according of gene expression levels.
In our system we applied non-supervised machine learning algorithms through the clustering algorithms: fuzzy c-means, self-organizing maps, vector quantization, hierarchical clustering and principal component analysis, for discovering similarities in the samples and/or behavior of genes.
Fuzzy C-means algorithm has been used as an alternative to k-means in the analysis of genes, while in k- means one gene must belong just to one cluster in C-means one element could be in two or more classes with different degree of belonging. For this algorithm must choose the number of clusters not less than 2, also select the distance metric and the degree to which possessions are shared in the clusters (diffusivity). With the centroids of each cluster fuzzy will get the diffuse array, the metric distance is applied to view the difference between the current diffusivity matrix with the previous and verified with a threshold value until the distance were less than or equal to threshold value. For our aim, in classification of samples this algorithm permits to find degrees of membership to few types of cancers for each case or person.
Algorithm of Fuzzy C-means
Input: md = the data set of gene expression profiles, c = number of clusters, u = threshold of fuzzy value
1) Randomly create c clusters
2) Repeat
3) For i = 1 to c do
4) Calculate the C-centroid
5) Get fuzziness matrix, md(n), with all centroids
6) calculate the distance d(md(n − 1), md(n))
7) End_For
8) Until u > d(md(n − 1), md(n))
Self-organizing maps (SOM) is a type of clustering is based on the Kohonen neural network [13] . Corresponds to the non-hierarchical clustering split because there is no dependency between the different clusters formed and begins with the full set of objects (genes or samples) to be grouped by their similarity. In SOM the objects are assigned to a partition group by their similarity, and the partitions are defined according to a geometric shape established at the beginning. With a lattice are created the number of neurons to classify for the NxM dimensions and initialize the weight vector for each neuron to the input vector, during the iterations the winner neuron is looking for, that represents the input vector, its weights and weights of its neighbours are updated; the iteration continue until a detention criteria is satisfied, when the map has minimal changes.
Algorithm of SOM:
Input: data set of genes expression profiles, u = threshold of difference
1) Create a lattice with N neurons,
2) Initialize randomly the weight vector for each k neuron, Wik, connected to input i
3) Repeat
4) Presents an input vector V to network
5) Compute distance d(Vi, Wik),
6) Get the winner neuron, with the smallest d(Vi, Wik), which represents the input vector,
7) Refresh the weight of the winner neuron and the neighbors
8) Until (d(Vi, Wik) < u).
Vector quantization method (VQ) is also applicable like non-hierarchical clustering, in the process the space is divided into several connected regions, also called Voronoi regions. Each region is represented by a centroid, which bind the closest input vectors according to a distance measure. Vector quantization used here is designed so that you can create any number of centroids not only in power of 2. We have N input vectors with G dimensions to be grouped into M centroids. The algorithm begins with an initial centroid, which is the average of all vectors, the current centroids are divided in each iteration, each vector is associated to its nearest centroid, the centroids are recalculated as the arithmetic mean of the vectors associated with it, and repeat the iterations until criteria of error are satisfied.
Algorithm of VQ:
Input: N is data set of genes expression profiles, ε = threshold of distortion
1) Find an initial codebook D1, with one centroid C1, by averaging all the vectors pj, with Lm = 1;
2) Repeat
3) For i = 1 to M do
4) Modify each Ci, Ci = Ci + φ of small magnitude to generate new centroids from each of them, generating a new codebook Dm + 1, Lm + 1;
5) End_For
6) Compute the difference d(pj, Ck), between the input vector pj and the cluster Rk whose centroid is Ck;
7) Assign each input vector into the cluster with minimal distortion measure;
8) Recompute the centroids Ck for each of the cluster, by averaging all the vectors pj that belong to Rk;
9) Compute the average distortion, A
10) Until Lm > codebook_size and A < ε
The codebook size is the number of clusters in the environment.
The principal component analysis (PCA) is a technique widely used in fields such as recognition of images and to find patterns in large data. Another use of PCA is the reduction of dimension of data vectors with no loss of information. Variables that represent those dimensions are chosen to collect the percentage of variability that is sufficient, called principal components. The algorithm for PCA gets the covariance matrix of the initial data with its eigenvalues and eigenvectors, the columns of the eigenvectors are sorted according to the values of the eigenvalues. Finally N columns take the eigenvector according to the N dimensions that you want to keep.
