Open Journal of Obstetrics and Gynecology
Vol.04 No.16(2014), Article ID:52138,30 pages
10.4236/ojog.2014.416141
Lethal Developmental Defects: An Overview
Ashutosh Halder
Department of Reproductive Biology, All India Institute of Medical Sciences, New Delhi, India
Email: ashutoshhalder@gmail.com
Academic Editor: Chris Constantinou, Stanford University Medical School, USA
Copyright © 2014 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 September 2014; revised 25 October 2014; accepted 18 November 2014
ABSTRACT
About 3% of all conceptions are associated with major congenital malformations, many of them are lethal developmental defect and genetic in origin or teratogenic (adverse effects of the envi- ronment during gametogenesis or early embryogenesis). Genetics with or without adverse envi- ronment has role in virtually every developmental defect/malformation disorders in causation, predisposition, susceptibility & modulation of disease. Advances in genetics, introduction of triple marker screening, routine obstetric ultrasound examination into obstetric practice & accesses to prenatal diagnosis helped in secondary prevention (early detection & termination) of lethal deve- lopmental defects. Ultrasound detection of fetal developmental defects/malformation is common now and often decision on pregnancy solely based on ultrasonic morphological description. This practice leads to difficulty in providing accurate counseling as well as preventing disorder in sub- sequent pregnancy, in particular early. Hence an understanding of reproductive genetics of major developmental disorders is important for today’s perinatal care specialists. This overview will outline the various lethal developmental defects observed in an advanced reproductive genetics set up and various approaches adopted to derive diagnosis. Detailed assessment of fetus after termination of pregnancy (spontaneous/induced) for fetal anomalies was carried out in most cases. As most cases was referred after termination in formalin routine chromosomal analysis was not possible however, in selected cases targeted FISH analysis with specific chromosomal probe was carried out to confirm clinical diagnosis. Detailed evaluation of fetus is important as this practice often helped in modification of genetic counseling, as well as course of management in the next pregnancy. No molecular diagnostic or screening work was carried out due to non availability of information and facility in past. However, this is important today as many of the lethal develop- mental defects are yet to be categorized etiopathologically, and hence immediate need is to start clinical registry along with biorepository of developmental defects cases for future research work on informative families, in particular with multiple affected fetuses/sibs, using genomics, proteo- mics, metabolomics, platforms.
Keywords:
Lethal Developmental Defect, Lethal Malformation, Neural Tube Defect, Chromosomal Abnormality, Amniotic Band Syndrome, Skeletal Dysplasia, Nonimmune Hydrops Fetalis

1. Introduction
Fecundity in human, in comparison to other mammals, is rather low. This is due to an enormous frequency of abnormalities in oogenesis and spermatogenesis. About 30% - 50% oocytes and 10% sperms are chromosomally abnormal. This results in high rate (50% - 60%) of abnormal conception as well as pregnancy wastage, either before, during or after implantation. Later on, in clinically recognized pregnancies, about 15% of conceptuses are lost as spontaneous abortion. The majority of early pregnancy wastage is caused by abnormalities of chro- mosome number, which is considered to be responsible for as many as 50% cases. Other genetic causes are suggested to influence early developmental genes leading to lethal developmental defects (lethal malforma- tions).
About 3% of all births are associated with major congenital malformations (many are lethal developmental defects). This load is more than the load of any other genetic disease. However, little is known about the causes of developmental defects, in particular lethal. Approximately 60 percent of developmental defects are of un- known origin [1] [2] . Environmental factors, medications, diet and personal behaviors have been identified as possible contributors to these defects besides genetic alterations. About 3% of all birth defects are caused by en- vironmental exposures and another 25% or more may be caused by an interaction between genes and the envi- ronment [1] [2] . The prevalence of lethal developmental defects with or without chromosomal disorders, com- bined with their severity, impose a great financial, social and emotional burden. This leads to distress not only at economical level but also at family (anxiety) & social level (stigma). These conditions are likely to repeat in subsequent pregnancy. Furthermore, incidence is bound to increase due to an increase in maternal age at con- ception as well as contamination of our food chain with toxic chemicals like pesticides, preservative, plastics, heavy metals, dioxin, etc besides radiations from household as well as personal electronic gadgets. Lethal deve- lopmental defects commonly encountered in reproductive genetics practice (Table 1) are neural tube defect (NTD), skeletal dysplasia (SD), amniotic band syndrome (ABS)/limb body wall complex (LBWC), non immune hydrops fetalis (NIHF) and other rare lethal developmental defects (Sirenomelia, OEIS, urorectal malformation sequence, Fetus in Fetu, Fetus Papyraceus, etc.). Congenital malformations and genetic disorders are important causes of morbidity and mortality in India too. In a multi-centric study (Mumbai, Delhi and Baroda) on 94,610 newborns major malformation frequency was found to be 2.03% and the commonest one was neural tube defects [3] . The frequency for down syndrome was 0.87 per 1000 or 1 per 1150 births.
Despite deciphering of the Human Genome challenges in diagnosis & management of lethal developmental defects still remain the same due to lack of interest in this field. The challenges are to understand pathophysiol- ogy of lethal developmental defects and to resolute phenotype & genotype heterogeneity. It is hoped that the in- formation generated on this field in coming years and later integrated in system biology will be of great service to answer following questions that are encountered everyday in teratology/dysmorphology practice viz., genetic/ genomic role, underlying pathophysiology, early prediction or prevent (primary/secondary) and feasibility of pharmacotherapy. Hence, it is essential to have detailed clinical information along with good biological mate- rials (bio-repository) as initiators and later screening through high throughput omics platforms. We should have system to preserve immense important cases of developmental defects. The most challenging area in develop- mental defect is accurate diagnosis, accurate counseling and early prediction and prevention and for this, it is very important to preserve these resources for research work. The aim to write this overview is to stimulate ob- stetrician, perinatal pathologist and reproductive geneticist to attempt contributing in coming years. This article outlines common lethal developmental defects in day to day practice experienced by author.
2. Neural Tube Defect
Neural tube defects (NTDs) are a group of malformations of the brain and spinal cord that originate from failure of closure of neural groove in early embryonic period. NTD is the commonest lethal developmental defects.

Table 1. Summarizes lethal developmental defects cases referred for necropsy.
Worldwide prevalence is one per thousand live births [4] . However, the incidence in India is more and in north- ern India the incidence is about 3.9/1000 births [5] . NTDs can be divided in various way viz., open NTD (result from failure of primary neurulation; no coverings for nerve tissue, leaving nerve tissue exposed and destroyed e.g., anencephaly, open spina bifida/meningomyelocele, craniorachischisis, anen-iniencephaly, etc.) or closed NTD (defective secondary neurulation; skin covered viz., spinal dysraphism, iniencephaly, etc.). Other NTD malformations, such as encephalocele, are likely to be postneurulation disorders. NTDs can also be divided as isolated NTDs (anencephaly, spina bifida, encephalocele, iniencephaly, craniorachischisis, etc.; Figures 1(A)- (W)) or syndromic NTD (NTD with trisomy 18 or 13, monosomy X, with Sirenomelia or with Meckel syn- drome, or with amniotic band syndrome, etc., Figures 2(A)-(V)). Chromosomes 13 and 18 aneuploidies are of- ten associated with NTD, viz., spina bifida [6] -[8] , anencephaly [9] [10] , encephalocele [11] , craniorachischisis [12] or syndromic NTD [13] . However, trisomy 13 is seen more commonly with holoprosencephaly [14] or ar- hinencephaly than NTDs. In spina bifida there is a bony defect in posterior vertebral arches through which her- niation of neural tissue and meninges occurs whereas in closed spina bifida herniated meninges and neural tissue are covered by skin (Figures 1(E)-(K)). A meningocele is a saccular herniation of meninges and cerebrospinal fluid through a bony defect of the spine. A myelomeningocele is characterized by herniation of the spinal cord through a bony defect of the spine.
The term iniencephaly implies deficiency of occiput in the region of foramen magnum, spina bifida of consi- derable extent and retroflexion (extreme extension) of the spine. In most cases the cranial cavity is well formed and skin over the retroflexed region is uninterrupted (Figures 1(R)-(T)). Howkins and Lawrie in 1939 [15] clas- sified iniencephaly into simple iniencephaly (clausus-no encephalocele and apertus with encephalocele) and an- encephaly with spinal retroflexion (Figures 1(U)-(W)). The incidence of iniencephaly has been estimated to be between 1 in 900 [16] to 1 in 2200 [17] deliveries; whereas from India it has been reported to be 1 in 60,000 de- liveries [18] . This low frequency could be due to wrong diagnosis as often diagnosed as anencephaly or as mul- tiple congenital anomalies. In my experience iniencephaly is not a rare malformation at all in northern India [19] . Iniencephaly again can be subdivided as simple iniencephaly and anen-iniencephaly (anencephaly with






