Open Journal of Immunology
Vol.2 No.4(2012), Article ID:26251,12 pages DOI:10.4236/oji.2012.24019
Intravenous immunoglobulin suppresses IL-10 production by activated B cells in vitro
![]()
1Department of Experimental Immunology, Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan; *Corresponding Author: tostakai@idac.tohoku.ac.jp
2Osaka Research Laboratory, Benesis Corporation, Osaka, Japan
3Center for General Education, Division of Human Sciences, Tohoku Institute of Technology, Sendai, Japan
4Department of Genetics, University of Erlangen, Erlangen, Germany
Received 21 August 2012; revised 23 September 2012; accepted 3 October 2012
Keywords: F(ab’)2 Fragment; Immune Inhibition; ITIM Receptors; Polyclonal IgG; SHP-1
ABSTRACT
A therapeutic preparation of polyclonal human IgG, i.e., intravenous immunoglobulin (IVIg), has been employed to treat several inflammatory and autoimmune disorders. B cells are supposed to be a target of IVIg, but the molecular mechanism is elusive because of the lack of a suitable experimental system. To gain an insight into the beneficial effect of IVIg on B cells, we first established an experimental setting in which IVIg modulates a murine B cell function in vitro, and then aimed at identifying the mechanistic features at the molecular level. Here we show that IVIg down-regulates IL-10 production by CpGactivated B cells in vitro. The responsible component of IVIg was identified as the F(ab’)2 portion, whose polyclonality is mandatory for the suppressive effect. IVIg, bound to the surface of activated B cells, was found to be co-localized with intracellular SHP-1 on confocal laser microscopy, suggesting that B cell-surface immunoreceptor tyrosine-based inhibitory motif-harboring receptors that recruit SHP-1 are target molecules for IVIg in our experimental setting. Overall, we postulate a scenario in which IVIg attenuates B cells by suppressing IL-10 production, a B cell growth factor, and thus down-regulates the production of pathogenic antibodies.
1. INTRODUCTION
Intravenous administration of a large quantity of polyclonal IgG antibodies purified from sera collected from a large number of healthy donors (IVIg) has been performed primarily for patients with antibody deficiencies, but nowadays is also being utilized for the treatment of several inflammatory and autoimmune disorders, such as Kawasaki disease and idiotypic immune thrombocytopenic purpura, respectively [1,2]. The underlying mechanisms of the actions of IVIg in such diseases have been proposed to involve blocking or neutralizing effects of Fc receptors, complements, pathogenic antibodies and cytokines, the former two effects being attributed to the Fc portion of IgG, and the latter two to Fab or F(ab’)2 [1,3]. The IVIg mechanisms have also been proposed to involve the initial recognition of sialylated IgG by a carbohydrate receptor on sensor macrophages indirectly leading to up-regulation of an inhibitory IgG Fc receptor, Fc RIIB, on effector macrophages [2]. Considering the pluripotential nature of IgG and the related molecules with which IgG molecules interact, it is reasonable to speculate that the IVIg mechanisms involve multiple scenarios for alleviation of diseases. It is mandatory, however, to precisely elucidate a molecular mechanism for improving IVIg administration or delineating more effective therapeutic alternatives [4].
While IVIg can directly neutralize pathogenic antibodies including autoantibodies via its anti-idiotypic activity in the preparation [5], the idea that activated B cells might also be a direct target for IVIg is attractive and has been examined rigorously [1,6]. In support of this notion, autoimmune patients such as one with systemic lupus erythematosus administered IVIg often show rapid and even sustained reduction of anti-nuclear autoantibodies in their serum [7,8]. While the cellular origin of pathogenic autoantibodies has long been debated, both innate B (B1) cells and conventional B (B2) cells contribute to their production in a murine system [9]. B1 cells are the main producer of IgM natural antibodies, which are poly-specific and even reactive with autologous molecules. It has not been established yet whether or not this is also the case for humans, because a human B1 population has only started to be defined recently [10]. On the other hand, B2 cells in mice and humans are believed to produce high-affinity IgG autoantibodies via activation and class switching with T cell help.
It is of note that B1 and B2 cells harbor intrinsic regulators, such as Fc RIIB [11,12], CD22 [13], and PirB [14,15], which suppress activation, proliferation, and excess production of antibodies. Thus, it is interesting to hypothesize that IVIg could bind to and trigger these inhibitory receptors on B1 and B2 cells through its intrinsic poly-reactivity, which might be related to its therapeutic effects including reduction of serum autoantibody levels. To test this hypothesis, we initially attempted to construct an experimental system in which IVIg can modulate any murine B cell function, and then tried to clarify whether or not the modulatory effect is related to inhibitory receptors, which will be described in detail in this paper.
2. MATERIALS AND METHODS
2.1. Mice
C57BL/6 (B6) mice were purchased from Charles River (Tokyo, Japan) and CLEA Japan (Tokyo, Japan). BXSB-Yaamice were purchased from SLC (Shizuoka, Japan). Pirb mice [16] and Fcgr2b mice [12] were backcrossed to B6 mice for 12 and 22 generations, respectively. Cd22 mice were generated by Lars Nitschke [17]. Mice were maintained and bred in the Animal Facility of The Institute of Development, Aging and Cancer, Tohoku University, an environmentally controlled and specific pathogen-free facility, according to guidelines for experimental animals defined by the University, and animal protocols were reviewed and approved by the Animal Studies Committee of the University. All experiments were performed on 7- to 10-week-old male and female mice.
2.2. Preparation of a 200 mg/ml IVIg Solution
The IVIg used in this study was Venoglobulin IH 5% I.V. (VGIH) (Benesis Co., Osaka, Japan), which was prepared from healthy volunteers. For in vitro experiments, 50 mg/ml VGIH was dialyzed against PBS prior to experiments, becauseVGIH is formulated as an acidic (pH 3.9 - 4.4) solution. The dialyzed IgG was concentrated to 200 mg/ml by ultrafiltration and used as IVIg.
2.3. Fragmentation of Human IgG
For preparation of F(ab’)2, 50 mg/ml VGIH was dialyzed against 100 mM acetate buffer, pH 4.5. Pepsin (Calbiochem, Darmstadt, Germany) was added to the solution (about 45 mg/ml IgG) in the ratio of 2 mg pepsin to 100 mg IgG, followed by incubation at 37˚C for 24 h [18]. Then the solution was dialyzed against PBS, concentrated, and purified by gel filtration using HiLoad 26/60 Superdex 200 pg (GE Healthcare, Buckinghamshire, England). The collected F(ab’)2 fractions were pooled and further purified withHiTrap Protein G (GE Healthcare) to remove residual undigested IgG or Fc fragments. The flow-through fraction was collected and concentrated to a 133.3 mg/ml F(ab’)2 solution. For preparation of Fab and Fc fragments, 50 mg/ml VGIH was first dialyzed against 5 mM phosphate buffer, pH 8.0. Papain (MP Biomedicals, Ohio, USA) digestion was carried out by the modified method of Snigurowicz et al. [19]. The dialyzed IgG solution in 5 mM phosphate buffer, pH 8.0, containing 0.1 mg/ml papain, 2mM EDTA, and 10 mM L-cysteine was incubated at 37˚C for 24 h. Then the solution was dialyzed against PBS and concentrated by ultrafiltration. To separate the Fab and Fc mixture from undigested IgG or smaller proteins, gel filtration was carried out on HiLoad 26/60 Superdex 200 pg (GE Healthcare). The fractions containing Fab and Fc were collected and dialyzed against a 1/50 PBS solution for a few days. Stillin 1/50 PBS, Fc fragments were crystallized according to Virella’s method [20]. The crystals were separated by centrifugation, washed with 1/50 PBS a few times to remove residual Fab, and then dissolved in 0.15 M NaCl. The Fc solution was adjusted to 66.7 mg/ml. The supernatant of the dialyzed solution was further purified with HiTrap Protein G (GE Healthcare) to remove residual Fc. The flow-through fraction was collected and the Fab concentration was adjusted to 66.7 mg/ml.