Hierarchical Clustering is an agglomerative clustering algorithm, where the genes that are separated in groups, forming a hierarchical tree with clusters of genes. The hierarchical clustering algorithm consists in calculating the distances between all pairs of objects (genes or samples), this is the same as assuming that every object is a cluster: {C1, ..., CN}, two closest clusters (Ci, Cj) are selected to form one, Cij, you have to repeat the selection of clusters until there are no pairs of comparison. For calculating the distance between clusters is calculated by 3 ways: simple linkage, corresponds to the minimum distance between elements in the clusters, emerging clusters are linked by very long branches may be less significant clusters; average linkage is the average distance between all elements of both clusters is complex for processing large amounts of data, like in the case of genes; complete linkage is the maximum distance between elements of the clusters. With the final group of clusters it is creating a dendogram or tree diagram that helps the visualization of clusters, you can find different levels of clusters and therefore relationship between the elements that comprise them.
Cluster validation was made with Cross-validation is very useful in gene expression analysis with microarrays because the number of samples is reduced by the cost and it is not possible have training and testing samples for all studies. Here the agents apply 10-fold cross-validation, for evaluating and comparing the results of clustering methods, where the samples are divided in two sets: training and testing. With 10-fold cross-validation the samples are divided into 10 equally sized segments, the groups are going to be used for 10 iterations of training and validation such that within each iteration 9 folds are used for learning while one fold of the data is held-out for validation. In sample classification the agent uses 10-fold cross-validation to measure the accuracy classification of samples, calculated according the number of right classified in testing sets.
2. MAS-GEN Architecture
The gene expression analysis through the microarray data could be improved by automation of many tasks. In the proposed system MAS-GEN, to reach the goals of gene identification and tumor classification is distributed in operational agents and many tasks could be solver in parallel with the knowledge of other experts. We intended to create a simple and transparent system for the user to do gene expression analysis without a lot experience. The biomedical experts for MAS-GEN integrate the team of Genomic Medicine in the General Hospital of México [14] .
MAS-GEN has four operational agents and one manager. In this multi-agent system the agents have some properties of individual intelligent agents and collaborate to reach a main goal. This is why communication and coordination between the agents is necessary. Some agents have autonomy to make own decisions according their internal state and the information received from the environment to solve his tasks. The autonomy of agents could be adjustable [15] in order to give part of the process control to the user the agent autonomy could be limited to respond to user needs.
Our operational agents are Pre-processing agent, Gene Identification agent, Sample Classification agent and Databases agent they are coordinated by a Manager agent.
Agents’ decision making is based on a system of production rules, where each agent has a base of facts and a set of rules implemented in the programming language Clips [16] .
The agent platform uses a set of functions or procedures to analyze the expression of thousands of genes in many experimental conditions. This set of statistical and machine learning methods is independent of the agents. The use of microarrays for gene expression analysis is a field that it is improving continuously with new or better methods and techniques, so, it is possible to increase the capacity of the system with the incorporation or modification of new functions for preprocessing or processing data, and presentation of data, without introducing conflicts in agents’ operation. In Figure 1, we present the MAS-GEN architecture.
With this independence processes and functions have been implemented in different programming languages such as Java, C++ and R.
MAS-GEN is integrated by four operational agents (Pre-processing agent, Gene Identification agent, Sample Classification agent and Database agent), which are coordinated by a master agent who interacts with the user. The set of statistical and machine learning methods are implemented out of platform agent and all agents can use them.
2.1. Pre-Processing Agent
The Pre-processing agent’s activity begins with the reading of data intensity files of expressed genes in experiments from microarrays. This agent uses functions for reading, normalizing and standardizing data, already implemented in the R language by Bioconductor [17] .

Figure 1. Architecture of MAS-GEN.
According the problem to be solves: sample classification or selection of genes the agent must give the format of data matrix, the matrix must be complete does not have missing values for all samples in all genes, and the data matrix has data normalized. To keep up with the dynamic field of Bioinformatics, the preprocessing agent can incorporate new methods for reading other gene Chips or data normalization, so the system facilitates the in- corporation of new pre-processing processes. This is an autonomous agent because have motivations to act.