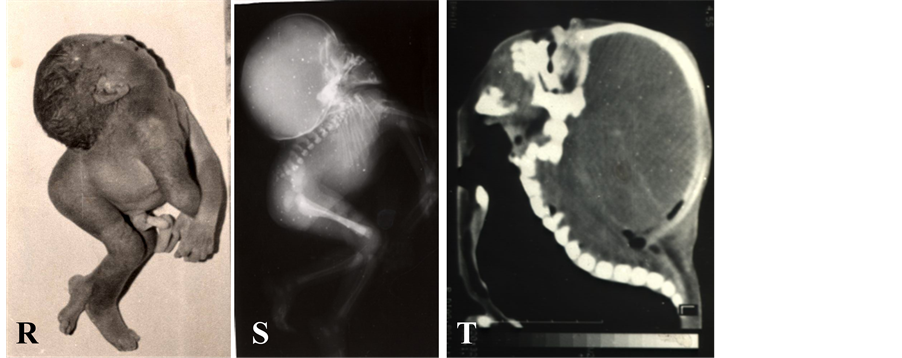

Figure 1. (A)-(W) showing isolated multifactorial neural tube defects (NTDs). Anencephaly (A-D): Clinical photographs (A, B) and X-ray (C, D) showing absent vault bones & brain. Tuft of hairs (B) is visible in lower back and corresponding to spina bifida oculta. Spina bifida (E-K): Clinical photographs showing spina bifida aperta, i.e., without any coverings (E, F) and spina bifida clauses, i.e., with coverings (I, J). Ultrasound examination show hydrocephalus (G) and spinal defect (H: longi- tudinal scan and K: transverse scan, spine defect is shown with arrow). Craniorachischisis (L-N): Clinical photographs (L), X ray (M) and ultrasound (N) showing complete destruction of spine (only wide spinal arch is visible, shown in arrow in N), spinal arches and skull vault leaving open spinal cord as well as brain for destruction (no proper brain & spinal tissue left). Encephalocele (O-Q): Clinical photograph (O), X ray (P) and ultrasound (Q) showing huge sac outside cranium containing most of brain tissue leading to apparent microcephaly (O). Note wide gap in occipital bone from where brain tissue herniated (P) and bone defect in occipital region of skull with herniation of brain (arrow) in early pregnancy (Q). Iniencephaly (R-T): Clinical photographs (R), X ray (S) and CT scan (T) showing extremely hyperextended head fixed with thoraco-lumber spine, compressed chest and abdomen along with hyperextension as well as disintegration of cervical & upper thoracic spines. Anen-Iniencephaly (U-W): Clinical photographs (U), X ray (V) and CT scan (W) showing anencephaly and extremely hy- perextended head along with hyperextension as well as disintegration of cervical & upper thoracic spines and absent vault bones.
spinal retroflexion). Iniencephaly is a developmental error occurring in early pregnancy prior to closure of ce- phalic neural folds at 24 days gestational age [20] . It is characterized by arrest as well as imperfect development of base of skull and vertebral column (cervicothoracic region mainly). At the early stage of embryo, paraverte- bral sclerotome (mesoderm) differentiates into two parts, a ventral mass (forms the vertebral bodies, pedicles and their cranial homologous) and a dorsal mass (forms neural arches and vault bones of the cranium). In inien- cephaly, one or both of these masses are hypoplastic or ill developed. The distinction between the two groups may relate to the time of onset and duration of insult. Primate and human neural tube closure was believed in past as a continuous process that begins in the cervical region of the embryo and proceeds rostally as well as
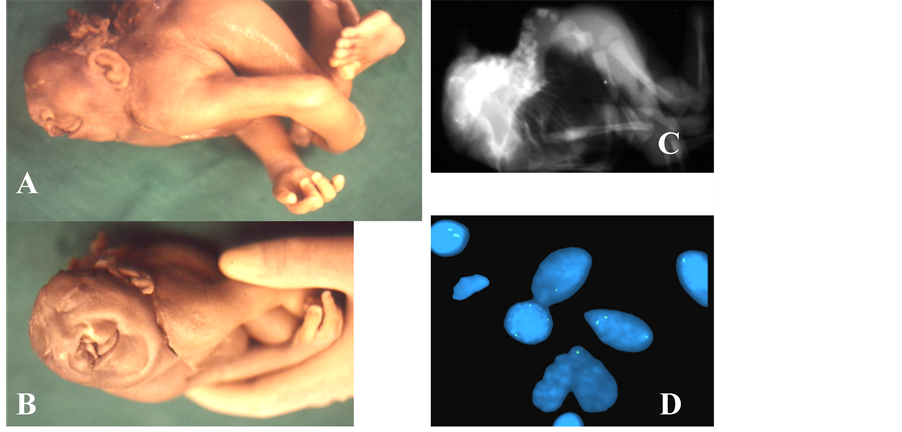


