Open Journal of Obstetrics and Gynecology
Vol.3 No.2(2013), Article ID:28658,4 pages DOI:10.4236/ojog.2013.32044
Ovarian desmoplastic small round cell tumors: Prognosis is poor! Case report*
![]()
1Department of Gynecology Obstetrics I, University Hospital Hassan II, Fez, Morocco
2Department of Pathology, University Hospital Hassan II, Fez, Morocco
Email: #saadihanane1@yahoo.fr
Received 3 January 2013; revised 6 February 2013; accepted 15 February 2013
Keywords: Small Round Cell Desmoplastic Tumor; Ovarian; Diagnosis; Treatment; Prognosis
ABSTRACT
The tumor desmoplastic small round cell in women is rare. Ovarian its location is exceptional. We report a patient of 20 years, the original North Africa who consulted for an increase in abdominal volume with impaired general condition. Pelvic ultrasonography and computed tomography were in favor of multiple peritoneal masses with ascites. After surgical exploration, the histological and immunohistochemical data for diagnosis small round cell desmoplastic tumor of ovarian the patient was a candidate for poly chemotherapy but she was died a month later. Diagnosis relies on histological and immunohistochemical data. The demonstration of a nonrandom translocation t (11; 22) (p13; q12) is specific for the disease. The management is multidisciplinary and combining surgery, a poly aggressive chemotherapy and radiotherapy. The prognosis remains poor.
1. INTRODUCTION
The small round cell desmoplastic tumor (SRCDT) was first described in 1989 by Rosai and Gerald [1]. This rare tumor, usually locating peritoneal, affects preferentially young subjects with a male predominance. The histological and immunohistochemical aspect is characteristic. Despite the multidisciplinary management, the prognosis remains reserved.
Through this observation and a literature review, we clarify the different epidemiological, clinical, biological, immunohistological and treatment of this rare tumor.
2. OBSERVATION
BF is 20 year old woman, the original North Africa, single, no remarkable medical history admitted in our department for treatment of abdominal-pelvic distension. The onset of symptoms began a month ago by an increase of abdominal volume associated with chronic abdominopelvic pain without signs of digestive or compression. All this operating in the context of impaired general condition and unencrypted weight loss. The review found a patient conscious, normotensive at 110/ 60 mmHg, polypneique with 20 cycles per minute at 37˚ afebrile, without lower limb edema. Abdominal examination was objectively a distended abdomen, unshirred umbilical without a collateral venous circulation with a diffuse dullness. The gynecological examination unrealized because the patient is blank. The remaining of the physical examination was unremarkable. Abdominopelvic ultrasound showed the presence of multiple polypoid heterogeneous confluent peritoneal lesions with solidocystic echotexture and very vascularized associated with ascites of medium abundance and left moderate ureterhydronephrosis. At the floor of the uterus without pelvic abnormalities the ovaries not seen. Abdominopelvic CT scan confirmed the presence of multiple peritoneal nodules of varied size from 1 to 16 cm spread throughout the whole peritoneum and the uterus pushed forward by the blurred diverse masses. A moderate left urétérohydronéphrose with light delay and secretion retention of index parenchyma (Figure 1). No liver damage, spleen, pancreas, stomach or lymph nodes. The rate of Ca125 was very high. Exploratory laparotomy revealed an average abundance of ascites with an aspect of diffuse peritoneal carcinomatosis, the greater omentum is a tumor of 15 cm at the expense of the right ovary with irregular surface and a second at the expense of the transverse colon. The uterus could not be viewed but only one part. The decision of tumor reduction was achieved. Histological results showed ovarian tissue site of a round cell tumor proliferation, diffuse architecture made of round cells provided with atypical nuclei, high mitotic index and large breakout of necrosis is also noted (Figures 2 and 3). An immunohistochemical study was performed,

Figure 1. Abdominopelvic CT scan confirmed the presence of multiple peritoneal nodules of varied size from 1 to 16 cm.

Figure 2. HES × 10: histological results showed ovarian tissue site of a round cell tumor proliferation and large breakout of necrosis is noted.