2.4. Cell Separation
For preparation of peritonealB1 cells and macrophages, peritoneal cells were harvested by washing the peritoneal cavity with ice-cold PBS. Then the cells were treated with unconjugated anti-Fc RIIB/III mAb (2.4G2; BD Biosciences, California, USA) to block Fc receptors, followed by staining with APC-conjugated anti-B220 (RA3-6B2;BioLegend, California, USA) and FITC-conjugated anti-CD11b (M1/70; BioLegend). B220 CD11b cells were sorted as peritoneal B1 cells and B220 CD11b cells as peritoneal macrophages with a FACS Aria III (BD Biosciences). For preparation of splenic B2 cells, spleen cells were treated with a mouse B cell isolation kit (#130-090-862; Miltenyi Biotec, Gladbach, Germany). The negative fraction (CD4 CD43 Tre-119) was sorted as splenic B2 cells with a AutoMACS Pro (Miltenyi Biotec). For separation of splenic T cells, spleen cells were stained with FITC-conjugated anti-CD5 (53-7.3; BioLegend) and PE-conjugated anti-CD3 (145-2C11; BD Biosciences) CD5 CD3 cells were sorted as splenic T cells with a FACS Aria III. For some experiments, B1 cells were isolated with a Auto MACS Pro. Briefly, peritoneal cells were stained with biotinylated anti-CD3 (145-2C11; BD Biosciences), anti-CD23 (B3B4; eBioscience), antiGr-1 (RB6-8C5; BioLegend), and anti-F4/80 (BM8; Serotec, Oxford, United Kingdom), followed by staining with anti-biotin microbeads (#130-090-485, MilteniBiotec). Then CD3 CD23 Gr-1 F4/80 cells were harvested by using an autoMACS Pro, and further stained with antiCD5 microbeads (MiltenyiBiotec). CD5 cells were purified with an auto MACS Pro and used as B1 cells. The purities of the sorted cells were consistently >95%.
2.5. Measurement of Antibodies and Cytokines
5 × 103 cells/100 l/well of peritoneal B1 cells and 1 × 105 cells/100 l/well of splenic B2 cells in the culture medium (RPMI1640 containing 10% FCS, 50 M 2-mercaptoethanol, 100 units/ml penicillin and 0.1 mg/ml streptomycin) were cultured in a 96-well plate and stimulated with 200 nMphosphothioate-CpG-B oligodeoxynucleotide (CpG) (ODN1826, 5’-TCCATGACGTTCCTGACGTT-3’, Invivogen, California, USA) or control phosphothioate-GpColigodeoxynucelotide (GpC) (ODN 1826 Control, 5’-TCCATGAGCTTCCTGAGCTT3’, Invivogen) with or without 10 mg/ml IVIg or BSA (#A 1470; Sigma-Aldrich, Missouri, USA). Forty-eight hours after the stimulation, supernatants were collected, and the amounts of IgM, IL-6 and IL-10 were determined with a mouse IgM ELISA quantification kit (BETHYL Laboratories Inc. Texas, USA), Mouse IL-6 ELISA MAX (BioLegend), and Mouse IL-10 ELISA MAX (BioLegend).
2.6. Immunoblot Analysis
Peritoneal B1 cells were stimulated with 200 nM CpG or control oligonucelotide GpC for 30 min at 37˚C with or without 10 mg/ml IVIg. Then the cells were lysed with a lysis buffer (1% NP-40, 20 mMTris, pH 7.3, 150 mM NaCl, 10 mM EDTA and 10% glycerol) supplemented with a protease inhibitors cocktail (#P8340; Sigma-Aldrich) and phosphatase inhibitor PhosSTOP (#4906845; Roche, Mannheim, Germany). The cell lysates were separated by SDS-PAGE and transferred to a PVDF membrane, and then treated with either antibodies anti-TAK1 (#4505; Cell Signaling Technology, Beverly, USA), anti-p-TAK1 (#9339; Cell Signaling Technology), anti-IRAK-1 (D51G7; Cell Signaling Technology), antiactin (AC-15; Sigma-Aldrich), anti-NF-B p65 (#3034; Cell Signaling Technology), anti-p-NF-B p65 (#3031; Cell Signaling Technology), anti-p38 MAPK (#9212; Cell Signaling Technology), anti-p-p38 MAPK (#9211; Cell Signaling Technology), anti-ERK1/2 (137F5; Cell Signaling Technology), or anti-p-ERK1/2 (D13.14.4E; Cell Signaling Technology). Anti-mouse IgG (#7076; Cell Signaling Technology) or anti-rabbit IgG (#7074; Cell Signaling Technology) was used as secondary antibodies. An ECL Western Blotting Detection System (GE Healthcare) was used for detection followed by development on X-ray film RX-U (Fujifilm Co., Tokyo, Japan). The signal intensity of each protein was estimated by densitometric scanning (Dolphin View Band Tool; KURABO, Osaka, Japan). The intensity of a phosphorylated protein was divided by the intensity of corresponding total protein for normalization and for determination of the relative phosphorylation as to unstimulated samples. To compare IRAK-1 degradation, the intensity of an IRAK-1 protein was divided by the intensity of -actin in a corresponding sample.
2.7. Viability Assay
5 × 103 cells/100 l/well of peritoneal B1 cells or 1 × 105 cells/100 l/well of splenic B2 cellsin the culture medium were cultured in a 96-well plate and then stimulated with 200 nMCpG (ODN1826, Nihon Gene Research Laboratories Inc., Sendai, Japan) or control GpC with or without 10 mg/ml IVIg or BSA. Forty-eight hours after the stimulation, the cells were collected, pooled, and then stained with FITC-conjugated Annexin V (#640906; BioLegend) and propidium iodide (PI) (#P4864; Sigma-Aldrich). Cell surfaces were stained by standard techniques, and flow cytometry was performed with a FACS Calibur (BD Biosciences). Annexin VPI cells were defined as live cells.
2.8. CpG Internalization Assay
5 × 103 cells/100 l/well of peritoneal B1 cells in the culture medium were cultured in a 96-well plate and stimulated with 200 nM Cy5-conjugated CpG (ODN1826, Nihon Gene Research Laboratories Inc., Sendai, Japan) with or without 10 mg/ml IVIg. Forty-eight hours after the stimulation, Cy5 fluorescence was measured with a FACS Calibur.