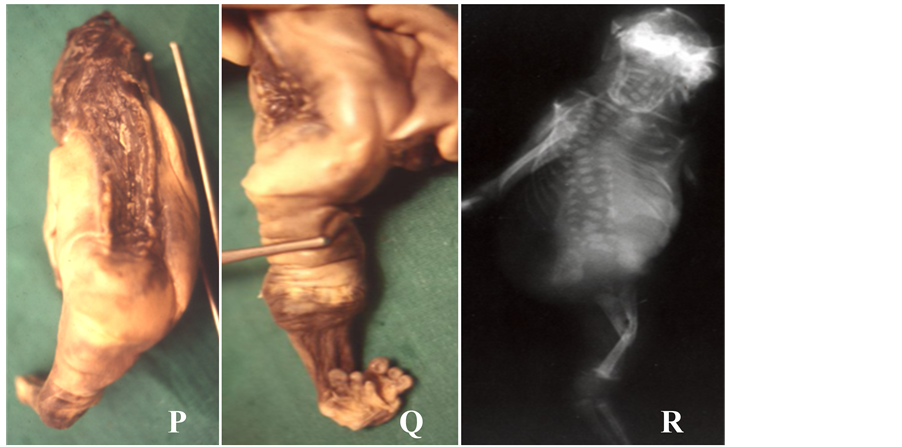
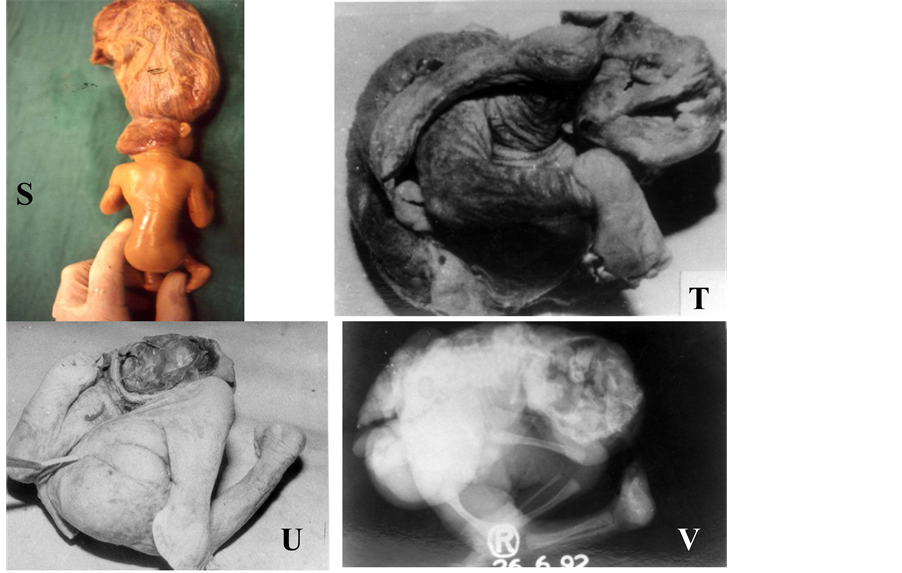
Figure 2. (A)-(V) showing syndromic neural tube defects (NTDs). NTD with chromosomal abnormalities (A-O): Aneneiniencephaly with mosaic trisomy 13 showing anencephaly, facial cleft (A, B), hyperextension of spine with cervical vertebral disintegration (C) and interphase FISH (D) with chromosome 13 probe showing mosaic trisomy 13 (3 as well as 2 copies of chromosome 13). Iniencephaly (also spina bifida aperta) with mosaic monosomy X showing extensive cystic hygroma, hydrops fetalis, spina bifida aperta (E, F), hyperextension of spine with cervical vertebral disintegration (G), septate cystic spaces/cystic hygroma on ultrasound (H) and interphase FISH (I) with chromosome X probe showing mosaic monosomy X (1 as well as 2 copies of chromosome X). Spina bifida with trisomy 18 showing terminated fetus with meningocele (J-L), omphalocele (J-L), antenatal ultrasound showing choroid plexus cyst (N) and omphalocele containing gut & liver (M) and interphase FISH with chromosome 18 showing 3 copies (O) thus proving trisomy 18 state. NTD with Sirenomelia (P-R): showing craniorachischisis & sirenomelia (P-R). X ray (R) shows no bony fusion of lower limbs (only soft tissue fusion). NTD with amniotic band syndrome/limb body wall complex (S-V): showing placenta attached to head leading to destruction of skull & brain (S), facial cleft with anenencephaly (T) and absent vault bones (U, V).
caudally [21] . However, evidences are accumulating in favor of multi-site initiation of neural tube closure in humans and NTD represents the failure of one or more of 5 closure sites [22] [23] . In simple iniencephaly, the defect found in occipitocervical region (more pronounced in apertus) indicates site 1 (mid cervical) and probably site 4 (caudal end of rhombencephalon) closure defect whereas in Group B (iniencephaly with anencephaly) where vault and facial bones as well as lower vertebrae, in addition to occipitocervical region, are involved, fa- vors all 5 sites (site 2―between prosencephalon and mesencephalon; site 3―at stomodeum; site 5―most caudal end of neural tube) closure defect hypothesis. The exaggerated spinal retroflexion observed with iniencephaly could be due to absence of neural arches. Associated malformations, deformations and hypoplasia, for example, pulmonary hypoplasia, diaphragmatic hernia, omphalocele, etc. could result from lack of space (increased pres- sure) in thoracic and abdominal cavity secondary to retroflexion and crowding of ribs. This condition is asso- ciated with female preponderance [19] however the cause is not known. It may be due to incomplete penetrance and/or male lethality. This need to be evaluated in future with special attention to chromosome X. Iniencephaly is clubbed with NTD [24] [25] . Most cases of iniencephaly are sporadic and are probably secondary to multi- factorial inheritance [24] . Chromosomal abnormality is rarely reported with iniencephaly. We have come across 39 cases of iniencephaly among 240 fetal autopsy referrals over a period of 10 years [19] [26] [27] . Only 3 cases had chromosomal abnormality or prompted suspicion for underlying chromosomal abnormality (Figures 2(A)- (O)). Literature search shows few reports of chromosomal aneuploidy (monosomy X, trisomy 2 and mosaic tri- somy 13) with iniencephaly [7] [27] -[29] . The first published case of iniencephaly with monosomy X was reported by Coerdt et al. [7] and later by Halder et al. [27] . Subsequently, Phadke & Thakur [28] and later Halder et al. [27] reported trisomy 13 mosaicism with iniencephaly. A case of trisomy 2 with iniencephaly in association of microcephaly, encephalocele and cervicothoracic spina bifida was reported by Seller et al. [29] . Iniencephaly may present as hydrops fetalis [19] . Whether chromosome abnormalities were causative or chance co- occurrence with iniencephaly is unknown.
Craniorachischisis is characterized by anencephaly and open spinal defect of the entire spine (Figures 1(L)- (N)) leading to exposure of entire neural tissue (brain & spinal cord). Craniorachischisis totalis with sirenomelia (Figures 2(P)-(R)) is a rare fetal malformation. Only few cases have been reported [30] -[32] . The association of craniorachischisis totalis and sirenomelia indicates the existence of a defect right from early embryogenesis. Formation of the neural tube first occurs on Day 22 at the level of somite 3; fusion proceeds both rostrally and caudally and the neural tube is closed by Day 26 whereas lower limb buds are recognizable between 28 - 32 days of developments [33] . From an embryological point of view, a condition that combines cephalic (craniospinalrachischisis) and caudal defects (fused lower limbs along with pelvic, lower vertebrae and genitourinary organ anomalies) are considered to be example of axial mesodermal dysplasia sequence [33] . A primary disturbance in the axial mesodermal may impair formation, elevation and approximation of neural folds leading to craniospinal rachischisis. Similarly, sirenomelia may occur due to failure in lateralization of lower limbs secondary to mesenchymal deficiency of the caudal eminence, i.e., axial mesoderm [33] . The caudal eminence is an active seat of cell proliferation which contributes to the formation of notochord, vertebrae, lower limb buds, perineum, neural plate, neural cord, hind gut and blood vessels. Although an extensive disturbance of axial mesoderm is likely to occur in the formation of neural tube defect, a milder alteration of neuroectoderm cannot be excluded in most sirenomelia as defect occurs in coordinated process of cell proliferation, migration and cell to cell adhesion.
Recurrence risk of isolated NTDs after one affected child is 3% - 5%, which is 10 times higher than the gen- eral population. It increases to about 10% after two affected children and to about 25% after three such births. After birth of one child with NTD a couple should be counseled about prevention in subsequent pregnancies; primary prevention by periconceptional folic acid supplementation and secondary prevention by antenatal ma- ternal serum alpha feto protein (MSAFP) and ultrasound examination along with selective termination of af- fected pregnancy. However, syndromic NTDs have different recurrence risk (sporadic to 25% as with Meckel syndrome). Hence, for accurate counseling gross examination & radiography may not be adequate in cases of NTD with multiple malformations. In these cases an expert opinion from geneticist with specialized tests may be required. MSAFP and ultrasound at 10 - 12 weeks (to exclude anencephaly) and again 16 - 18 weeks (to exclude all other types) should be advised in subsequent pregnancy. MSAFP (taking cut-off > 2.5 MOM using BPD for estimating gestation age) has a detection rate of 85% at 16 - 17 weeks. In skilled hand and in high-risk women ultrasound has a detection rate of 98% not only for NTD but also for most major/lethal malformations.
3. Chromosomal Abnormality
Normal chromosome complement in human is 46, XX (female) or 46, XY (male) and known as diploid state, while one set of 23 chromosomes seen in gametes (23, X in female or 23, Y and 23, X in male) is known as hap- loid. Error in segregation of chromosomes during cell division leads to gain or loss of chromosome, which is called aneuploidy. It includes missing of a member of the pair (monosomy) or presence of more than two chro- mosomes in a pair (trisomy for 3 numbers, tetrasomy for 4 numbers, etc.). Errors in mitosis result in mosaicism, i.e. two types of cells originated from single cell. Rarely, admixture of 2 or more zygote results in chimerism, i.e., two or more types of cells originated from two or more zygote in early embryonic life. Sometimes an indi- vidual may have an additional set of haploid (triploidy; 69) or diploid (tetraploidy; 92) chromosomes. The phe- notype due to chromosomal abnormality depends on gain or loss, fusion, rearrangement, or altered parental in- heritance of genes located on the chromosome. Most aneuploidies of autosomes, unbalanced structural abnor- malities of autosomes, triploidies, tetraploidies and other polyploidies are spontaneously aborted or produces le- thal developmental defect. Remaining aneuploidies and unbalanced structural abnormalities of autosomes pro- duce mental retardation and varying degree of dysmorphism. Sex chromosome aneuploidies, in particular mo- nosomy X is lethal in most however other types are mild and often produce hypogonadism. Chromosomal ab- normality is one of the major causes of lethal malformation in perinatal period. The incidence of chromosomal abnormalities at birth is about 0.5%. However, in pregnancy with ultrasound detected malformations, its inci- dence is much higher [34] .
3.1. Trisomy 21
Trisomy 21 is caused by presence of all or part (21q21-22) of extra chromosome 21 leading to over expression of chromosome 21 genes (amyloid beta A4 precursor protein, microRNAs, etc.). Trisomy 21 is the commonest autosomal aneuploidy in live births (one in 800 to 1000) however in early pregnancy its incidence is much more. Over 85% of trisomy 21 conceptuses abort spontaneously. In early fetal stage it is characterized by cardiac mal- formations, duodenal atresia, facial dysmorphism, etc. (Figures 3(A)-(E)). Majority of cases of trisomy 21 are due to three copies of chromosome 21 where the risk of recurrence is negligible. About 4% cases are due to translocation. The risk of recurrence in translocation cases varies depending on the chromosomes involved and sex of the carrier parent (more if mother is the carrier and may be up to 100% with chromosome 21:21 translo- cation). Birth of a child with trisomy 21 is preventable through maternal serum screening, prenatal ultrasonography, chromosome analysis and selective termination of pregnancy. Parental age at conception, in particular maternal age above 35 years, increases the risk of trisomy 21. In future clinical manifestation of trisomy 21 due to gene imbalance across extra chromosome 21 can be corrected by inserting a single gene, XIST (the X-inac- tivation gene) to one chromosome 21 and to form a chromosome 21 Barr body thus inactivating one chromo- some 21 as naturally occurs with one of X chromosome in normal female.
3.2. Trisomy 18
It is the second most common autosomal trisomy (3 copies of chromosome 18, instead 2 copies) in live births (one in 3000). It is more in female births. The incidence increases with maternal age. Most of trisomy 18 con- ceptuses abort spontaneously. The syndrome is characterized by multiple congenital malformations of heart, kidneys, central nervous system (choroid plexus cyst, spina bifida, etc.), gastrointestinal tract (omphalocele, etc.), etc. (Figures 2(J)-(O)) besides dysmorphism (low-set malformed ears, hypertelorism, etc.), cleft lip/palate, clench- ed hands, underdeveloped thumbs, rocker bottom feet and joint contractures. Ultrasound can detect fetal mal- formations like omphalocele, choroid plexus cyst & polyhydramnios (Figures 2(M)-(N)). About 30% - 40% of antenataly diagnosed omphalocele has abnormal karyotype [35] and most commonly trisomy 18. Trisomy 18 is also common with choroid plexus cyst [36] . Most fetal malformations are detected around 18 - 22 weeks of ges- tation, when a rapid diagnosis is required to make decision particularly for termination of pregnancy, as in many countries termination is not permitted after 20 - 22 weeks gestation. In this situation, parental anxiety is also considerable and most of them prefer speedy decision. Traditionally chromosomal analysis of fetus is carried out by conventional cytogenetics that takes 10 - 20 days. This is not the ideal situation and hence there is a need for alternative rapid technique. Interphase FISH is one such technique (Figure 2(O)) [6] .