2.2. Gene Identification Agent
This agent makes one of the main tasks of MAS-GEN, has the goal of creating lists of genes from which genes potentially involved in the diseases investigated can be identified. It also requires the collaboration of the databases agent to characterize the genes, and the collaboration of the sample classification agent to review the capacity of classification samples with the selected lists. The use case diagram for gene identification is in Figure 2.
In the aim of identification of genes all the agents of MAS-GEN collaborate. Identification of genes begins with a user request to manager agent, which presents the requests to pre-processing agent and gene identification agent to create lists of genes and manager agent to the user system presents the results.
This agent applies filters based in the experience of the experts in this task. The agent works with statistical and machine-learning methods, like vector quantization and self-organizing maps, for creation of lists of genes. The agent has to make different clusters of genes to decide what clusters are promising in order to include the most important genes to the research. Gene Identification agent evaluates the lists of genes generated according the knowledge given by sample classification agent and databases agent. In the selection of a list of genes the gene identification agent looks upon if the genes in the list can make a correct classification with the samples and the previous knowledge about relation between the genes and the disease from external databases.
2.3. Sample Classification Agent
The agent of sample classification makes groups with the microarray samples based in the quantitative data of gene expression profiles. The results of this agent help to do the difference between healthy and diseased samples, as well as the identification of cancer type and tumor variants. The creation of groups of samples is using machine learning through clustering methods: 1) self-organizing maps (SOM); 2) vector quantization (VQ) [18] , and 3) c-means and principal component analysis (PCA). In Figure 3 is the use case for sample classification task.
Sample Classification agent responds to request and applies clustering methods to make the groups of samples
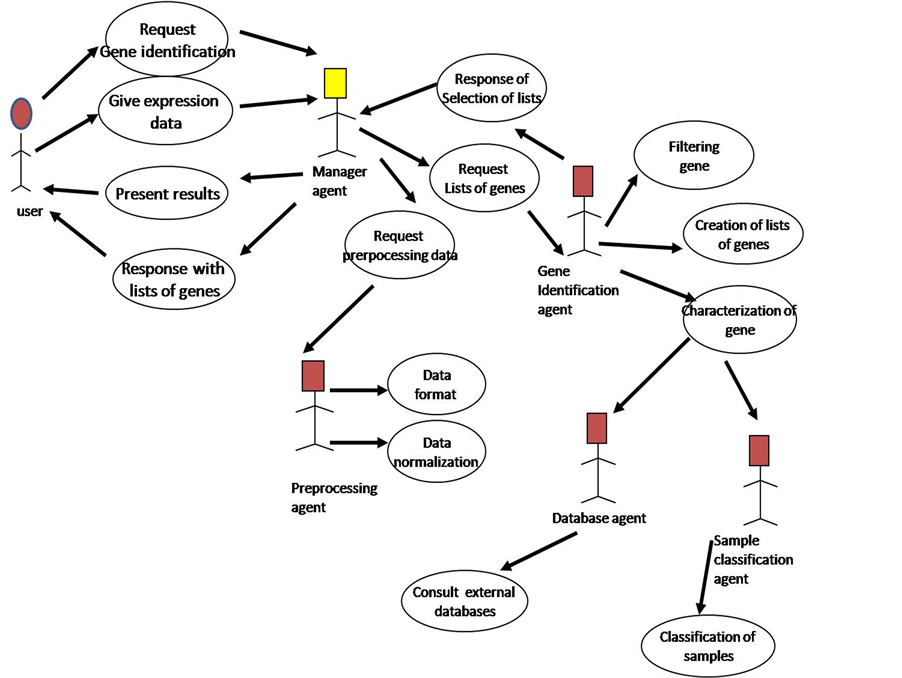
Figure 2. Use case diagram for identification of genes.

Figure 3. Use case diagram for classification of samples.
using the numerical data of gene expression. With the knowledge about the groups of samples, another task for this agent is to classify new samples, if the sample is a tumor or not, or what kind of tumor represents, this knowledge is relevant in medicine to determine the state and accurate treatment for a diseases.
Also, Sample Classification agent collaborates in the goal of identification of relevant genes, this agent tests if the lists of genes given by the gene identification agent can classify properly the samples.