2.9. Confocal Laser-Scanning Microscopic Analysis
For cell-surface staining, MACS-sorted splenic B2 cells, and FACS Aria III-sorted B1, T cells and macrophages from B6 mice were treated with IVIg, anti-tetanus toxin human IgG1 (SA13), or BSA for 30 min at 4˚C. The cells were fixed with 4% paraformaldehyde for 15 min at room temperature (RT), treated with primary antibodies in PBS containing 1% BSA for 60 min at RT, permeabilized with PBS containing 0.2% Triton X-100 for 5 min at RT, and then stained with the second antibodies in PBS containing 1% BSA for 60 min at RT.
For detection of incorporated IgG or its fragments, MACS-sorted peritoneal B1 cells from B6 mice were stimulated with CpG in the presence of IVIg, F(ab’)2 or Fab for 30 min or 24 h at 37˚C. The cells were fixed and permeabilized as above, and treated with primary antibodies in PBS containing 1% BSA for 60 minutes at RT.
For detection of SHP-1 and SHIP, splenic B2 cells were incubated with Alexa Fluor488-conjugated human IgG (10 mg/ml) instead of IVIg for 30 min at 4˚C, and treated with CpG and/or anti-human IgG for 10 min at 37˚C. The cells were fixed with 4% paraformaldehyde for 15 min at RT, treated with the primary antibodies in PBS containing 1% BSA for 60 min at RT, permeabilized with PBS containing 0.2% Triton X-100 for 10 min at RT, and then stained with the secondary antibodies in PBS containing 1% BSA for 60 min at RT. Confocal laserscanning microscopic analysis was performed with Alexa Fluor488-conjugated human IgG instead of IVIg, SYTOX-Orange (#S11368; Life Technologies) for nuclei, Alexa546-conjugated anti-SHP-1 (C-19; SantaCruz, California, USA), Alexa546-conjugated anti-SHIP (Santa Cruz), or DyLight649-conjugated anti-IgMmAbs (#115- 496-075; Jackson Immuno Research, Pennsylvania, USA). Both anti-SHP-1 and anti-SHIP antibodies were labeled with Alexa488 or 546 using an Alexa Fluor® Monoclonal Antibody Labeling Kit (Life Technologies), according to the manufacturer’s protocol. Fluorescence signals were observed under a confocal laser-scanning microscope (Fluoview FV1000; Olympus, Tokyo, Japan).
For detection of colocalization of IVIg and early endosome antigen-1 (EEA-1), MACS-sorted peritoneal B1 cells from B6 mice were stimulated with CpG in the presence of IVIg, F(ab’)2 or Fab for 30 min at 37˚C. The cells were fixed, permeabilized as above, and then treated with primary and secondary antibodies in PBS containing 1% BSA for 60 min at RT. IVIg or its fragments was detected with anti-human IgG (#A21445; Life Technologies, Maryland, USA). EEA-1 was visualized with anti-EEA-1 antibodies (#2411; Cell Signaling Technology) and Fluor488-conjugated anti-rabbit IgG (#A1- 1070; Life Technologies). Fluorescence signals were observed under a confocal laser-scanning microscope (Fluoview FV1000, Olympus).
The correlation of the distributions of two fluorescence signals was evaluated by linearization analysis, the location of the molecules observed in one plane of the cell versus the whole cell being determined. Scans of adjacent to the cell-surface were obtained by drawing a ring-shaped area on the mid-focal plane of each cell. The fluorescence intensities of the two target molecules were normalized as to the highest peak (y axis).
2.10. Surface Plasmon Resonance (SPR) Analysis of Anti-Human IgG Binding to IVIg, F(ab’)2 and Fab
SPR analysis was performed with a BIAcore 2000 (BIAcore, Uppsala, Sweden). In brief, ligand proteins [IVIg, F(ab’)2 and Fab] were covalently immobilizedat 1500 to 2500 response units on research-grade CM5 chips (BIAcore) by the amine-coupling method. After buffer exchange to HBS-EP buffer comprising 10 mM HEPES, pH 7.4, 150 mM NaCl, 3.4 mM EDTA, and 0.005% Surfactant P20 (BIAcore), analyte proteins were injected over the immobilizedflow cells at 25˚C. The binding interactions wereassayed at 20 l/min. The binding responses were subtracted from the nonspecific responses to an empty flow cell. Kinetic constants were calculated from sensor gram data with the BIA evaluation program (version 3.0.2; BIAcore).
2.11. Statistical Analysis
Statistical analyses were performed using Student’s t-test. P < 0.05 was considered as statistically significant.
3. RESULTS
3.1. IVIg Suppresses IL-10 Production by B1 and B2 Cells Stimulated via TLR9
To test our hypothesis that a direct target of IVIg could be B cell-surface inhibitory receptors, we first attempted to construct an in vitro experimental system in which IVIg modulates any B cell-mediated function. Since the B cell population in a murine system can be subdivided into B1 and B2 cells, we isolated each population from peritoneal cavities or spleens, respectively, of C57BL/6 mice, and compared their responsiveness to CpG, a tolllike receptor (TLR) 9 ligand, in the presence or absence of 10 mg/ml IVIg or BSA as a control in vitro. As shown in Figures 1(a)-(d), inclusion of such a high concentration of exogenous BSA in the culture medium caused a slight, presumably non-specific, suppression of IgM, IL- 6, and IL-10 production by B1 or B2 cells or both. We did not observe a significant IVIg-specific reduction in IgM and IL-6 production or in cell viability, compared to those parameters of BSA as a control in both B1 and B2 cells (Figures 1(a), (b) and (d)). We found, however, that IL-10 production by both CpG-activated B1 and B2 cells was significantly reduced in the presence of IVIg (Figure 1(c)). This suppression was dose-dependent as to IVIg (see Figure 3(a)). We thus concluded that IVIg suppresses IL-10 production by CpG-activated B1 and B2 cells in vitro. This finding is further examined as to the mechanistic details as well as the implications hereafter.