Figure 3. (A)-(E) showing trisomy 21 with facial dysmorphism (A); sandal gap (B); duodenal atresia (C); double bubble sign on antenatal ultrasound examination (D) and interphase FISH with chromosome 21 (showing three copies of chromosome 21).
3.3. Trisomy 13
Trisomy 13 affects approximately one in 10,000 live births. It is associated with severe growth retardation and major congenital anomalies including NTD (Figures 2(A)-(D)). The typical features are holoprosencephaly, microcephaly, microphthalmia, spinal defect, omphalocele, cleft palate, cleft lip, polydactyly, overlapping of fingers and rocker-bottom feet [14] .
3.4. Monosomy X
Monosomy X (Turner syndrome) occurs one in 2500 female births. Approximately 98% of Turner syndrome conceptuses end up with miscarriage. In early pregnancy may present as cystic hygroma with hydrops fetalis (Figures 2(E)-(H), Figures 4(A)-(C)) [27] . Turner syndrome cases present with short stature, broad chest, low hairline, low-set ears, webbed neck, increased carrying angle of the elbow and gonadal dysfunction. Major sys- temic problems are also frequently present, including congenital heart defect (bicuspid aortic valve, coarctation of the aorta), kidney defect (horse shoe kidney; Figure 4(B)), hypothyroidism, diabetes mellitus, vision prob- lems, hearing defect, etc.
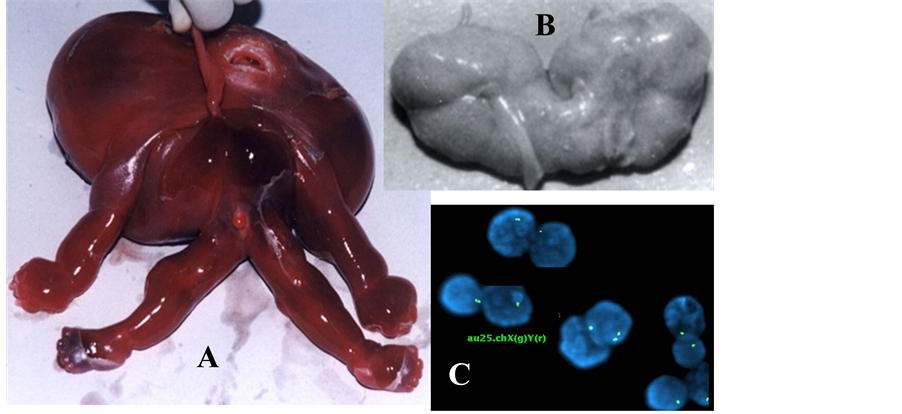
Figure 4. (A)-(C) showing monosomy X (C; XY FISH showing mostly 1 signal of chromosome X & no Y) with cystic hygroma (A), hydrops fetalis (A) and horse-shoe kidney (B).
3.5. Triploidy
Triploidy (Figures 5(A)-(F)) accounts for 2% of all conceptions, but only one in 50,000 are live births as mostly present as missed abortion (Figure 5(A) and Figure 5(B)) or aborted early in fetal life (Figure 5(C) and Figure 5(D)). Triploid newborns die immediately after birth. They present with growth retardation, relative macroce- phaly, cleft lip, low-set ears, agenesis of corpus callosum, heart defects, dysgenetic kidney and omphalocele. Triploidy and other polyploidies generally lead to miscarriage, still birth or partial hydatidiform mole and are often associated with cancer.
4. Amniotic Band Syndrome (ABS)/Limb Body Wall Complex (LBWC)
Amniotic band syndrome (ABS) and/or limb body wall complex (LBWC) is considered to be caused by rupture of the amnion with secondary effects on the fetus producing malformation due to interruption of normal mor- phogenesis, deformation due to distortion of established structures and disruption of structures already formed [37] [38] It may be associated with loss of amniotic fluid, producing secondary effects due to oligo-hydramnios [39] . It is seen in 1 in 70 spontaneous abortions [40] . The syndrome is under diagnosed and its presentation is so variable that no two cases are exactly alike. Pathogenesis of this defect is probably heterogeneous [41] . Mecha- nisms invoked have included germ disc disruption [42] [43] , genetic disruption [44] , vascular disruption [45] [46] and amniotic disruption [47] .
The diagnosis of ABS/LBWC is based on two out of three manifestations viz., craniofacial clefts; limb body wall defects and amniotic band attachment (Figures 6(A)-(F)). Single phenotype (resulting from placenta/ membrane attachment with embryo followed by effects due to fetal movement that result in pulling, disruption, and entanglement of fetal parts, with/or without oligohydramnios leading to compression effect as well as pul- monary hypoplasia) with different terminology by different investigators, indicates only etiopathologic hetero- geneity.
Despite recognition for more than 300 years, fetal disruption complex is a constellation of congenital malfor- mation that lacks precise definition and a satisfactory pathogenetic explanation. It is characterized by fetal ano- malies caused by various mechanisms including aberrant embryonic morphogenesis, fetal/embryonal vascular compromise, mechanical deformations and secondary disruption of normally developed structures. Occurrence of amniotic bands would appear to be a requirement for the diagnosis; however, the presence of fetal malforma- tion in a non-embryogenic distribution can allow the diagnosis even in the absence of bands. It appears that ABS/LBWC is a syndrome of etiopathogenetic heterogeneity where the above pathogenetic modes produce some or most of the clinical picture of ABS/LBWC.

Figure 5. (A)-(F) showing triploidy (3 copies of chromosome 1 & sex chromosome suggestive of triploidy state) in early ges- tation/missed abortions (A, B) and mid gestation with extreme growth restriction and relative macrocephaly (C, D) and in- terphase FISH with chromosome X/Y (E) and 1 (F).
5. Nonimmune Hydrops Fetalis (NIHF)
Hydrops fetalis is defined as fluid accumulation in some or all serous cavities of the fetus with variable degree of generalized skin edema along with or without an edematous placenta [48] . Non-immunologic hydrops fetalis (NIHF) is due to causes other than fetomaternal blood group incompatibility [49] . It was first identified by Potter [50] and its reported incidence is 1 in 3000 birth [51] . Numerous maternal, fetal and placental pathologies are known to cause NIHF [52] . However, in most published series, 10% - 60% of such causes have been termed idiopathic [53] [54] . The increasing availability of obstetric ultrasound has resulted in early diagnosis even at asymtomatic stage which enables a wider range of investigatory scope. It is suggested that a complete perinatal workup including necropsy can lead to a precise diagnosis in most cases.
From the myriad of maternal, fetal and placental problems which are known to cause non-immune hydrops fetalis (NIHF), many different causes of the disorder could be identified by extensive perinatal workup. However, despite full effort I found cases without clinical, biochemical and post mortem findings justifying the develop- ment of hydrops fetalis (unexplained). The common demonstrable causes of the disorder are skeletal dysplasia, iniencephaly [19] , chromosome abnormality (monosomy X ), cyst adenomatoid malformation of lung (Figures 7(A)-(F)), umbilical cord true tight knot (Figures 7(G)-(H)) [55] , Twin-Twin transfusion syndrome (Figures 7(I)-(K)), diaphragmatic hernia and multiple bilateral renal cysts. Polyhydramnios, anemia, and pregnancy in- duced hypertension are frequent maternal complications. Ultrasonography is always successful in detecting hy- dropic state. Whenever NIHF is diagnosed, one should look for congenital malformations as well as fetal hy- poxia and umbilical vessels blood flow for evaluation of umbilical cord and placental pathology besides ne- cropsy.
The hydropic state results from the combination of a total increase in body water and a relative increase in fluid in the interstitial space (Figure 7(D)). When the latter becomes severe there is usually free fluid in body cavities as well (Figure 7(E) and Figure 7(F)). Maintenance of normal fluid distribution requires a balance of osmotic and hydrostatic forces on either side of cell membrane as well as an intact sodium pump. The common mechanisms are severe chronic fetal anemia (leading to hypoxemia), fetal heart failure (leading to reduction of arterial hydrostatic pressure) and obstruction to venous return from the placenta (leading to villous edema & hypoxemia-hypo nutrition to fetus) and they act through a common pathway at the cellular level, i.e., hypoxia [56] . There are many factors that modify the effect of primary abnormality viz., stage of gestation (in late preg- nancy fetal tolerance is reduced and hence more hydrops in late pregnancy). In my experience I did not find any case of hydrops before 18 weeks of pregnancy and severe hydrops before 20 weeks of pregnancy. This is ace- lerated in second half of pregnancy by rapid fetal growth.
A wide range of disorders have been reported in association with NIHF including a diverse range of malfor-

Figure 6. (A)-(F) showing limb body wall complex/amniotic band syndrome cases featuring disruption of upper half of body including abdomen, thorax and head (A), constriction bands (A, F; arrow), membrane/amniotic band attachment (E; arrow), limb amputation, necrosis (D), facial asymmetric cleft (C) and placental attachment with head (A) and back (B; arrow).
mations, chromosomal anomaly, genetic metabolic disease, infection, twin pregnancy, etc. [56] . Among 15 cases of NIHF presented to me (unpublished data), there were 2 cases in whom no primary abnormalities could be found. In one there was a similar history of hydropic state in previous pregnancy thus indicating a monogenic recessive disorder. In the remaining cases a possible pathogenetic mechanism were derived through autopsy and related investigations. Most of the cases had shown lesions that had caused impairment of venous return to fetal heart (skeletal dysplasia, NTD/craniorachischisis, diaphragmatic hernia, umbilical cord knot, etc.) or heart failure (Twin-Twin transfusion syndrome). The obstruction to venous return to heart secondary to increased intrathoracic pressure in skeletal dysplasia (compromised thorax) & diaphragmatic hernia seems a major factor in development of hydrops in eight cases, i.e., over 50% of cases. The situation can be described as a fetal val- salva maneuver and here fetal development of thoracic viscera is constricted by skeletal dysplasia or by thoracic vertebral rachaischisis. Cases like thanatophoric dysplasia, achondrogenesis, osteogenesis imperfecta, hydrolethalus syndrome, anen-iniencephaly and right sided diaphragmatic hernia. In all cases there were significant thoracic