2.4. Database Agent
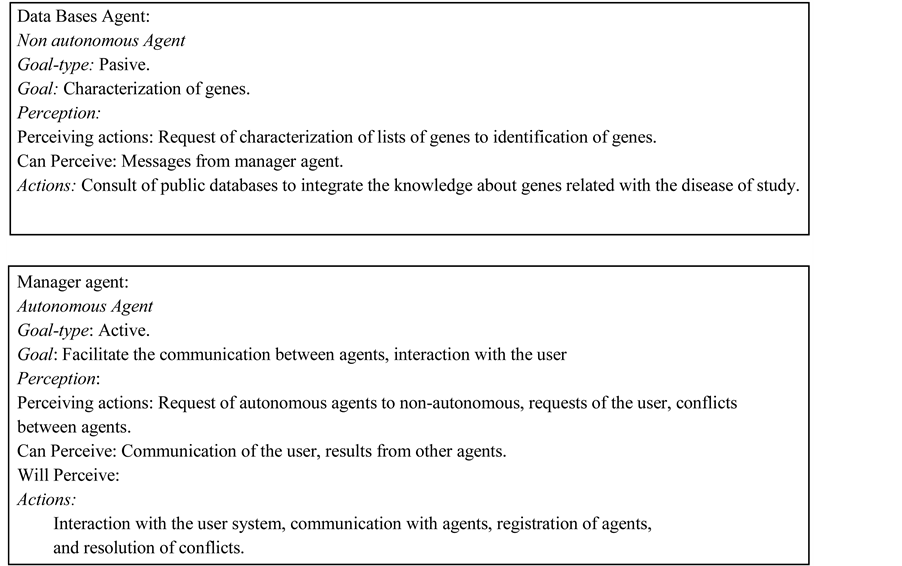
The gene expression analysis to obtain a list of important genes requires the characterization of selected genes. Database agent is specialized in selecting and gathering information of genes using several resources of databases with genomic information through the Internet, through NCBI [19] and other tools like DAVID [20] . Database agent interacts with external databases. Databases agent must select only the relevant information about genes that could be important in the user decision. Finally that agent standardizes and prepares the information about genes to present it to the user. This agent is not autonomous because it works by requisition of other agent, gene identification agent or sample classification agent.
2.5. Manager Agent
MAS-GEN has a manager agent, which coordinates all activities. This agent interacts with the user, gets his requests and gives him the results, coordinates the interaction among agents, has the register of the agents into the system, where the agents presents their abilities to others to respond of their desires.
According with Luck [21] we can classify the entities in an environment in objects, agents and autonomous agent. Autonomous agents are the agents motivated by self to act in the environment, like the agents sample classification, preprocessing and gene identification are autonomous. The database agent is not autonomous because other agents to make its tasks call it. Then in our system we have agents and autonomous agents, since we think all the entities have a goal to reach. This technique has been utilized with success for the organization of agents in different subjects like: 1) a pedagogical context; 2) geothermal wells; 3) video games; and 4) planning and schedule [9] [10] [22] -[25] .



3. Case Study-Cervical Cancer
3.1. Data
For this paper we use MAS-GEN for identifying relevant genes for cervical cancer. The data of gene expression was generated using the Affymetrix HGFocus Gen Chip of mRNA that contains ~8600 genes. The data are from 42 Mexican women with a diagnosis of cervical cancer with Human Papillomavirus 16 (HPV16). Biological samples were collected during the course of routine clinical practice at the oncology service at the General Hospital of Mexico (Mexico City) and also from 12 controls (non-malignant conditions or for non-cervical cancer were included). The tumor samples are of two cervical cancer subtypes: 14 adenocarcinoma (ACC) and 28 squamous cell carcinoma (SCC), given the histopathological point of view. The assay microarrays were performance in the unit of Genomic Medicine in the General Hospital of Mexico [14] .
3.2. Process for Selecting Relevant Genes
The selection of relevant genes is with minimal intervention of the user. At the beginning the user selects the files with the intensity of expression of ~8600 genes for 52 samples and requests the goal of selecting relevant genes. This instruction generates the user query for initializing the work of manager agent, which creates a plan to reach the goal and the operational agents execute the plan. Pre-processing agent reads files with expression of thousands of genes, review the state of data, if the data matrix is complete, apply tests of normalization data and gives the format to data in order to other agent can analysis the gene expression profiles.
The gene identification agent with the data like a matrix with a dimension of N rows of genes and M columns of samples applies filters to reduce the number of genes. With this task the number of genes is reduced only, which have differential expression for the study. The filtering methods, like SAM and t-test, use a threshold to select a list, this threshold is a parameter recommended by an expert but that the user can modify and the agent could propound one according the data. The user could view and use this first a priori list, that also other agents could use it to solve a problem.