3.2. IVIg Binds to B1 and B2 Cell Surfaces
Before examining possible IVIg binding to B cell in
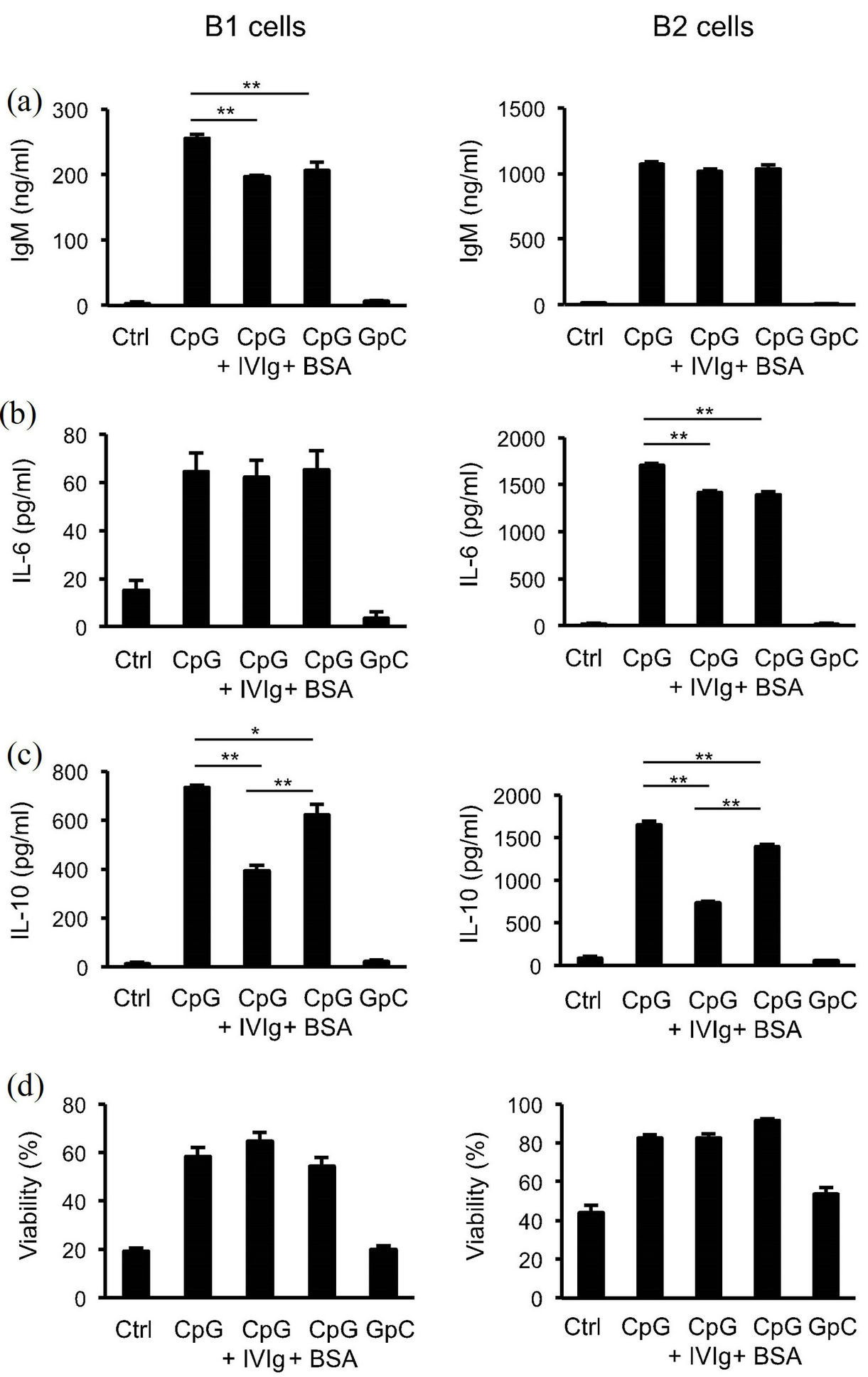
Figure 1. Modulation of cytokine and antibody production by peritoneal B1 cells andsplenic B2 cells by IVIg. Peritoneal B1 cells and splenic B2 cells were stimulated with 200 nM CpG or control GpC for 48 h with or without 10 mg/ml IVIg or BSA, and then the production of IgM (a), IL-6 (b), and IL-10 (c) was measured by ELISA, and cell viability (d) byflow cytometric analysis. Data are shown as means for 4 to 8 samples ± SEM. The results are representative of more than three independent experiments. **P < 0.01; *P < 0.05.
hibitory receptors, we wanted to see the kinetics of IVIgbinding to B cells on a confocal laser microscopy. To this end, freshly isolated B1 and B2 cells, and T cells and macrophages for comparison, were incubated for 60 min at 4˚C in the presence of 10 mg/ml IVIg or BSA as a control, and the bound human IgG was detected with fluorochrome-labeled anti-human IgG (H L). Figure 2(a) shows the binding of a substantial amount of IVIg on the surface of the B1 and B2 cells as well as T cells and macrophages. The signal intensities of the bound IgG did not differ significantly among the cells tested (Figure 2(b)). Nonspecific binding of FITC-labeled anti-human IgG was negligible, as BSA inclusion instead of IVIg did not yield blight fluorescence (Figures 2(a) and (b)). Interestingly, when we employed anti-tetanus monoclonal human IgG instead of IVIg, we did not observe its binding to B cells (Figure 2(c)). These results indicate that IVIg, namely a human polyclonal IgG preparation, binds to any structure(s) on murine B cells as well as T cells and macrophages, and the binding was suggested to be dependent on its polyclonality.
3.3. F(ab’)2 Fragment of IVIg Recapitulates B Cell Suppression
Next we aimed at identifying which portion of IgG has the binding and suppressive effects on IL-10 production by CpG-activated B cells in vitro. Peritoneal B1 cells in culture were stimulated with CpG at 37˚C in the presence of various doses of whole IVIg or the digested fragments, namely F(ab’)2, Fab, or Fc, and then the production of IL-10 was assessed after 48 h (Figure3(a)). The suppressive effect was dose-dependent, 10 mg/ml of whole IVIg being found to be most effective. We verified that incorporation of fluorochrome-labeled CpG into B cells was not inhibited by the presence of such a high dose of IgG (Figure 3(b)). We found that F(ab’)2 at 6.6 mg/ml, an equimolar concentration to that of whole IVIg, caused similar suppression, but an equimolar Fab or Fc at 3.3 mg/ml failed to do so (Figure 3(a)). These results indicate that the F(ab’)2 fragment of IVIg is the effective portion of IgG responsible for the suppression of CpGinduced B cell activation. It is of note that the suppression was almost completely abolished when F(ab’)2 was further split into monovalent Fab, suggesting that the bivalent nature of F(ab’)2 is somehow important for the suppression.
We then examined the fate of whole IVIg and its fragments in peritoneal B1 cells incubated at 37˚C. As shown in Figures 3(c) and (d), the signal of whole IVIg detected with fluorochrome-labeled anti-human IgG (H L) was tightly associated with B cells after 30 min and even 24 h later. We found this was also the case for F(ab’)2. In the case of Fab, however, the signal was faint (Figures 3(c) and (d)). At least a part of the IVIg, or F(ab’)2 signal was co-localized with EEA-1, an early endosomal marker, suggesting that a substantial portion of the IVIg or F(ab’)2 was incorporated into the endosomal fraction (Figure 3(e)). To exclude the possibility that these differences in signal intensities could be due to attenuated binding of anti-human IgG to the Fab fragment, we verified that the anti-human IgG (H L) was able to bind to the Fab fragment of IVIg with an affinity roughly comparable to that in the case of F(ab’)2, although it was lower than that for intact IVIg (Figure 3(f)). Based on these results, we concluded that IVIg and its F(ab’)2 portion bind to the B cell surface and then are internalized into the cytosol upon incubation at 37˚C.