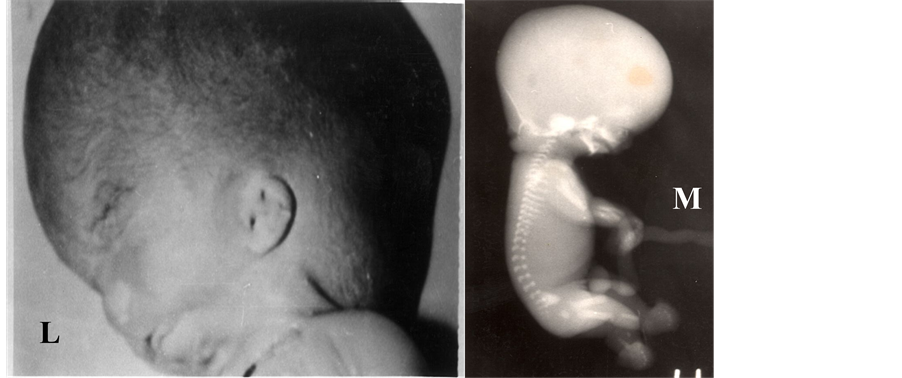
Figure 7. (A)-(F) showing cases of hydrops fetalis from cyst adenomatoid malformation of lung (A-F: honey comb appear- ance on cut section-C, skin edema-D, ascites-E, hydramnios & placental edema-F), umbilical cord true tight knot (G, H), twin-twin transfusion syndrome (I-K; donor twin hydropic and recipient twin plethoric) and hydrolethalus syndrome (L-M: hydrocephalus, polydactyly, hallux duplex, clubbed feet, small mandible, poorly formed nose; hypoplastic eyes, etc.).
space compromise and all fetuses had hydrops and pulmonary hypoplasia.
The medical literature contains little specific data on cord true knot (Figures 7(G)-(H)) and hydropic state [55] . Hydrops in monozygotic twin gestation may affect recipient, donor or both, in that order of frequency (Figures 7(I)-(K)). Twin transfusion syndrome has been diagnosed as early as 18 weeks of gestation and often has severe effects in mid gestation. The donor fetus presents with severe anemia and hydrops whereas recipient fetus plethoric (rarely hydropic). In twin transfusion syndrome the mother often presented with severe hydrom- nios, pregnancy induced hypertension, antepartum hemorrhage and preterm delivery. Cyst adenomatoid mal- formation of lung may present with severe hydrops fetalis (Figures 7(A)-(C)). Other causes of NIHF are chro- mosomal anomalies, some inborn error of metablism, fetal infection, visceral malformations (cardiovascular, res- piratory, urinary, gastrointestinal, hepatic, etc.), placental disorders (e.g. chorioangioma) and umbilical cord pa- thology (knot, thrombosis, entanglement etc.). This disorder often recurs and observed recurrence in achondro- genesis (recurred in subsequent pregnancy), hydrolethalus syndrome (precurred in 2 previous pregnancies in one case; Figures 7(L)-(M)) and idiopathic hydrops fetalis (precurred in previous pregnancy). It is felt that detail examination of placenta and umbilical cord for its pathology (leading to hypoxia and hypo-nutrition) may reveal etiological basis in large number of idiopathic hydrops. At present these organs are being neglected from detail examination and now it demands its importance.
6. Lethal Skeletal Dysplasia
Lethal skeletal dysplasia commonly encountered in early pregnancy are thanatophoric dysplasia, osteogenesis imperfecta, achondrogenesis, short rib polydactyly syndrome, Roberts syndrome, hypophosphatasia, campomelic dysplasia, chondrodysplasia punctata (rhizomelic type), atelosteogenesis, etc. Thanatophoric dysplasia is the commonest lethal skeletal dysplasia followed by osteogenesis imperfecta [57] [58] . Thanatophoric dysplasia type I (Figures 8(A)-(C)) is characterized by severe shortening of limbs with redundant skin folds, large head, frontal bossing, depressed nasal bridge, and narrow thoracic cage, short ribs, platyspondyly, hypoplastic iliac bones, marked shortening of long bones including bowing of femora, etc. Thanatophoric dysplasia type II is characterized by straight short femora. The condition is caused by mutation (usually de novo) in fibroblast growth factor receptor 3 (FGFR3) and hence can be diagnosed in first trimester by molecular analysis [59] -[61] . Osteogenesis imperfecta (also known as brittle bone disease) is a connective tissue & skeletal disorder caused by defects in type I collagen (COL1A1 and COL1A2) synthesis secondary to mutation in the COL1A2 gene [57] [58] [62] . Osteogenesis imperfecta type II (Figures 8(D)-(G)) is the most severe among Osteogenesis imperfectas and detectable early prenatal life by sonography. Presence of blue sclerae besides bone fracture supports diagnosis. Achondrogenesis (Figures 8(H)-(M)) is an autosomal recessive lethal skeletal dysplasia. It presents as
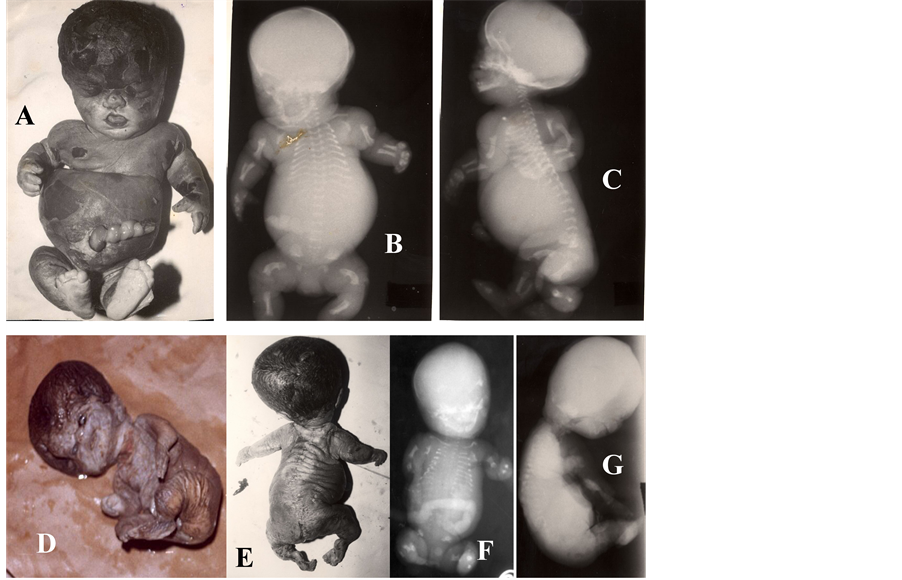
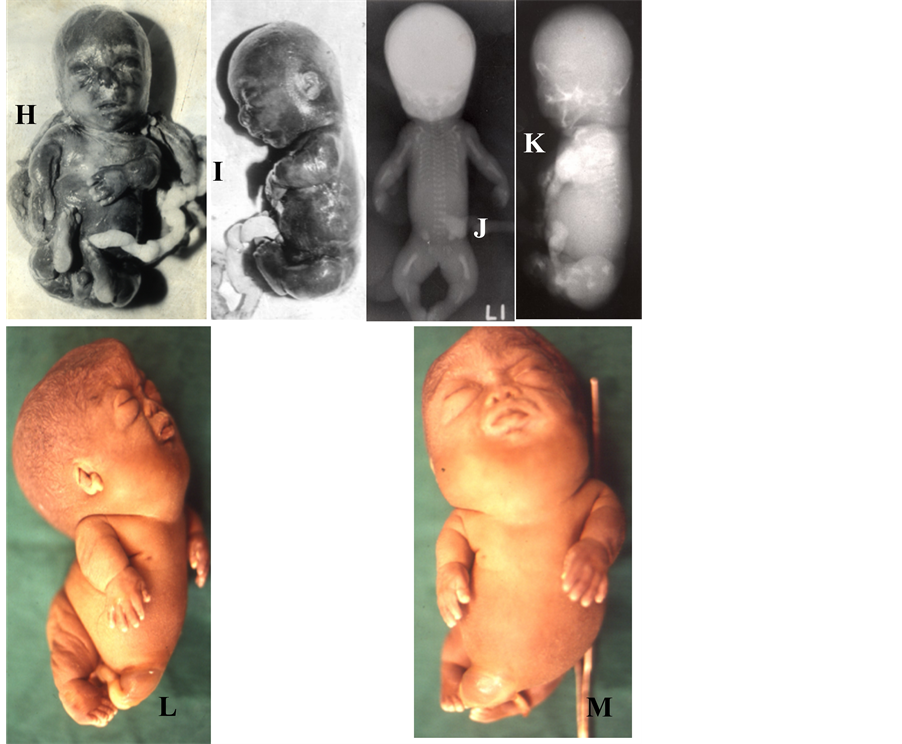