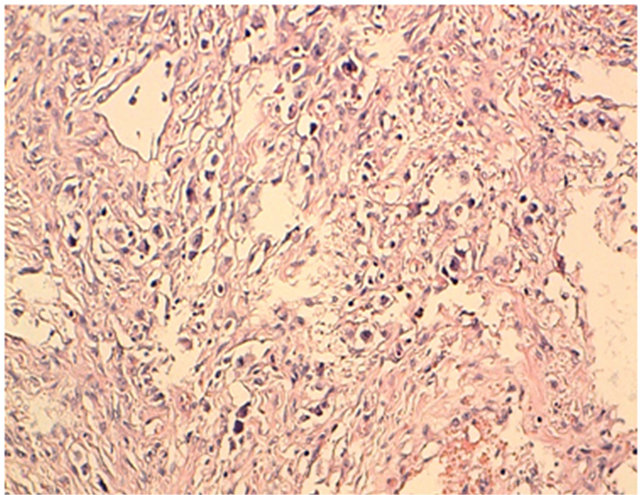
Figure 3. Hes × 20: tumor proliferation made of layers of cells with oval clear cytoplasm in a lymphocyte stroma.
all in favor of a small round cell desmoplastic tumor ovarian cancer with positive staining for antibodies to desmin (Figure 4) and very focally with the anti CKAE1/ AE3 (Figure 5) on the contrary the marking was negative for antibodies to CD99, anti-CD20, anti-CD3, antiinhibin and myogenin antibodies. The patient was a candidate for combination chemotherapy but the patient was dead a month later.
3. DISCUSSION
The SRCDT belong to the group of little differentiated malignant small cells. This group includes Ewing sarcoma, embryonic rhabdomyosarcoma, neuroblastoma, peripheral neuroectodermal tumors (PNET) and desmoplastic tumors [2].
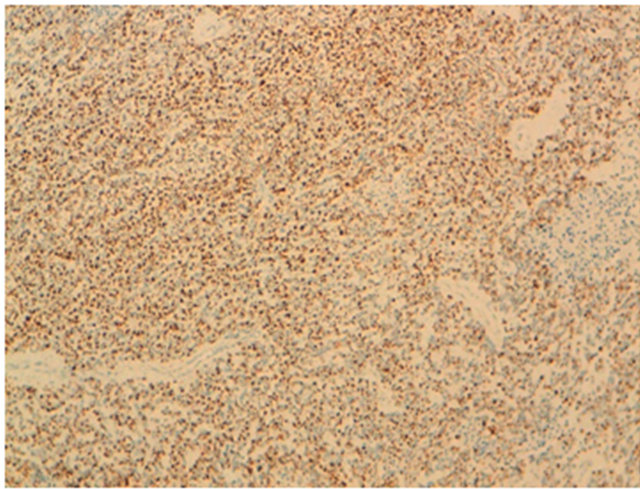
Figure 4. Immunohistochemistry: small round cell desmoplastic tumor ovarian cancer with positive staining for antibodies to desmin.

Figure 5. Immunohistochemistry: small round cell desmoplastic tumor ovarian cancer with positive staining very focally with the anti CKAE1/AE3.
It primarily affects adolescents and young adults with a male predominance (sex ratio 5/1) [3]. The median age of diagnosis is 20 years and generally less than 40 years, ranging between 5 years and 50 years [4]. Its histogenesis is uncertain, but his predilection for serous suggests with mesothelial origin [5]. It grows at the expense of serous especially the abdominal and/or pelvic peritoneum. Less commonly, it affects the pleura, lung, bone, neck, paratesticular region and ovaries. In the latter location, the symptomatology is revealing a large mass abdominopelvic pain associated with satellite nodules. All this operating in the context of impaired general condition, weight loss and unexplained fever. Laboratory tests showed an inflammatory syndrome with a blood serum marker Ca125 and NSE (neuron-specific enolase) is increased [6]. Imaging does not provide specific diagnostic feature of SRCDT. Indeed, the CT often shows a large abdominopelvic mass, heterogeneous, seat of hemorrhage, necrosis and/or calcification. This mass is usually associated with peritoneal dissemination and the metastatic lesions, especially ganglion. Of peritoneal effusions and/or pleural and cuts of intra-abdominal organs can be seen [7].