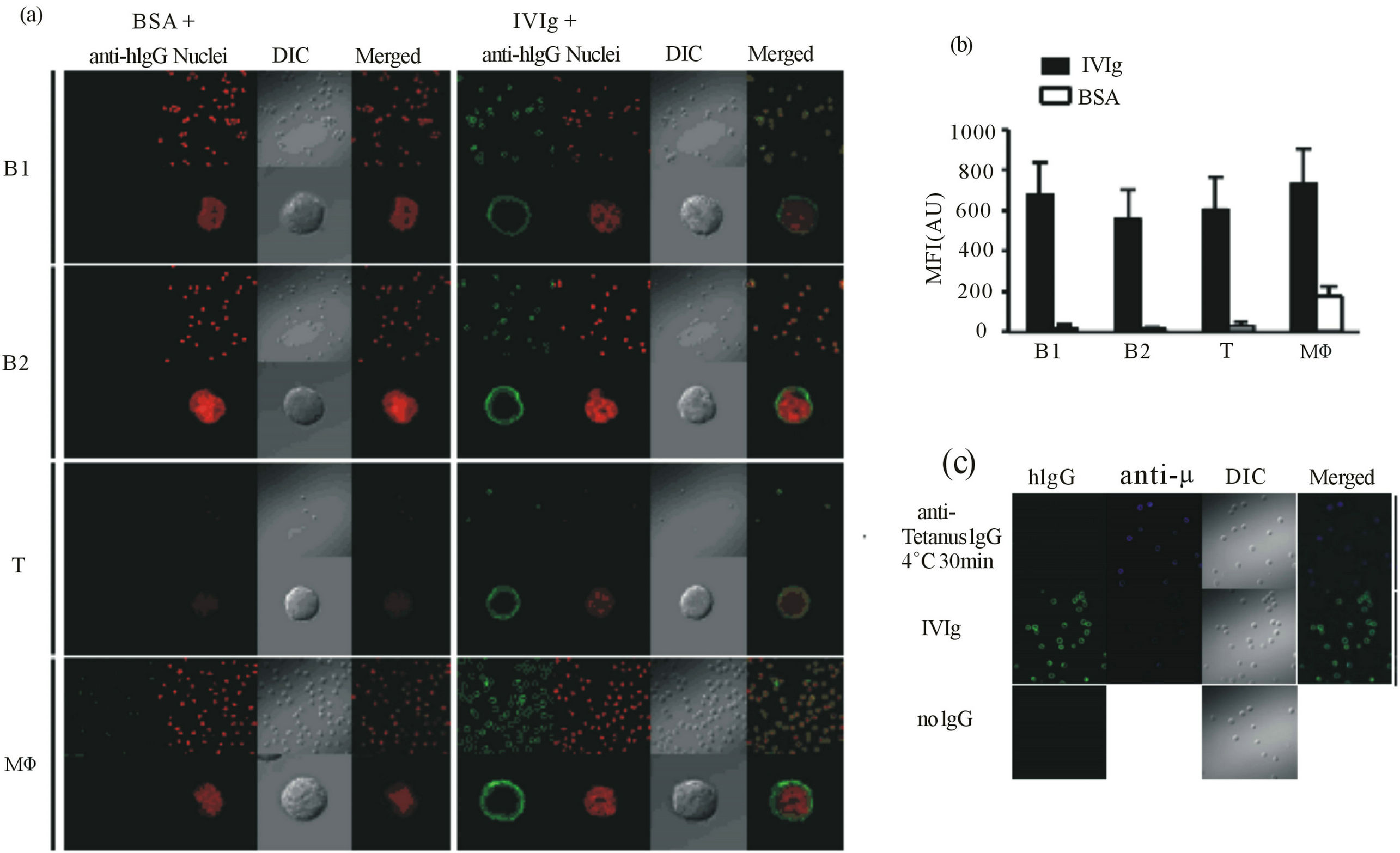
Figure 2. Binding of IVIg to the B cell surface. (a) and (b) Peritoneal B1 cells, splenic B2 and T cells, and peritoneal macrophages were treated with IVIg or BSA for 30 min at 4˚C, fixed, and then stained with Alexa488-conjugated anti-human IgG antibodies and SYTOX-Orange. Acquisition of images at low (upper row) or high (lower row) magnification (a), and measurement of fluorescence intensities (b) were performed under a Fluoview FV1000 confocal laser scanning microscope. In (b), each point represents the mean ± SD for 41 to 62 images (IVIg) or 8 to 30 images (BSA). Green, IVIg; red, nucleus; DIC, differential interference contrast; Merged, merged image of IVIg and nucleus. (c) Peritoneal B1 cells were treated with IVIg, anti-tetanus toxin monoclonal human IgG or no antibody for 30 min at 4˚C, fixed, and then stained with Alexa488-conjugated anti-human IgG andAlexa546-conjugated anti-m antibodies. Green, IVIg; blue, B cell receptor as a marker for cell membrane; DIC, differential interference contrast; Merged: merged image of IVIg and B cell receptor.Acquisition of the images was performed under a Fluoview FV1000 confocal laser scanning microscope. MFI, mean fluorescence intensity.
3.4. TLR9-mediated Activation Signal Is Suppressed Partly by IVIg
Upon binding to CpG, TLR9 evokes several signaling pathways culminating in activation of four key molecules, namely NF-B, ERK, JNK, and p38 MAPK, that are translocated into the nucleus and activate transcription of proinflammatory cytokines. However, little is known about how IVIg is involved in the TLR9-initiated signaling. To determine the mechanistic features of IVIg-mediated suppression of IL-10 production by B cells, we examined TAK1, IRAK-1, the NF-B p65 subunit, p38 MAPK, and ERK by immunoblot analysis 30 min after B1 cell activation with CpG in the presence or absence of IVIg. As shown in Figures 4(a) and (b), the phospho-TAK1, phospho-p65, and phospho-ERK levels were decreased significantly, while IRAK-1 degradation and the phospho-p38 level did not change in the presence of IVIg, suggesting that TAK1, NFB, and ERK were vulnerable to IVIg. Thus, downstream of TLR9-initiated signaling was partly suppressed by IVIg, although we did not identify the key molecule that IVIg targets.
3.5. IVIg Is Co-localized with SHP-1 upon Activation of B Cells
Finally, we wanted to specify the molecular link between the rigorous IVIg binding to membranes shown in Figure 2 and the inhibition of TLR9 signaling in Figure 4. In our hypothesis, we assumed at this point the involvement of inhibitory receptors on B cells. In support of this notion, we recently pointed out that PirB, an immunoreceptor tyrosine-based inhibitory motif (ITIM)- harboring B cell inhibitory receptor that recruits src homology 2 domain-containing tyrosine phosphatase (SHP)- 1, suppresses TLR9 signaling in B1 cells stimulated with CpG [21]. Therefore, we next attempted to clarify whether or not IVIg binding modulates localization of intracellular SHP-1 or src homology 2 domain-containing inositol 5’-phosphatase (SHIP), two versatile inhibitory

Figure 3. Effects of IVIg and its fragments on peritoneal B1 cells.(a) Modulation of IL-10 production by CpG-stimulated B1 cells by IVIg or its fragments. Data are means for quadruplicate samples ± SEM. The results are representative of three independent experiments. Statistical analyses were performed using Student’s t test. **P < 0.01; (b) Flow cytometric detection ofintracellular CpG in peritoneal B1 cells. Peritoneal B1 cells were stimulated with 200 nMCy5-conjugated CpGin the presence or absence of 10 mg/ml IVIg. Forty-eight hours after the stimulation, Cy5 fluorescencewas measured by flow cytometry; (c) and (d) Incorporation (c), (d) and intracellular localization (e) of IVIg or its fragments into peritoneal B1 cells. The cells were stimulated with 200 nMCpG for 30 min or 24 h in the presence of IVIg, F(ab’)2, or Fab, fixed, permeabilized, and then stained with (c), (d) Alexa488-conjugated anti-human IgG and SYTOX-Orange or (E)Alexa647-conjugated anti-human IgG and Alexa488-conjugated anti-EEA-1 antibodies. Acquisition of images (c), (e) and measurement of fluorescent intensities (d) were performed under a confocal laser-scanning microscope. In C, green, IVIg or one of its fragments; red, nucleus; DIC, differential interference contrast; Merged, merged image of IVIg or one of its fragments and nucleus; Merged-3ch, merged image of IVIg or one of its fragments, nucleus, and DIC. In (d), each data point represents the mean ± SEM for 30 images. In E, green, EEA-1; red, IVIg or one of its fragments; DIC, differential interference contrast; Merged, merged image of IVIg or one of its fragments and EEA-1; Merged-3ch, merged image of IVIg or one of its fragments, EEA-1, and DIC. (f) Surface plasmon resonance (SPR) analysis of the binding of anti-human IgG antibodies to IVIg (blue), F(ab’)2 (green), or Fab (red) for verification of the binding ability of the second antibody as to IgG fragments. Representative SPR profiles are shown for IVIg, F(ab’)2, and Fab bound to anti-human IgG, as well as for an unimmobilized flow cell with bound anti-human IgG as a reference flow cell.