Figure 8. (A)-(R) showing lethal skeletal dysplasia. Thanatophoric dysplasia type I (A-C) showing shortened limbs, large head, frontal bossing, depressed nasal bridge, narrow thoracic cage, short ribs, platyspondyly, hypoplastic iliac bones, marked shortening of long bones and bowing of femora. Osteogenesis imperfecta type II (D-G) showing blue sclera, cataract, bone fracture (long bones & ribs) and short limbs due to deformed/crumbled bones. Achondrogenesis (H-M) showing micromelia, defective ossification of the skull and spine, short beaded ribs and deformed & short long bones. Short rib polydactyly syn- drome (N-P) showing short limb, polydactyly, thoracic hypoplasia, and extremely short ribs on X ray necrogram (P). Roberts syndrome (Q-R) showing tetraphocomelia or hypomelia, cleft lip/palate, widely spaced eyes, exophthalmos, underdeveloped ala nasi, ear malformations, oligodactyly, thumb hypoplasia, syndactyly, flexion contractures, etc.
intrauterine growth failure, micromelia, defective ossification of the skull and spine, short beaded ribs and de- formed as well as short long bones. Achondrogenesis Type 1A is caused by mutations in the TRIP11 (thyroid hormone receptor interactor 11) gene, resulting in deficiency of the golgi microtubule associated protein 210 [63] whereas achondrogenesis type 2 is caused by mutation in collagen type II gene (COL2A1). COL2A1 mutation also associated with Kniest dysplasia and spondyloepiphyseal dysplasia congenital [64] . Short rib polydactyly syndrome (Figures 8(N)-(P)) is a heterogeneous lethal skeletal dysplasia. It is characterized by short limb, po- lydactyly, thoracic hypoplasia, heart defects and renal dysplasia. There are 4 types of the short rib polydactyly syndrome, e.g., type I (Saldino-Noonan); type II (Majewski); type III (Verma-Naumoff) and type IV (Beemar- Langer). However, there is phenotypic overlapping between the types. Roberts syndrome (Figures 8(Q)-(R)) is a rare autosomal recessive disorder characterized by symmetrical defects of limbs (tetraphocomelia or hypome- lia), craniofacial abnormalities (microcephaly, cleft lip and/or palate, down slanted palpebral fissures, widely spaced eyes, shallow orbits, underdeveloped ala nasi, ear malformations, etc.), growth retardation, oligodactyly, thumb aplasia or hypoplasia, syndactyly, clinodactyly, flexion contractures, etc. Cohesion 1 homolog 2 (ESCO 2) mutation leads to lack of cohesin acetylation resulting nucleolar defects and translational inhibition and respon- sible for Roberts syndrome [65] . Camptomelic dysplasia is also heterogeneous lethal skeletal dysplasias and characterized by abnormal development of bones and cartilages. It is caused by a mutation in gene Sox9 on chromosome 17 and it is transmitted as an autosomal dominant trait. The clinical features are shortening and bowing of the long bones besides sex reversal. The majority of cases die during the fetal or early neonatal pe- riods. Atelosteogenesis (I and III) is autosomal dominant lethal skeletal dysplasias. It is characterized by verte- bral abnormalities, hypoplastic long bones and joint dislocations. It is caused by mutations in the gene encoding filamin B (FLNB). Hypophosphatasia is a rare autosomal recessive disorder caused by deficient activity of tis- sue nonspecific alkaline phosphatase (TNSALP) and characterized by defective bone mineralization [66] . Peri- natal lethal cases are stillborns, without mineralization of bones, hypercalcemic and dies from respiratory com- plications due to rachitic deformities of the thoracic cage and associated pulmonary hypoplasia. Lethal hypo- phosphatasia are inherited in autosomal recessive or dominant manner. Molecular diagnosis is possible with mu- tational analysis of TNSALP gene. Now panel of genes known to be associated with the skeletal dysplasias (con- sisting of approximately 100 genes) can be screened quickly for molecular confirmation through the use of next- generation sequencing platform [67] .
7. Sirenomelia
Sirenomelia is characterized by single fused lower limb (Figure 9(A) and Figure 9(B)) and multiple other de- fects that include defects of contiguous organs such as urogenital system, rectum and lower vertebrae, which constitute a single developmental field. This sequence is known to be associated with noncontiguous anomalies, such as CNS defect and radial dysplasia, which do not lie in the same field. The pathogenesis of sirenomelia is uncertain. Some of the proposed theories include posterior axial mesodermal defect [68] [69] , vascular steal [70] [71] , teratogenic effect [72] , axial mesodermal dysplasia sequence [33] and midline developmental field defect [30] . Absence of familial cases and discordance in monozygotic twin make a genetic mechanism unlikely [73] . Some of the manifestations of Sirenomelia are secondary to oligohydramnios and vascular insufficiency.
8. OEIS Complex
A combination of severe congenital anomalies of the cranial region (corresponding to the oculo-auriculo-verte- bral) and caudal region (resembling the caudal deficiency sequence) was described [74] [75] . The term “axial mesodermal dysplasia” was used by Russell et al. [74] to describe this condition. However, cases was described with combinations of the cranial anomalies suggestive of oculo-auriculo-vertebral sequence and caudal anoma- lies resembling the omphalocele-extrophy of bladder, imperforate anus, and spine defects (OEIS) complex that suggests a defect during blastogenesis may be responsible (Figures 10(A)-(E)) [76] . Oculo-auriculo-vertebral sequence is known to be variable in its clinical presentation, the mildest of which may be the presence of a un- ilateral ear tag only [77] . The caudal anomalies may resemble the caudal deficiency sequence or the OEIS com- plex [78] or defect of blastogenesis [76] . During blastogenesis the entire embryo represents a single develop- mental field and any “hit” during this period can result in widespread anomalies which are severe in nature, usually of the midline and frequently incompatible with life [79] . A generalized alteration in mesodermal cell migration during the primitive streak period has been postulated to cause the malformations described as “axial
mesodermal dysplasia” and OEIS [74] [80] . However, these malformations can also be adequately explained as defects of blastogenesis [76] .
9. Urorectal Septum Malformation Sequence
The malformations/abnormalities associated with urorectal septum malformation sequence (URSMS) are ambiguous genitalia in the form of a phallic-like structure (in 46, XX individuals), absence of urethral and vaginal openings, imperforate anus, hypoplastic bladder/rectum/urethra/uterus, uro-rectal-vaginal fistulae, Müllerian duct defects, and impaired kidney function (Figures 11(A)-(C)). Although most of URSMS has been described as a sporadic condition, several cases reported [81] or unreported (one of my case was repeated in subsequent pregnancy and abnormalities observed were ambiguous genitalia, cloacal deformity, renal dysplasia, Müllerian duct dysplasia, tracheo-esophageal fistula, asplenia, normal adrenal and normal 46, XX) in sibs. URSMS can be

Figure 9. (A) and (B) sirenomelia showing front and back view of fused lower limbs, absent external genitalia, urethral opening and anus.
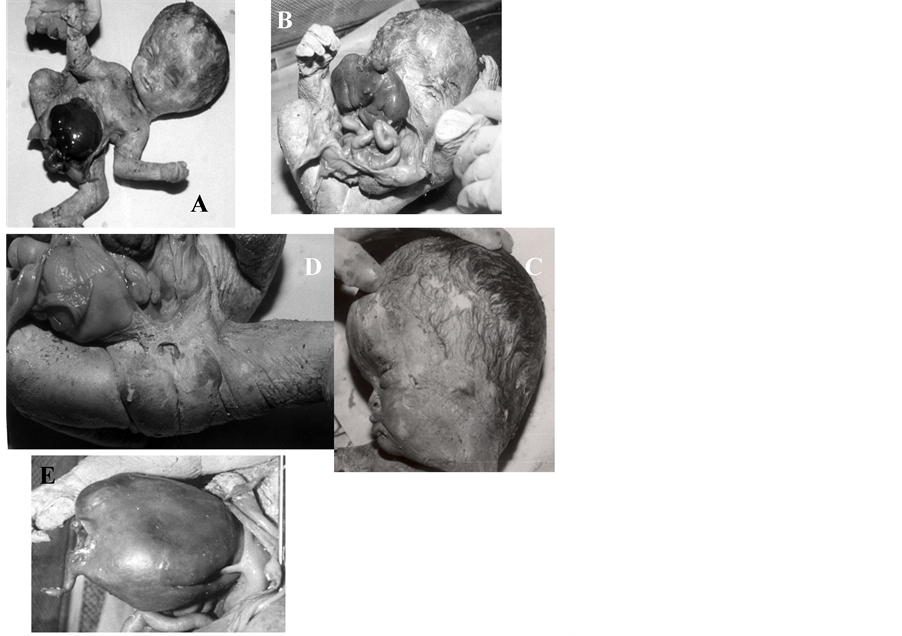
Figure 10. (A)-(E) OEIS complex (omphalocele, extrophy of bladder, imperforate anus and spine defects) showing ompha- locele (A), extrophy of bladder (B), microtia (C), imperforate anus (D) and globular/unlobulated liver (E).

Figure 11. (A)-(C) urorectal septum malformation sequence showing ambiguous genitalia (C); absence of urethral and va- ginal openings (C), imperforate anus (C); hypoplastic bladder/rectum/uterus, uro-recto-vaginal fistulae (B); Müllerian duct defects (B), etc.
divided as complete or partial. Absent perineal and anal openings with ambiguous genitalia, renal agenesis or dysplasia, and mullerian duct maldevelopment are included under complete URSMS, and a single perineal or anal opening draining a common cloaca with an imperforate anus is called partial URSMS. Complete URSMS is usually a lethal anomaly. The pathogenesis of URSMS appears to be defective caudal mesoderm resulting in in- terference of normal hindgut development and failure of breakdown of the cloacal membrane [82] .
10. Arthrogryposis Multiplex Congenita
Arthrogryposis is defined as multiple contractures involving more than one area of the body. Arthrogryposis multiplex congenita (AMC) is a malformation with various etiology and complex clinical features, including multiple congenital contractures in different body areas. Its etiology still remains unclear but generally any cause that leads to reduced fetal movement may lead to congenital contractures and in severe cases to fetal akinesia deformation sequence. The underlying cause may be neurogenic or myopathic, or connective tissue disorder, in- trauterine compression, teratogenic exposure or vascular insult or even genetic. Genetic etiology cannot be ruled out in some cases including one of my case who had three consecutive affected pregnancy (all 3 male; likely X linked; Figures 12(A)-(C)). The disease can be diagnosed in early pregnancy when a lack of mo- bility and an abnormal position is noted in ultrasound scanning along with polyhydramnios and fixed limb position.
11. Fetus-in-Fetu
Fetus in fetu is a rare abnormality where malformed fetus grows within the body of its twin (Figure 13(A) and Figure 13(B)). It results from abnormal embryogenesis and a parasitic twin of a diamniotic, monozygotic twin. It should be differentiated from teratoma which has no axial arrangement. Unlike teratomas, fetus in fetu is a benign disorder. It is usually intraperitoneal or retroperitoneal but may also present in other parts of body. It is usually surrounded by a membrane analogous to amniotic sac and supplied by a single feeding vessel. A true placenta is usually absent. Absence of an independent circulatory system explains the subsequent growth retar- dation. It is usually malformed due to pressure from host organ [83] . It has vertebral body and organs arranged around axis. This differentiates it from teratoma. Spencer [84] has suggested that a Fetus in fetu must have one or more of the following conditions: enclosed within a distinct sac; partially or completely covered by normal skin; grossly recognizable anatomic parts; attached to the host by only a few relatively large blood vessels; and associated with the neural tube or the gastrointestinal system. A teratoma, on the other hand, shows lesser or- ganization of microscopically identifiable tissues. Teratomas have a broader attachment site with multiple small blood vessels.