A magnetic resonance imaging (MRI), the tumor has heterogeneous high signal intensity and for a signal sequence and T2 hypo-or is intense in the T1 sequence [8]. The diagnosis is confirmed by histological examination complemented by immunohistochemical study. At the macroscopic, the tumor is often lobulated, with graywhite color including areas of necrosis or hemorrhagic suffusions [9]. Histologically, the appearance is characteristic: it is a tumor composed of massive proliferation of cells that are undifferentiated, uniformed, and small, with scanty cytoplasm and hyperchromatic nucleus with a high nucleo-cytoplasmic ratio and a high mitotic index [10]. In half the cases there are rhabdoid eosinophilic intracytoplasmic inclusions. The stroma is typically desmoplastic, hypocellular, consisting of fibroblasts or myofibroblasts. Central necrosis or cystic degeneration may be observed. The immunohistochemical profile of this tumor is characteristic. It allows the differential diagnosis with other types of small round cell tumors, it combines the simultaneous expression of epithelial antigens (cytokeratin: 87A 91%), muscle (desmin: 81% to 91%), mesenchymal (vimentin: 97%) and neuroendocrine (NSE, CD57, synaptophysin +/−, chromogranin +/−) [4,11]. Myogenin and myo D1 are negative. The nuclear expression of WT1 (Wilm’s tumor) is almost always observed. This immunohistochemical profile can distinguish the SRCDT among all the small round cell tumors [9]. The demonstration of a nonrandom translocation t (11; 22) (p13; q12) that is particular to the disease [12]. This cytogenetic abnormality was clarified at the molecular level by the technique of reserve transcriptase polymerase chain reaction (RT-PCR), it reflects the fusion of the N-terminal domain of the gene EWS (Ewing sarcoma gene) located on chromosome 22 with the field C-terminal gene WT1 (Wilm’s tumor) located on chromosome 11. Freezing of a surgical specimen is essential to enable this study [6].
The therapeutic management is multidisciplinary. It combines: A wide surgical excision of the initial tumor mass to improve the progression-free survival. This was demonstrated by Schwartz, who found a complete response or judged as very good in 13 of 15 achieved for which excision of at least 90% of the tumor was possible combined with chemotherapy and radiotherapy [13]. An preor post-operatively that of aggressive chemotherapy. Indeed, two protocols have been proposed: the first is Kushner at Memorial Sloan Kettering Cancer Center who used cyclophosphamide, doxorubicin, vincristine, etoposide and ifosfamide (P6 protocol) at high doses. These treatments were followed by local radiotherapy or new treatments of chemotherapy-based on myeloablative dose thiotepa with stem cells preservation. Concerning of a series of 12 patients, 7 were alive and disease not affected with a decline ranging from 21 to 60 months, the other 5 patients had disease progression within 13 to 26 months [14]. The second protocol is the FARHAT at the Gustave Roussy Institute, which reported a series of 5 cases of SRCDT treated with a combination of cisplatin, etoposide, cyclophosphamide and doxorubicin or epirubicin. Stabilization of the tumor was observed for a period of 4 to 9 months in patients with intraperitoneal location, but all died because of their disease [15]. A complete response was observed in the patient with a tumor initially localized at the para testicular region. This form of TDPCR would be a better prognosis due to earlier treatment [15].
The place of radiotherapy is not defined for these rare tumors. A total dose of 30 Gy in the abdomen or the pelvis was proposed for palliative purposes [16]. Its prognosis remains poor with a poor overall survival; hence the need for prospective studies of adequate therapeutic assessment currently based mainly on surgery, chemotherapy and targeted therapies.
4. CONCLUSION
The SRCDT are rare tumors whose diagnosis is based on pathologic, immunohistochemical data and cytogenetic study by the detection by RT-PCR of the fusion transcript EWS-WT1. The management is multidisciplinary involving extensive surgery, aggressive chemotherapy and possibly radiotherapy seems to improve survival even though the prognosis is poor. Prospects based on targeted therapies remain the hope of management.
5. AUTHORS’ CONTRIBUTIONS
HS; HF and NM were the principal authors and major contributors in writing the manuscript. SE and CB analyzed and interpreted the patient data and reviewed the literature. AA and AB read and corrected the manuscript.
All authors read and approved the final manuscript.
REFERENCES
- Gerald, W.L. and Rosai, J. (1989) Desmoplastic small cell tumor with divergent differentiation. Pediatric Pathology, 9, 177-183. doi:10.3109/15513818909022347
- Battisti, S., Renaudin, K., Rigaud, J., Hetet, J.F., Braud, G., Kapfer, J., et al. (2005) Tumeurs desmoplastiques à petites cellules rondes paratesticulaires. Progres En Urologie, 15, 333-336.