phosphatases recruited to ITIM-harboring inhibitory receptors in activated B cells. We isolated splenic B2 cells, incubated them with IVIg for 30 min at 4˚C, stimulated them with CpG only or CpG and anti-human IgG for 10 min at 37˚C to cross-link bound IgG molecules, and fixed them, and then examined the localization of bound IgG and SHP-1 or SHIP with specific fluorochrome-labeled antibodies under a confocal laser microscope. In the absence of CpG stimulation, the bound IgG molecules were distributed evenly on the membranes (Figure 5(a) left, top and third rows). We noted that stimulation of IVIgbound B cells with CpG induced aggregation of the bound IgG molecules into a few discrete patches like the capping of B cell antigen receptors with cognate antigens (Figure 5(a) left, second and bottom rows). Interestingly, the IgG aggregates on the surface were well co-localized with SHP-1 but not SHIP (Figure 5(a) left, second and bottom rows). Forced cross-linking of the bound IgG with anti-human IgG resulted in clearer co-localization of SHP-1 but not SHIP than that in the absence of crosslinking (Figure 5(b) right, second and bottom rows). Therefore, a possible molecular link between IgG binding to B cell surface and TLR9 inhibition was suggested to be a B-cell surface ITIMharboring inhibitory recaptor(s) that recruits SHP-1, in line with our initial hypothesis, in which IVIg targets one or several ITIM-
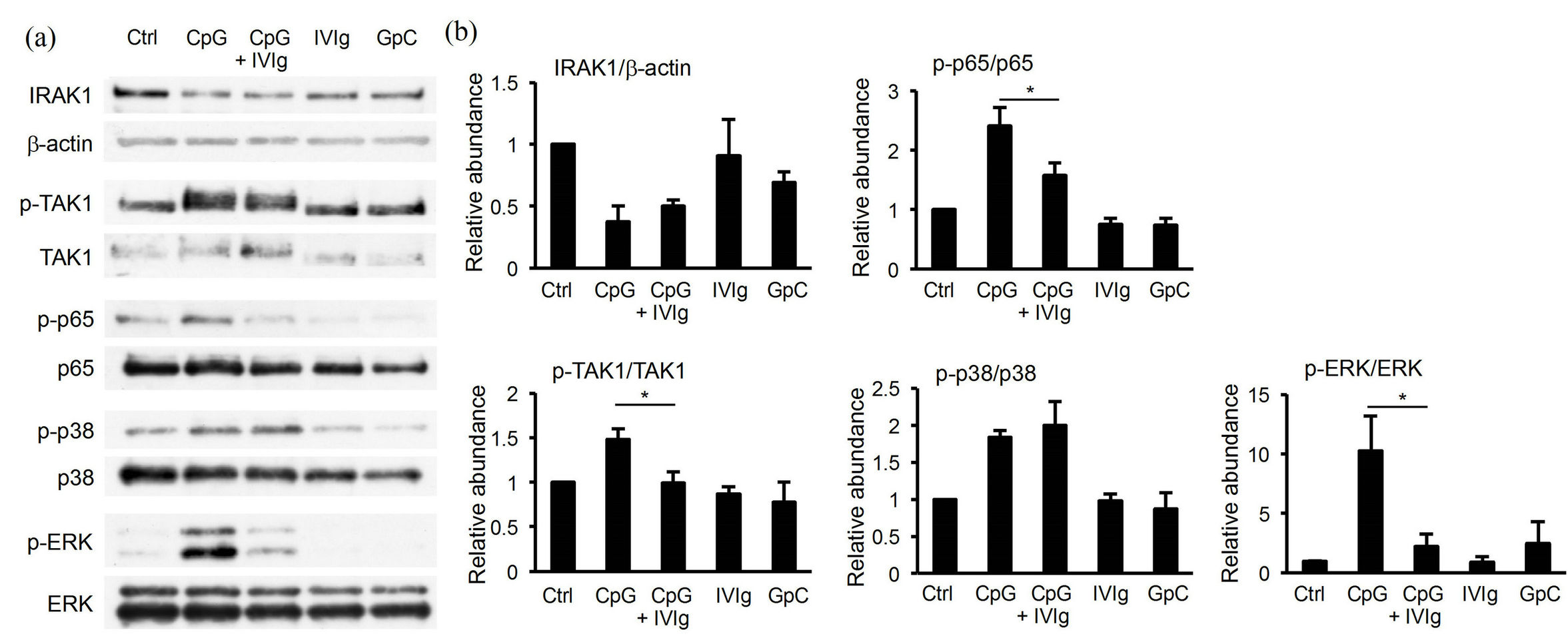
Figure 4. Modulation of TLR9 signaling in peritoneal B1 cells by IVIg. Peritoneal B1 cells were stimulated with 200 nM CpG for 30 min with or without 10 mg/ml IVIg, and then analyzed for TLR9 signaling by Western blotting (a) of IRAK-1 degradation, and phosphorylated TAK1, NF-kB p65, p38 MAPK, and ERK1/2. Each blot is representative of three separate experiments. Each signal intensity was estimated by densitometric scanning (b) and shown as a relative value as to an unstimulated sample. Data shown are the means of three separate experiments ± SEM. *P < 0.05.
harboring membrane-bound inhibitory receptor(s), and induces SHP-1 recruitment to the target receptor(s) upon B cell activation with CpG.
3.6. Neither PirB nor CD22 Solely Mediates Suppression
Our very final goal was identification of the key inhibitory receptor that serves as a major target for IVIg and recruits SHP-1 upon B cell activation. B cells are known to express a series of SHP-1-recruiting inhibitory receptors on their surface. These include CD22 [or sialoglycoprotein-binding lectin (Siglec)-2] [17], CD72 [22, 23], Siglec-G [24], and PirB [16]. On the other hand, a major SHIP-recruiting receptor is Fc RIIB [25]. We isolated peritoneal B1 cells from gene-targeted mice deficient in CD22 or PirB, or in Fc RIIB as a control, and tested them for their IL-10 responses to CpG stimulation in the presence or absence of IVIg. We observed that the suppressive effect of IVIg was not abolished even in the absence of each inhibitory receptor (Figures 6(a) and (b)), or in Fc RIIB deficiency (Figure 6(c)). We concluded that the suppression of CpG-induced IL-10 production by B cells by IVIg is not solely mediated through PirB or CD22.