Figure 12. (A)-(C) arthrogryposis multiplex congenita showing multiple congenital contractures, fixed limb position, etc.

Figure 13. (A), (B) Fetus in fetu showing grossly recognizable but malformed fetal anatomic parts viz., limbs, trunk, buttock, etc enclosed within a distinct sac (cut opened) and covered by skin.
12. Fetus Papyraceus
The term fetus papyraceus (vanishing twin or fetal resorption) is used to describe a mummified fetus associated with multiple gestations where one fetus dies and flattened (Figure 14(A) and Figure 14(B)) between the mem- branes of living fetus and uterine wall. The dead fetus is compressed by its growing twin to a flattened, parch- ment like state known as fetus papyraceus [85] .
13. Hydrocephalus, Hypertricosis & Mullerian Abnormality
Congenital severe hydrocephalus is an important birth defect. The causes are heterogeneous as so clinical pres- entation. Although genetics play an important role in the aetiology of isolated congenital hydrocephalus, genetic etiology (mutations in L1CAM, MPDZ, etc.) is detected only in few cases [86] . I have across a case with severe hydrocephalus (Figures 15(A)-(D)) occupying skull cavity completely leading to papery thin brain parenchyma (Figures 15(C)). Fetus was born alive at 35+ weeks and expired within 12 hours of birth. Baby had bifid nose (due to extreme enlargement of skull with separation of sutures; Figure 15(A)), hypertrichosis (Figure 15(C)) and Mullerian duct abnormality (double vagina & cervix).

Figure 14. (A), (B) Fetus papyraceus showing mummified, flattened, parchment like fetus.

Figure 15. (A)-(D) hydrocephalus, hypertricosis & mullerian abnormality syndrome showing severe hydrocephalus, i.e., large head (A, D), bifid nose (due to separation of sutures by extensive hydrocephalus), hypertrichosis (B) and papery thin brain parenchyma (C).
14. Prune Belly Syndrome with Diaphragmatic Hernia
Prune-belly syndrome is a rare disease. The clinical manifestations are deficiency of abdominal wall muscles & genitourinary malformations. The pathogenesis is not fully understood but often associated with urinary tract obstruction, in particular posterior urethral valve. Other theories like mesodermal or allantois abnormalities are also proposed. Prune belly syndrome is often associated with pulmonary, cardiovascular, gastrointestinal and musculo skeletal abnormalities in addition to consequences of oligohydramnios viz., telepes equinovarus defor- mity, pulmonary hypoplasia, scoliosis, etc. In early pregnancy severe urethral obstruction may cause extreme distension of abdomen, and later herniation of abdominal viscera into thorax leading to hypoplasia of thoracic viscera (lungs, heart) & very rarely umbilical hernia or even herniation of thoracic as well as abdominal viscera through thoracic wall defect (Figure 16(A) and Figure 16(B))
15. Critical Analysis
Lethal developmental defects are one of the leading causes of perinatal mortality. A detailed examination of every lethal developmental defect case is necessary for proper reproductive counseling and management. Post- mortem studies have shown that antenatal diagnosis by ultrasound although is fairly accurate however often misses associated anomalies and final diagnosis. The accuracy of prenatal malformation ultrasonography is also likely to vary with the experience of the observer, time spent on the ultrasound examination and quality of the equipment used [26] . With increased availability of ultrasonography, many major malformations are detected early in pregnancy and pregnancies terminated without detailed work up in developing countries, including India. Hence, examination of the terminated fetus for detailed examination and specific test is essential for accurate diagnosis and genetic counseling and planning for next pregnancy. In our experience we observed additional malformations in as many as 50% cases as well as revision of diagnosis in over 30% cases [26] . This is also ob- served by others [87] - [89] . However, in developing countries perinatal autopsy is not a routine practice because of constraints in the form of expertise, finances, time, space and social issues. As a result diagnosis remains doubtful; hence a correct recurrence risk and possibility of early prenatal diagnosis to the parents. Post mortem fetal radiography (and photography) of selected cases can be a positive step in this direction [90] .
Chromosomal analysis of malformed fetus is an important investigation for a correct diagnosis. Although a proportion of fetal anomalies can be explained by chromosomal abnormalities (aneuploidy, unbalanced translo-