- Chang, F. (2006) Desmoplastic small round cell tumors. Cytologic, histologic and immunihistochemical features. Archives of Pathology & Laboratory Medicine, 130, 728- 732
- Rais, H., El Mansouri, F., Belaabidia, B., Essadki, O., Oussehal, A. and Saif, I. (2010) Tumeur desmoplastique à petites cellules rondes paratesticulaires: A propos d’une observation avec revue de la littérature. Cancer/Radiothérapie, 14, 111-114.
- Gerald W.L., Miller, H.K., Battifora, H., Miettiner, M., Silva, E.G. and Rosai, J. (1999) Intra abdominal desmoplastic Small round cell tumor. Report of 19 cases of distinctive type of high-grade polyphenotypic malignancy affecting young individuals. The American Journal of Surgical Pathology, 15, 499-513.
- Roques, B., Pichon, A., De Laroche, G., Garnier, J.F. and Jacquin, J.P. (2000) Tumeurs desmoplastiques intra abdominales à petites cellules rondes: Une entité parmi les carcinoses péritonéales de l’adulte jeune. Gastroentérologie Clinique et Biologique, 24, 359-360.
- Jellouli, M., Mongi, M., Krichene, I. M., Zakhama, A., Belghith, M., Jouini, R., Sahnoun, L., Rammeh, S., Harbi, A. and Nouri, A. (2003) Tumeur desmoplastique à petite cellules rondes intra abdominal de l’enfant: A propos d’une observation avec revue de la littérature. Bull Cancer, 90, 1063-1066.
- Chouli, M., Viala, J., Dromain, C., Fizazi, K., Duvillard, P. and Vanel, D. (2005) Intra-abdominal desmoplastic small round cell tumors: CT findings and clinicopathological correlations in 13 cases. European Journal of Radiology, 5, 438-442. doi:10.1016/j.ejrad.2004.09.002
- Kallel, R., Ayadi, L., Masmoudi, A., Makni, S., Beyrouti, I., Frikha, M. and Boudawara, T. (2008) Tumeur intraabdominale inhabituelle. Revue Medicale de Bruxelles, 29, 180-183.
- Eaton, S.H. and Cendron, M.A. (2006) Primary desmoplastic small round cell tumor of the kidney in a 7-yearold girl. Journal of Pediatric Urology, 2, 52-54. doi:10.1016/j.jpurol.2005.05.008
- Maliki, M., Mahassini, N., Zouaidia, F., Bernoussi, Z., Mansouri, F. and El Hachimi, A. (2004) Tumeur desmoplastique à petites cellules rondes: A case report. Revue Médicale de Liège, 59, 451-454.
- Afif, H., Benouhoud, N., Aichane, A., Trombati, N. and Bouayad, Z. (2006) Métastases pulmonaires d’une tumeur desmoplastique à petites cellules rondes. Revue de Pneumologie Clinique, 62, 53-55. doi:10.1016/S0761-8417(06)75415-3
- Schwartz, R.E., Gerald, W.L., Kushner, B.H., Coit, D.G., Brennan, M.F. and Laquaglia M.P. (1998) Desmoplastic small round cell tumors: Pronostic indicators and results of surgical management. Annals of Surgical Oncology, 5, 416-422. doi:10.1007/BF02303860
- Kushner, B.H., Laquaglia, M.P. and Wollner, N. (1996) Desmoplastic small round cell tumor: Prolonged progression-free survival with agressive multimodality therapy. Journal of Clinical Oncology, 14, 1526-1531.
- Farha, T.F., Culine, S., Lhomme, C., Duvillard, P., Terrierlacombe, M.J., Michel, G., Soulie, P., Theodore, C., Lotz, J.P. and Droz, J.P. (1996) Desmoplastic small round cell tumor. Result of a four-drug chemotherapy regimen in five adult patients. Cancer, 77, 1363-1366. doi:10.1002/(SICI)1097-0142(19960401)77:7<1363::AID-CNCR21>3.0.CO;2-Z
- Goodman, K.A., Wolden, S.L., La Quaglia, M.P. and Kushner, B.H. (2002) Whole abdominopelvic radiotherapy of desmoplastic small round cell tumor. International Journal of Radiation, Oncology, Biology Physics, 54, 170-176. doi:10.1016/S0360-3016(02)02871-7
NOTES
*The authors declare that they have no competing interests.
#Corresponding author.