Open Journal of Clinical Diagnostics
Vol.3 No.4(2013), Article ID:40269,3 pages DOI:10.4236/ojcd.2013.34028
A case of hemophagocytic syndrome as a complication of Plasmodium vivax malaria
![]()
Padmashree Dr. D. Y. Patil Hospital and Research Centre, Nerul, Mumbai
Email: drmonika257@gmail.com, fmcbom@gmail.com
Copyright © 2013 Ananya Mukharjee et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 29 August 2013; revised 29 September 2013; accepted 6 October 2013
Keywords: Hemophagocytic Syndrome; Plasmodium vivax Malaria
ABSTRACT
Hemophagocytic lymphohistiocytosis syndrome (HPS) is a potentially fatal hyperinflammatory response characterized by a generalized histiocytic proliferation with marked hemophagocytosis in bone marrow [1]. Hemophagocytic syndrome has been associated with genetic mutations, autoimmune diseases, hematological malignancies or infections [2,3]. According to the data from Centre for Disease Control and prevention (CDC) Plasmodium falciparum has been associated with HPS but not the Plasmodium vivax [4-7]. We report a case of hemophagocytic syndrome as a complication of Plasmodium vivax malaria which is a rare presentation according to the data. This patient presented with high grade fever with chills (P. vivax positive), fever however did not respond to anti-malarials. The patient continued to have high grade fever with altered sensorium and deranged liver function with pancytopenia. Since she fulfilled the criteria of (HPS), patient was put on injectable steroids and responded dramatically. Hemophagocytic syndrome is a potentially fatal syndrome and therefore high index suspicion and early treatment is the key to reduce the mortatlity.
1. CASE REPORT
A 21 years old female presented to the emergency department with high grade intermittent fever associated with chills and headache for last 20 days. Being an endemic area the patient had received antimalarials outside in the initial days of fever. Yet the fever continued. When the patient presented to us, she was febrile [Temperature: 39.5˚C], pale and had a moderate splenomegaly.
On admission with us she was further evaluated and her blood investigations on the very first day of admission were suggestive of pancytopenia viz. Hemoglobin: 8.7 gm%; Total leucocyte count: 0.9 × 109/lit; Platelets: 52 × 109/lit, Neutrophils: 0.4 × 109/lit.
The peripheral blood smear revealed the presence of ring forms of developing trophozoites and mature schizonts of Plasmodium vivax malarial parasite. Abdominal ultrasound confirmed moderate splenomegaly.
The patient received injectable artesunate for five days. Despite this the fever did not subside and the patient started deteriorating further. Her sensorium got altered and she was shifted to Intensive care unit. Her liver function tests were repeated which were derranged and suggestive of hepatitis. On further evaluation ferritin levels were found to be more than 2000 µg/lit and the fibrin degradation product (FDP) was 400, with normal reticulocyte count of 0.5% and the pancytopenia continued. The laboratory reports are as follows (see Table 1).
The lumbar puncture was done in view of altered sensorium which showed pleocytosis. The bone marrow examination was done to investigate the cause of pancytopenia. The bone marrow did not reveal any significant abnormality. At this juncture, hematological opinion was taken as haemophagocytic syndrome was suspected. Her serum triglecerides were found to be 446 mg/dl.
Almost seven out of nine criteria for the diagnosis of Hemophagocytic Syndrome were fulfilled in our case and the diagnosis of HPS was confirmed (see Table 2). The patient was treated with injectable methyleprednisolone on the depicted lines of treatment of HPS and showed dramatical improvement. In three days treatment with systemic steroids the patient was shifted out of ICU with remarkable improvement clinically and hemodynamically. The patient was then discharged after four days with oral steroids in tapering dosages for next 6 weeks.
2. DISCUSSION
Hemophagocytic syndrome is a reactive disorder of mono-
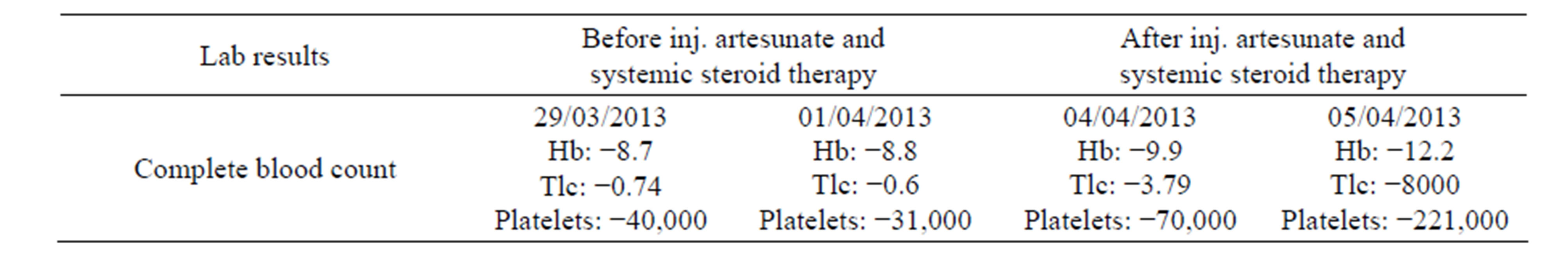
Table 1. Laboratory reports.