Figure 16. (A), (B) Prune belly syndrome showing extremely distended abdomen secondary to urethral obstruction (posterior urethral valve) along with herniation of abdominal & thoracic viscera through right lateral upper thoracic wall defect (A, B).
cation, deletions, or duplications), it may not be possible to do chromosomal analysis after termination in many cases as fresh tissue is unavailable as fetus usually sent in formalin. Formalin makes chromosomal analysis im- possible by the conventional chromosome culture method. Molecular techniques like interphase fluorescence in sit hybridization [91] or comparative genomic hybridization (CGH) or array CGH have been employed success- fully to determine chromosome ploidy in formalin fixed or dead/disintegrated fetal tissues [92] . These newer molecular techniques are capable to detect other genetic problems viz., microdeletion/microduplication syn- dromes. The lack of chromosomal, virological, biochemical and molecular analysis may hinder in making an ae- tiological diagnosis in many occasions, in particular with hydrops fetalis. Post termination radiological and his- topathological examination is important for skeletal dysplasia besides molecular confirmation. The commonest congenital malformation seen in practice is neural tube defects. Although multifactorial NTDs are common, one may see syndromic NTD like Mekel syndrome, amniotic band syndrome, trisomy 18, trisomy 13, etc. Hence, diagnosis and counseling should be based on the post-termination diagnosis and not solely on ultrasound exa- mination. Mistake in diagnosis & thus counseling may lead to birth of another malformed baby with lifelong burden and medico-legal consequences; hence an understanding of reproductive genetics is important for obste- tricians in coming years.
Although it is impossible to prevent all malformations, major common malformations like common aneuploi- dies and open fetal defects e.g., neural tube defect, abdominal wall defects, etc can be easily be prevented (sec- ondary prevention) by maternal serum triple test screening & ultrasound examinations. Fortunately, many of the major anomalies such as multifactorial NTDs, conotruncal anomalies of the heart such as double outlet right ventricle, anterior abdominal wall defects such as omphalocele and oral clefts are somehow (50% - 60%) pre- ventable (primary prevention) by supplementation of periconceptional folic acid alone. As scientific knowledge and medicine advance so do the expectations of the public. Advances in molecular biology through Human Ge- nome Project, introduction of triple marker screening & routine obstetric ultrasound examination into obstetrics practice, easy access to early gestational sac and prenatal diagnosis have helped both drive and meet public ex- pectations in preventing lethal developmental disorders. All pregnant women should be offered serum screening & ultrasound examination at 16 - 18 weeks for fetal malformation as a component of routine antenatal care. All abnormal serum screening and/or ultrasound examination should be followed with prenatal diagnosis (chromo- somal/molecular/level 2 ultrasound examination) to confirm whether the fetus is affected or not.
Reproductive genetic counseling is progressively becoming an integral part of obstetric practice. Availability of prenatal diagnosis for many malformations has added new dimension to genetic counseling. The advent of prenatal & preimplantation diagnosis has allowed these couple the option of having unaffected offspring and this has immensely increased the scope of genetic counseling in obstetric patient especially in women with previous child affected with a lethal malformation/developmental defect. The genetic counseling is also important to pre- vent future risk for medico legal problems (law suits) between obstetricians and patient.
16. Conclusions
The cause of developmental defects is unknown in majority cases until now. The possible association between environmental contaminants and developmental defects is an important concern, besides chromosomal disorders, genomic disorders (microdeletion/duplication disorders), monogenic disorders, epigenomic disorders and multi- factorial disorders. Without etiologic diagnosis, there is no way to provide accurate counseling and management strategy, most importantly predictive & preventive medicine approach. Therefore, research is essential in this field to identify underlying etiology in cases as many as possible for more accurate prediction or prevention of another case. Research with families affected by several pregnancies is absolute essential as these likely to pro- vide positive associations. Studying (morphologic, genomic, epigenomic, proteomic, etc.) embryos from the pe- riod when most developmental defects arise will be an important step towards understanding and perhaps even- tually predicting and preventing fetal abnormality in future. Malformed fetal repository (whole, tissue, organ, blood, etc from affected, parents, unaffected, etc. members) as well as clinical details is first step towards achiev- ing this goal.
In summary when a fetal lethal developmental defect is identified in pregnancy, the parents should be offered a reproductive genetic counseling, should be referred to a tertiary ultrasound unit for repeat assessment and at- tempt to detect other anomalies, should be offered other imaging techniques such as fetal echocardiography, ul- trafast fetal MRI, etc. in specific cases, if required. Every step should be ensured for prenatal diagnosis from pa- rental & fetal testing, even if involves invasive testing (chromosomal, specific molecular, etc.) and should be encouraged for complete fetal autopsy as well as stored in biorepository. And finally but most importantly, fam- ily with multiple affected fetus should be referred to a genetic centre to find out underlying genetic cause which is most likely contribute for better future in this field of medicine.
Acknowledgements
The author wishes to thank the Department of Medical Genetics, Pathology and Radiology, SGPGIMS, Luck- now for work-up of cases, including fetal autopsy.
References
- Kalter, I.T. and Warkany, J. (1983) Congenital Malformation Etiologic Factors and Their Role in Prevention. Parts I and II. The New England Journal of Medicine, 308, 424-431, 491-497. http://dx.doi.org/10.1056/NEJM198302243080804
- Nelson, K. and Holmes, L.B. (1989) Malformations Due to Presumed Spontaneous Mutations in Newborn Infants. The New England Journal of Medicine, 320, 19-23. http://dx.doi.org/10.1056/NEJM198901053200104
- Verma, I.C. (2000) Burden of Genetic Disorders in India. Indian Journal of Pediatrics, 67, 893-898. http://dx.doi.org/10.1007/BF02723953
- Mitchell, L.E. (2005) Epidemiology of Neural Tube Defects. American Journal of Medical Genetics Part C―Seminars in Medical Genetics, 135, 88-94. http://dx.doi.org/10.1002/ajmg.c.30057
- Sharma, A.K., Upreti, M., Kamboj, M., Mehra, P., Das, K., Misra, A., Dhasmana, S. and Agarwal, S.S. (1994) Inci- dence of Neural Tube Defects of Lucknow over a 10-Year Period from 1982-1991. Indian Journal of Medical Research, 99, 223-226.
- Agarwal, S., Chadda, V., Sharan, R. and Halder, A. (2003) Rapid Prenatal Diagnosis of Trisomy 18 by Interphase FISH. Asian Journal of Obs & Gynae Practice, 7, 14-15, 23.
- Coerdt, W., Miller, K., Holzgreve, W., Rauskolb, R., Schwinger, E. and Rehder, H. (1997) Neural Tube Defects in Chro- mosomally Normal and Abnormal Human Embryos. Ultrasound in Obstetrics & Gynecology, 10, 410-415. http://dx.doi.org/10.1046/j.1469-0705.1997.10060410.x
- Luo, J., Balkin, N., Stewart, J.F., Sarwark, J.F., Charrow, J. and Nye, J.S. (2000) Neural Tube Defects and the 13q Deletion Syndrome: Evidence for a Critical Region in 13q33-34. American Journal of Medical Genetics, 91, 227-230. http://dx.doi.org/10.1002/(SICI)1096-8628(20000320)91:3<227::AID-AJMG14>3.0.CO;2-I
- Bird, L.M., Pretorius, D.H., Mendoza, A.E. and Jones, M.C. (1997) Anencephaly with Holoprosencephalic Facies Due to Ring Chromosome 18. Clinical Dysmorphology, 6, 351-358. http://dx.doi.org/10.1097/00019605-199710000-00009
- Chen, C.P., Chern, S.R., Lee, C.C., Chen, W.L. and Wang, W. (2001) Prenatal Diagnosis of Mosaic Ring Chromosome 13 with Anencephaly. Prenatal Diagnosis, 21, 102-105. http://dx.doi.org/10.1002/1097-0223(200102)21:2<102::AID-PD992>3.0.CO;2-5
-
- Donaldson,
-
- Jauniaux, E., Halder, A. and Partington, C. (1998) A Case of Partial Mole Associated with Trisomy 13. Ultrasound in Obstetrics Gynecology, 11, 62-64. http://dx.doi.org/10.1046/j.1469-0705.1998.11010062.x
- Howkins, J. and Lawrie, R.S. (1939) Iniencephalus. BJOG: An International Journal of Obstetrics & Gynaecology, 46, 25-31. http://dx.doi.org/10.1111/j.1471-0528.1939.tb05313.x
-
- Stark, A.M. (1951) A Report of Two Cases of Iniencephalus. BJOG: An International Journal of Obstetrics & Gyna- ecology, 58, 462-464. http://dx.doi.org/10.1111/j.1471-0528.1951.tb04027.x
- Jayant, K., Mehta, A. and Sanghvi,
- Halder, A., Pahi, J., Pradhan, M., Pandey, A., Gujral, R. and Agarwal,
-
- Streeter, G.L. (1951) Developmental Horizons in Human Embryos. Age Group XI to XXIII. In: Heuser, C.H. and Corner, G.W., Eds., Embryology Reprint, Vol. II, Carnegie Institute, Washington DC, 165-196.
- Scherrer,
-
- Casbay, L., Szabo, I.,
- Erdincler, P., Kaynar, M.Y., Canbaz, B., Kocer, N., Kuday, C. and Ciplak, N. (1998) Iniencephaly: Neuroradiological and Surgical Features. Case Report and Review of the Literature. Journal of Neurosurgery, 89, 317-320. http://dx.doi.org/10.3171/jns.1998.89.2.0317
- Pahi, J.,
- Halder, A., Agarwal, S. and Pandey, A. (2005) Iniencephaly and Chromosome Mosaicism: A Report of Two Cases. Con- genital Anomalies, 45, 102-105. http://dx.doi.org/10.1111/j.1741-4520.2005.00076.x
-
- Seller,
- Opitz,
-
- Halder, A., Pahi, J., Chadda, V. and Agarwal,
-
- Rizzo, N., Pittalis,
-
- Gross,
- Torpin, R. (1965) Amniochorionic Mesoblastic Fibrous Strings and Amnionic Bands: Associated Constricting Fetal Malformations or Fetal Death. American Journal of Obstetrics & Gynecology, 91, 65-75.
- Higginbottom,
- Torpin, R. (1968) Fetal Malformations. In: Thomas, C.C., Ed., Amniotic Bands, WB Saunders Co., Springfield, 130- 137.
- Kalousek,
- Halder, A. (2010) Amniotic Band Syndrome and/or Limb Body Wall Complex: Split or Lump. The Application of Clinical Genetics, 3, 7-15. http://dx.doi.org/10.2147/TACG.S8894
- Streeter,
- Bamforth, J.S. (1992) Amniotic Band Sequence: Streeter’s Hypothesis Reexamined. American Journal of Medical Genetics, 44, 280-287. http://dx.doi.org/10.1002/ajmg.1320440304
- Donnai, D. and Winter, R.M. (1989) Disorganisation: A Model for Early Amnion Rupture? Journal of Medical Genetics, 26, 421-425. http://dx.doi.org/10.1136/jmg.26.7.421
-
-
- Miller, M.E.,
- Holzgreve, W., Curry,
- Hutchison, A.A., Drew, J.H., Yu, V.Y., Williams, M.L., Fortune, D.W. and Beischer, N.A. (1982) Nonimmunologic Hy- drops Fetalis: A Review of 61 Cases. Obstetrics & Gynecology, 59, 347-352.
- Potter, E.L. (1943) Universal Edema of Fetus Unassociated with Erythroblastosis. American Journal of Obstetrics & Gynecology, 46, 130.
- Maidman,
- Ballantyne,
- Villaespesa,
- Santolaya, J., Alley, D., Jalfe, R. and Wersof, S.L. (1992) Antenatal Classification of
- Halder, A. (2007) Nonimmunologic
- Keeling, J.W. (1991)
-
- Abdulkadir,
- Wattanasirichaigoon, D. and Charoenpipop, D. (2006) Thanatophoric Dysplasia: Roentgenographic Findings and Detection of a
- Chen, C.P., Chern, S.R., Shih, J.C., Wang, W., Yeh, L.F., Chang, T.Y. and Tzen, C.Y. (2001) Prenatal Diagnosis and Genetic Analysis of Type I and Type II Thanatophoric Dysplasia. Prenatal Diagnosis, 21, 89-95. http://dx.doi.org/10.1002/1097-0223(200102)21:2<89::AID-PD21>3.0.CO;2-9
- Kitoh, H., Brodie, S.G., Kupke, K.G., Lachman, R.S. and Wilcox, W.R. (1998) Lys650Met Substitution in the Tyrosine Kinase Domain of the Fibroblast Growth Factor Receptor Gene Causes Thanatophoric Dysplasia Type I. Mutations in Brief No. 199. Human Mutation, 12, 362-363.
- Bodian, D.L., Chan, T.F., Poon, A., Schwarze, U., Yang, K., Byers, P.H., Kwok, P.Y. and Klein, T.E. (2009) Mutation and Polymorphism Spectrum in Osteogenesis Imperfecta Type II: Implications for Genotype-Phenotype Relationships. Human Molecular Genetics, 18, 463-471. http://dx.doi.org/10.1093/hmg/ddn374
- Grigelioniene, G., Geiberger, S., Papadogiannakis, N., Mäkitie, O., Nishimura, G., Nordgren, A. and Conner, P. (2013) The Phenotype range of Achondrogenesis
- Nagendran, S.,
- Xu, B., Lu, S. and Gerton,
-
- Grody,
-
- Young,
-
- Talamo,
- Schinzel, A.A.G.L., Smith,
- Perez-Aytes, A., Montero, L.,
-
-
- Haldar, A., Sharma,
- Rollnick,
-
- Optiz, J.M. (1993) Blastogenesis and the “Primary Field” in Human Development. Alan R. Liss, Inc., New York, for the National Foundation-March of Dines. BD: OAS, 29, 3-37.
- Moore, C.A. and Weaver, D.D. (1990) Exstrophy of Cloaca Sequence. In: Buyse, M.L., Ed., Birth Defects Encyclo- pedia, Blackwell Scientific Publications, Oxford, 648-649.
- Aggarwal, S. and
-
-
-
- Landy,
- Al-Dosari, M.S., Al-Owain, M., Tulbah, M., Kurdi, W., Adly, N., Al-Hemidan, A., Masoodi, T.A., Albash, B. and Alkuray,
- Manchester,
-
- Clayton-Smith, J., Farndon,
- Sharma,
- Halder, A. and Park, Y.K. (1999) Identification of the Appropriate Tissue from Formalin Fixed Perinatal Autopsy Material for Chromosomal Ploidy Detection by Interphase FISH. Indian Journal of Medical Research, 110, 102-106.
- Halder, A. and Fauzdar, A. (2006) Extreme Skewing of Sex Ratio and Low Aneuploidy in Recurrent Early Missed Abortion. Indian Journal of Medical Research, 124, 41-50.