For this study case were selected the best fifty genes upregulated from the genes with differential expression. After, for generating sub lists, the gene identification agent creates clusters of genes using machine learning like VQ and SOM algorithms. To evaluate the adequacy of a selected list of genes, the agents test if a list of genes has the capacity to divide the samples in the basic groups: control and cases. After, the list of genes is selected to continue with the characterization of genes using external databases. Data base agent consults information about the disease or a related problem, biological functions and biological processes in which the genes are involved. In Table 1 are the 3 best lists of genes generated by MAS-GEN with the clusters of genes according the results from the tasks of gene identification, sample classification and database agents. Some of the genes had been reported already about cervical cancer according PubMed [14] . In Figure 4 the patterns of genes expression of the three best lists selected compare with the expression pattern of the reported genes.
The expression patterns of four lists of genes: a) genes already reported with cervical cancer; b) genes of selected list-1; c) genes of selected list-2; and d) genes of selected list-3. In b), c) and d) are the three best-selected lists of genes by MAS-GEN. The three lists selected by MAS-GEN were grouped by clustering methods so their genes have very similar expression in each one.
The user can review the results through some tables and graphics presented by manager agent with the knowledge obtained by the operational agents. The knowledge given by MAS-GEN could support decision making by the user about a set of genes. In Table 2 show the principal biological process obtained by data base

 (a) (b)
(a) (b)
 (c) (d)
(c) (d)
Figure 4. Expression patterns with the lists of genes.

Table 1. Genes of the best selected lists.
Table 2. Participation of genes in biological processes.
agent for some gene lists. The knowledge from Internet databases about important biological processes of genes selected by MAS-GEN is reported with the percentage of genes involved of the three selected lists. Table 3 has information about important biological functions of genes selected by MAS-GEN, these results were gotten also by Database agent by consulting databases in internet.
3.3. Process for Sample Classification
For this aim the principal actor is sample classification agent, which applies, clustering methods and cross validation with 10-fold. In this paper, the sample classification agent makes two kind of classification of samples: a) classification in 2 basic classes: tumors and controls, and b) in subtypes of cervical cancer: SCC and ACC. We use a list of genes already reported with relation of cervical cancer and one list obtained by gene identification agent. After pre-processing data, the sample classification agent gets a matrix with N rows of samples and M columns of genes; to create groups of samples.
The classification of subtypes given by the agent is comparing to histopathological classification, which is according the subjective direct observation of the cellular tissue made by the pathology expert. However, the use of molecular biology through the intensity of gene expression is not equals in many cases of cancers but it could give new forms of classification [26] . The aptitude of dividing the samples with the list of already reported genes in PubMed (CDKN2A, TOP2A, CDC2, PCNA, CCNB2, CDC20, TYMS, RFC4, ZWINT, SYCP2, CKS2) and the 3 lists of selected genes obtained by Gene Identification agent are in Table 4, the results of the clustering methods validated with 10-fold cross-validation for the basic classification: tumors and controls, and for two types of cervical cancer: ASC and SCC. Figure 5 presents the graphics of PCA and Figure 6 has the dendograms for sample classification between tumors and controls given by MAS-GEN with the 3 selected lists of genes and with the list of reported genes. Using reported genes and the best lists of genes selected by MAS-GEN, a) visualization with PCA graphics of 2 principal components; and b) visualization with dendograms, both for classification of control and tumor samples.
4. Discussion and Future Work
The results achieved in the classification of samples between cases and controls have a minimal misclassifica-

Figure 5. Visualization with dendograms of sample classification between tumors and controls.

Figure 6. Visualization with PCA of sample classification between tumors and controls.
Table 3. Biological functions of genes selected in gene identification.
Table 4. Accurate classification samples with selected lists and reported genes.
tion, so the clusters could be used to the determinate if a sample is healthy or not. In the classification of samples into subtypes of cervical cancer like adenocarcinoma and squamous cell carcinoma the rate of misclassification is high due to know disagreement between the histological classifications cytologic morphology of the tumor and the quantitative data of the microarrays. For deterministic sub classification it is necessary to do more processing and research to correctly interpret the results of classification of tumors by molecular biology.