4. DISCUSSION
4.1. F(ab’)2-Mediated B Cell Suppression
IVIg exhibits idiotype-binding activity and an autoreactive nature toward various autologous molecules in humans, such as Fas, MHC class 1 and 2, Fc receptors, CD5 and CD40 [4,26], presumably because of the fact that IVIg is derived from thousands of healthy volunteers and largely comprises polyclonal, poly-reactive natural antibodies. Interestingly, IVIg preferentially binds to human B cells rather than T cells, because it binds to the B cell antigen receptor (BCR) in addition to various other molecules on the human B cell surface via its antiidiotypic activity [27]. Since the experiments in this study employed a combination of a human IVIg preparation and murine B cells, namely a somewhat artificial setting, we observed comparable IVIg binding to B cells, T cells and macrophages (Figures 2(a) and (b)), suggesting that the binding does not occur solely to BCR. Interestingly, however, this binding was dependent on IVIg’s polyclonal F(ab’)2 (Figure 2(c) and Figures 3(c) and (e)), suggesting that our experimental system partly mimics human IVIg binding to human B cells. Therefore, we consider that the observations in this study can be extrapolated well to those possibly occurring in human B cells.
It has been shown that IVIg can be swiftly internalized into the human B cell cytoplasm via an as yet unknown mechanism [27]. As shown in Figure 3(e) in this study, we observed that IVIg was internalized shortly after its binding to B cells and some of the IgG molecules were found in the early endosomal fraction. We do not know, however, whether the internalization is necessary or dis pensable for IVIg’s inhibitory effect on TLR9 signaling shown in Figure 4, which will be one of the next subjects to be clarified.
4.2. A Possible Role of SHP-1 in IVIg-Mediated Suppression
Our current observation, i.e., that the TLR9 signal and IL-10 production by CpG-activated B cells is inhibited
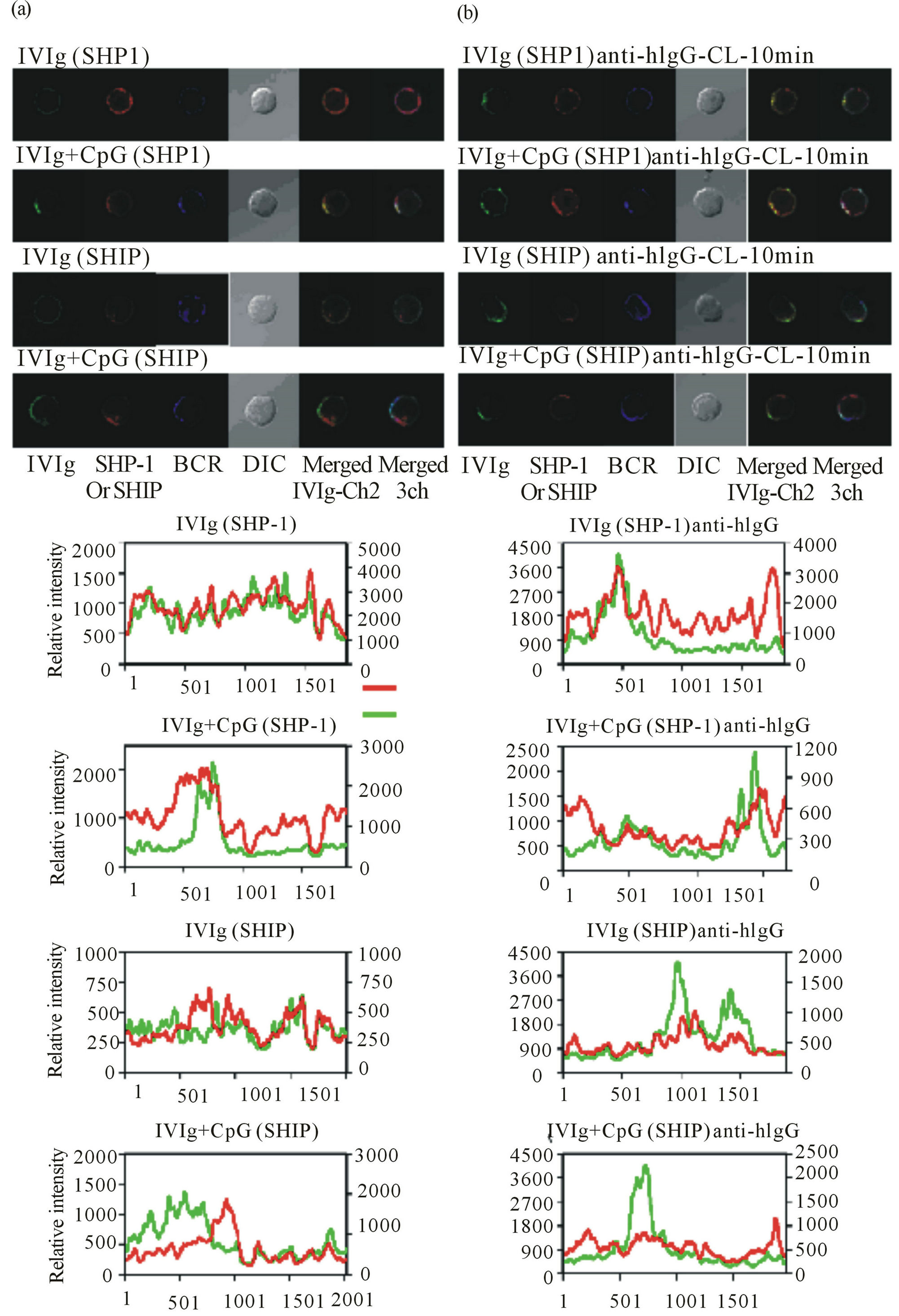
Figure 5. Co-localization of the IVIg and SHP-1 but not SHIP signals in B cells. Splenic B2 cells were treated withIVIg for 30 min at 4˚C, and then treated with CpG alone (a) or CpG and Alexa488-conjugated anti-human IgG as a cross linker (anti-hIgG-CL) of the bound IVIg (b) for 10 min at 37˚C, fixed, stained with DyLight647-conjugated anti-mouse IgMfor 60 min at RT, permeabilized, and then stained again with Alexa546-conjugated anti-SHP-1 or anti-SHIP. Green, IVIg; red, SHP-1 or SHIP; BCR, B cell receptor; DIC, differential interference contrast; Merged IVIg-Ch2, merged image of IVIg and SHP-1 or SHIP; Merged 3ch, merged image of IVIg, BCR, and SHP-1 or SHIP. Lower graphs present data obtained on linear analysis of the fluorescence images shown above. Red, SHP-1 or SHIP signal; green, IVIg.