Table 2. Diagnostic criteria for hemophagocytic syndrome.
nuclear phagocytic system. The incidence is 1.2 cases per million individuals per year [8]. It is a potentially fatal disorder involving an exaggerated but ineffective immune response [9] in which there is increase in the levels of IFN-γ, Interleukin 6, 10, soluble interleukin 2 receptors, TNF-α, with inappropriate functioning of Natural killer (NK) cells and cytotoxic T lymphocytes [10].
The main mechanism of HPS is the lack of performin dependent cytotoxicity which is an essential function of NK cells and cytotoxic T lymphocytes [10]. These cells generally store performin and granzyme proteins within them in a specialized secretory lysozomes which are secreted when they come across an infected or a tumour derived target cell leading to apoptosis. This mechanism of performin mediated cytotoxicity is diminished or absent as a result of genetic mutations or following an infectious or immune stimulous [11]. Thus cytokine dysfunction results in uncontrolled accumulation of activated T-lymphocytes and activated histiocytes in many organs which ultimately leads to hemophagocytosis and organ damage [11].
The HPS is a fatal syndrome. The Histiocyte society trials, HLH 94 and HLH 2004 have defined the diagnostic criteria for the diagnosis of this rare syndrome [8,9].
The reported mortality in secondary HPS vary from 8% - 22% highest being in Ebstein Barr virus related HPS [12].
In our case, fever lasting for more than 7 days, splenomegaly, Cytopenia involving more than 2 cell lines, hypertriglyceridemia, Elevated serum ferritin levels, hepatitis and hemophagocytosis as demonstrated on bone marrow i.e seven out of nine criteria were fulfilled with supportive evidences of altered sensorium and deranged LFTs.
Also this very rare syndrome is generally seen with Plasodium falciparum but not with Plasmodium vivax per say [4-7] which forms the hallmark of this case.
This patient was treated on the depicted lines of management of HPS with immunosuppressive therapy viz injectable methyleprednisolone 1 gm once a day. Intravenously for 5 days followed by maintenance dose of oral prednisolone 1 mg/kg tapered over a period of 6 weeks with remarkable improvement clinically and hemodynamically.
3. CONCLUSION
Hemophagocytic syndrome is a potentially life threatening complication of Malaria. It is generally seen in Plasmodium falciparum malaria but very rarely it can even occur in Plasmodium vivax malaria. Therefore high suspicion of index and early treatment is the key to reduce the mortality with this fatal syndrome.
REFERENCES
- Imashuku, S. (1997) Differential diagnosis of hemophagocytic syndrome: Underlying disorders and selection of the most effective treatment. International Journal of Hematology, 66, 135-151. http://dx.doi.org/10.1016/S0925-5710(97)00584-7
- Fisman, D.N. (2000) Hemophagocytic syndromes and infection. Emerging Infectious Diseases, 6, 601-608. http://dx.doi.org/10.3201/eid0606.000608
- Vinoth, P.N., Thomas, K.A., Selvan, S.M., Suman, D.F. and Scott, J.X. (2011) Hemophagocytic syndrome associated with plasmodium falciparum infection. Indian Journal of Pathology and Microbiology, 54, 594-596. http://dx.doi.org/10.4103/0377-4929.85105
- Anwar, M., Saleem, M. and Malik, I.A. (1995) Severe haemophagocytic syndromein falciparum malaria. Journal of Pakistan Medical Association, 45, 302-303.
- Ohno, T., Shirasaka, A., Sugiyama, T. and Furukawa, H. (1996) Hemophagocyticsyndrome induced by plasmodium falciparum malaria infection. International Journal of Hematology, 64, 263-266. http://dx.doi.org/10.1016/0925-5710(96)00495-1
- Retornaz, F., Seux, V., Arnoulet, C., Durand, J.M., Sainty, D. and Soubeyrand, J. (2000) Plasmodium falciparum malaria infection complicated byhaemophagocytic syndrome in an old man. Acta Haematologica, 103, 224-225. http://dx.doi.org/10.1159/000041055
- Sermet-Gaudelus, I., Abadie, V., Stambouli, F., Hennequin, C., Lenoir, G. and Gendrel, D. (2000) Haemophagocytic syndrome in Plasmodiumfalciparum malaria. Acta Paediatrica, 89, 368-369. http://dx.doi.org/10.1111/j.1651-2227.2000.tb01343.x
- Henter, J.I., Elinder, G., Ost, A. and the FHL Study Group of the Histiocyte Society (1991) Diagnostic guidelines for hemophagocytic lymphohistiocytosis. Seminars in Oncology, 18, 29-33.
- Janke, G., Henter, J.I. and Imashuku, S. (2005) Clinical aspects and therapy of hemophagocyticlymphohistiocytosis. In: Weitzman, S. and Egler, R.M., Eds., Histiocytic Disorders of Children and Adults. Cambridge University Press, Cambridge, 353-379.
- Fukaya, S., Yasuda, S., Hashimoto, T., et al. (2008) Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: Analysis of 30 cases. Rheumatology (Oxford), 47, 1686. http://dx.doi.org/10.1093/rheumatology/ken342
- Risma, K. and Jordan, M.B. (2012) Hemophagocytic lymphohistiocytosis: Updates and evolving concepts. Current Opinion in Pediatrics, 24, 9. http://dx.doi.org/10.1097/MOP.0b013e32834ec9c1
- Henter, J.-I., Samuelsson-Horne, A., Aricò, M., et al. (2002) Treatment of hemophagocyticlymphohistiocytosis with HLH-94 immunochemotherapyand bone marrow transplantation. Blood, 100, 2367-2373. http://dx.doi.org/10.1182/blood-2002-01-0172