MAS-GEN is an intelligent system to help biologists and medical teams in the analysis of gene expression to understand the genetic details of disease, represents a significant improvement in the state of gene expression analysis. Like a multi-agent system MAS-GEN, provides a flexible tool, using coordinated and specific goal- directed agents that naturally allow decomposition of the complex problem in a single software system. Key advantages over using several different software applications are the time saved and error reduction in managing the process through different data format and reduced amount of user intervention.
For future work we propose implementing more learning and statistical methods to improve gene identification and tumor classification. We propose to create a method to combine the current presumptive results with validating qRT-PCR for selecting genes.
5. Conclusions
In MAS-GEN through the interaction of the agents we can contribute to giving solution of two present questions of genomic medicine: selecting a list of relevant genes and identification and classification of samples, applying many methods in different steps like an expert with the minimal intervention of the user. The advantage for the user of MAS-GEN is that he does not need to do himself all the steps for the analysis of the matrix going through different applications nor to know all the parameters and methods for pre-processing the data expression of genes. All this knowledge is part of the agents’ expertise. The results generated by MAS-GEN about the identification of genes is a proposal of relevant genes based on the quantitative evaluation of gene expression levels; after that the user might apply biological methods to validate them like quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and immunochemistry, in order to define marker genes. MAS-GEN is a tool for supporting the decisions.
The distribution in several specialized agents doing the tasks of gene expression analysis (pre-processing methods, data filtering, machine learning, and presentation and display of results) provides a versatile system that can be conveniently adapted to the rapidly changing area of bioinformatics.
The analysis of gene expression is feasible through multi-agent collaboration of operational agents (of data pre-processing, gene identification, classification of samples, and database management), and a single management agent assigns tasks and controls data flow. Operational agent can execute in parallel some tasks because the agents have sufficient independence.
Also, we achieved great flexibility with the implementation of the agents using Java for the platform and Clips in the use of decision rules. By keeping separated the machine learning technics and other methods of the agents’ platform, we can modify, add or delete these components without affecting the operation, functionality or design of agents involved in the gene expression analysis. This is important in Bioinformatics field where, to avoid obsolescence, a system must be easy to adapt to frequently emerging new or improved procedures.
Multi-agent technology allows gathering several tasks into one software tool that can simplify, with the minimal user intervention, to automate the selection of relevant genes from large amount of microarray data, and classify tumor samples based on principles of molecular biology, supported by statistical and machine learning techniques and genetic information available on the Internet.
Acknowledgements
This paper is part of the research being carried out by Edna Márquez―to obtain her PhD in Posgrado en Ciencia e Ingeniería de la Computación at the Universidad Nacional Autónoma de México. It is supported by CONACYT, by PAPIIT-DGAPA UNAM under Grant IN-117612. Also the authors thank to Hospital General de México.
References
- Tinker, A.V., Boussioutas, A. and Bowtell, D.D.L. (2006) The Challenges of Gene Expression Microarrays for the Study of Human Cancer. Cancer Cell, 9, 333-339.
- Merelli, E. and Luck, M. (2005) Agents in Bioinformatic. Knowledge Engineering Review, 20, 117-125.
- Merelli, E., Armano, G., Cannata, N., Corradini, F., d’Inverno, M., Doms, A., Lord, P., Martin, A., Milanesi, L., M öller, S., Schroeder, M. and Luck, M. (2006) Agents in Bioinformatics, Computational and Systems Biology. Briefings in Bioinformatics, 8, 45-59.
- Bryson, K., Luck, M., Joy, M. and Jones, D. (2000) Applying Agents to Bioinformatics in GeneWeaver. Lecture Notes in Computer Science, 1860, 60-71. http://dx.doi.org/10.1007/978-3-540-45012-2_7
- Lam, H.C., Garcia, M.V., Juneja, B., Fahrenkrug, S. and Boley, D. (2006) Gene Expression Analysis in Multi-Agent Environment. International Transactions on Systems Science and Applications, 1.
- Jin, L., Steiner, K., Schmidt, C. and Situ, G. (2005) A Multiagent Framework to Integrate and Visualize Gene Expression Information. IEEE ICDM Workshop on MADW & MADM, 1-7. http://www.eecis.udel.edu/~kamboj/pubs/kamboj.madw.05.pdf
- Štiglic, G. and Kokol, P. (2004) Using Multi-Agent System for Gene Expression Classification. Proceedings of the 26th Annual International Conference of the IEEE EMBS, San Francisco, 1-5 September 2004, 2952-2955.
- Wooldridge, M. (2002) An Introduction to Multiagent Systems. John Wiley, USA.
- Laureano-Cruces, A.L., Ramírez-González, T., Sánchez-Guerrero, L. and Ramírez-Rodríguez, J. (2014) Multi-Agent System for Real Time Planning Using Collaborative Agents. International Journal of Intelligence Science, 4, 91-103.
- Laureano-Cruces, A.L. and Espinoza-Paredes, G. (2005) Behavioral Design to Model a Reactive Decision of an Expert in Geothermal Wells. International Journal of Approximate Reasoning, 39, 1-28. http://dx.doi.org/10.1016/j.ijar.2004.08.002
- Jennings, N. (2001) An Agent-Based Approach for Building Complex Software Systems. Communications of the ACM, 44, 35-41. http://dx.doi.org/10.1145/367211.367250
- Koutkias, V., Malousi, A. and Maglaveras, N. (2007) Engineering Agent-Mediated Integration of Bioinformatics Analysis Tools. Multiagent and Grid Systems, 3, 245-258.
- Kohonen, T. (1995) Self Organizing Maps. Springer, Berlin.
- Espinosa, A., Alfaro, A., Roman-Basaure, E., Guardado-Estrada, M., Palma, Í., Serralde, C., et al. (2013) Mitosis Is a Source of Potential Markers for Screening and Survival and Therapeutic Targets in Cervical Cancer. PLoS ONE, 8, e55975. http://dx.doi.org/10.1371/journal.pone.0055975
- Karasavvas, K., Burger, A. and Baldock, R. (2004) Bioinformatics Integration and Agent Technology. Journal of Biomedical Informatics, 37, 205-219. http://dx.doi.org/10.1016/j.jbi.2004.04.003
- Clips (2014) http://clipsrules.sourceforge.net/
- Gentleman, R., Carey, V., Huber, W., Irizarry, R. and Dudoit, S. (2005) Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer. http://www.bioconductor.org/
- Márquez, E., Savage, J., Espinosa, A., Berumen, J. and Lemaitre, C. (2008) Gene Expression Analysis for Tumor Classification Using Vector Quantization. 3rd IAPR International Conference on Pattern Recognition in Bioinformatics (PRIB 2008), Melbourne, 15-17 October 2008, 95-103.
- National Center for Biotechnology Information (2014) http://www.ncbi.nlm.nih.gov/
- DAVID Bioinformatics Resources (2014) http://david.abcc.ncifcrf.gov/
- Luck, M. and d’Inverno, M. (2001) A Conceptual Framework for Agent Definition and Development. The Computer Journal, 44, 1-20.
- Sánchez-Guerrero, L., Laureano-Cruces, A.L., Mora-Torres, M., Ramírez-Rodríguez, J. and Silva-López, R. (2013) A Multi-Agent Intelligent Learning System: An Application with a Pedagogical Agent and Learning Objects. Creative Education, 4, 181-190. http://www.scirp.org/journal/ce
- Laureano-Cruces, A.L., Acevedo-Moreno, D.A., Mora-Torres, M. and Ramirez-Rodriguez, J. (2012) A Reactive Behavior Agent: Including Emotions for a Video Game. Journal of Applied Research and Technology (CCADET-UNAM), 10, 651-672.
- Sánchez-Guerrero, L., Laureano-Cruces, A.L., Mora-Torres, M. and Ramìrez-Rodríguez, J. (2011) Multiagent Architecture for Errors Management in Content Organized in Learning Objects. Proceedings of World Conference on E-Learning in Corporate, Government, Healthcare, and Higher Education 2011, Hawai, 17-21 October 2011, 2462- 2467. www.EdiTLib.org
- Laureano-Cruces, A.L. and Verduga-Palencia, D.O. (2010) Simulación de un juego de futbol utilizando una arquitectura Multiagente-Reactiva. In Libro: Desarrollo Tecnológico. (Alfa-Omega), XXIII Congreso Nacional y XI Congreso Internacional de Informática y Computación de la ANIEI, Puerto Vallarta, 11-15 de octubre, 485-493.
- Berman, J. (2004) Tumor Classification: Molecular Analysis Meets Aristotle. BMC Cancer, 4, 10.