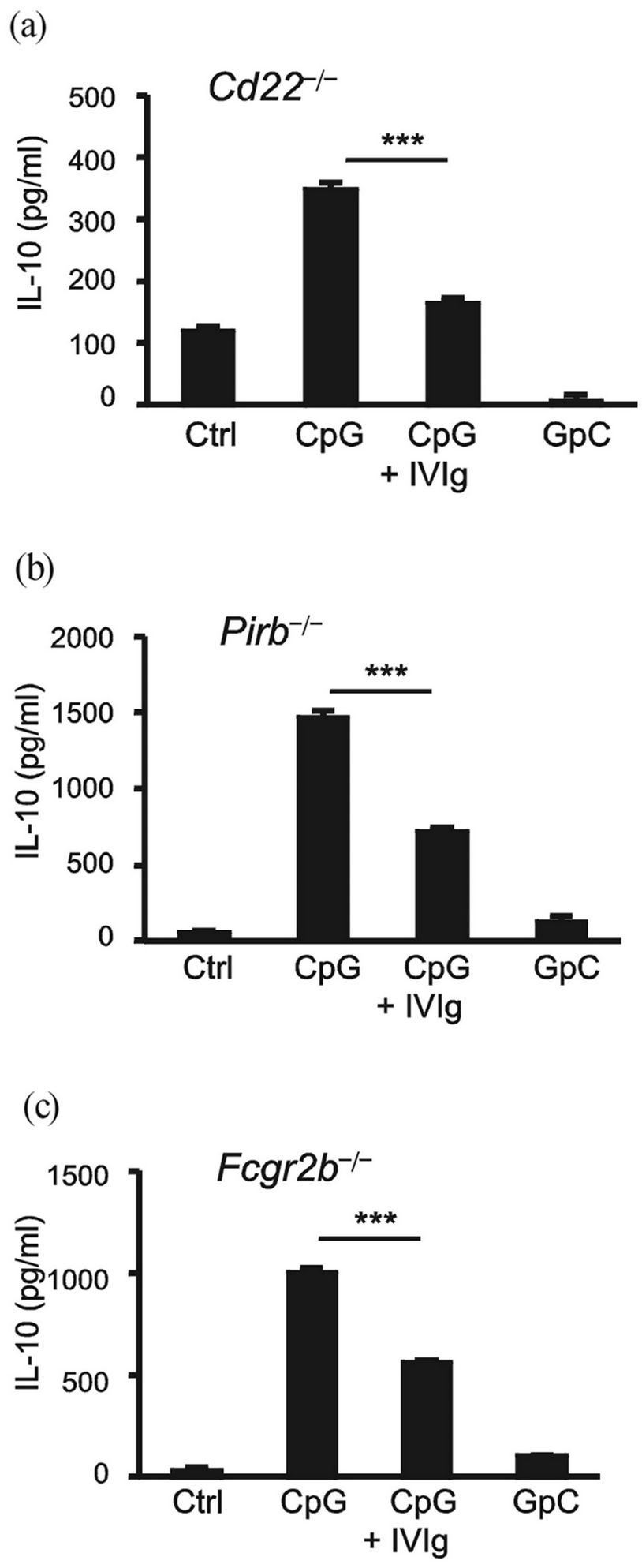
Figure 6. Inhibition of IL-10 production by CpG-activated B cells by IVIg even in the absence of PirB or CD22. Peritoneal B1 cells from Cd22-/- (a) Pirb-/-; (b) or Fcgr2b-/-; (c) mice were stimulated with 200 nMCpG or control GpC for 48 h in the presence or absence of 10 mg/ml IVIg, and then IL-10 was measured by ELISA. The results are representative of three independent experiments. ***P < 0.001.
via IVIg, is a novel finding, but the mechanistic rationale is largely unknown at present. One interesting clue as to this issue is that SHP-1 was recruited to the membrane proximal region upon CpG stimulation and was co-localized with IVIg, which forms several patches on the plasma membrane of CpG-stimulated B cells (Figure 5(a)). We suppose that one, but not all, of the target molecules of IVIg on B cells could be a member of the group of inhibitory receptors that recruit SHP-1 upon B cell activation, although the candidates of such inhibitory receptors are multiple. A preceding report showed that human CD22 plays an important role as a target molecule of IVIg in suppression of B cells [28,29]. Taking advantage of murine B cells genetically deficient in CD22 [17], we tested CD22’s involvement in our experimental setting. However, murine CD22 was found to be dispensable for the inhibition of IL-10 production (Figure 6(a)). It is still possible, however, that CD22 and other inhibitory receptors, such as PirB, CD72, or SiglecG, could collaboratively suppress B cell activation.
4.3. IVIg-Mediated Downregulation of IL-10
The roles of IL-10 in immune responses are considered to be multiple, IL-10 playing an inhibitory role in inflammation in some cases, while it can become harmful in inflammation in others [30,31]. In particular, IL-10 produced by B cells can be a growth factor for B cells themselves, and induce proliferation and differentiation. It is tempting to speculate that one of the IVIg-mediated therapeutic effects is the reduction of IL-10 production by B cells, which is obligatorily the next important subject for clarification in the human system. Interestingly, Kessel et al. [32] showed recently that IL-10 and IL-6 production was reduced by IVIg treatment in CpG-stimulated human B cells in culture, which were isolated from healthy volunteers as well as from SLE patients.
5. CONCLUSION
In this study, we initially attempted to find a suitable experimental system for examining an effect of IVIg on B cells in vitro, and then tried to clarify the molecular mechanism. Although the effect of IVIg on murine B cells was unexpectedly mild, we unequivocally found that IL-10 production by CpG-activated B1 and B2 cells was reproducibly inhibited by IVIg in vitro. Based on this observation, we were able to suggest the significance of SHP-1-recruiting inhibitory receptor(s) on membranes in the suppression of TLR9 signaling. We postulate a scenario in which IVIg attenuates B cells by suppressing IL-10 production, a B cell growth factor, and thus downregulates the production of pathogenic antibodies. Identification of such inhibitory receptor(s) targeted by IVIg on B cells could be the next challenging step toward elucidation of the mechanism underlying alleviation of inflammation and autoimmune diseases by this enigmatic but versatile therapeutic IgG preparation.
6. ACKNOWLEDGEMENTS
We thank Nicholas Halewood for the editorial assistance. This work was supported in part by the Core Research for Evolutional Science and Technology Program of the Japan Science and Technology Agency, a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a grant from the Global Center of Excellence Program for Innovative Therapeutic Development Towards the Conquest of Signal Transduction Diseases with Network Medicine. The authors report no conflict of interest
REFERENCES
- Ballow, M. (2011) The IgG molecule as a biological immune response modifier: mechanisms of action of intravenous immune serum globulin in autoimmune and inflammatory disorders. Journal of Allergy and Clinical Immunology, 127, 315-323. doi:10.1016/j.jaci.2010.10.030
- Anthony, R.M., Wermeling, F. and Ravetch, J.V. (2012) Novel roles for the IgG Fc glycan. Annals of the New York Academy of Sciences, 1253, 170-180. doi:10.1111/j.1749-6632.2011.06305.x
- Ibáñez, C. and Montoro-Ronsano, J.B. (2003) Intravenous immunoglobulin preparations and autoimmune disorders: Mechanisms of action. Current Pharmaceutical Biotechnology, 4, 239-247. doi:10.2174/1389201033489775
- Lemieux, R., Bazin, R. and Néron, S. (2005) Therapuetic intravenous immunoglobulins. Molecular Immunology, 42, 839-848.doi:10.1016/j.molimm.2004.07.046
- Hartung, H.P. (2008) Advances in the understanding of the mechanism of action of IVIg. Journal of Neurology, 255, 3-6. doi:10.1007/s00415-008-3002-0
- Néron, S., Boire, G., Dussault, N., Racine, C., de BrumFernandes, A.J., Côté, S. and Jacques, A. (2009) CD40- activated B cells from patients with systemic lupus erythematosus can be modulated by therapeutic immunoglobulins in vitro. Archivum Immunologiae et Therapiae Experimentalis, 57, 447-458. doi:10.1007/s00005-009-0048-3
- Branch, D.W., Porter, T.F., Paidas, M.J., Belfort, M.A. and Gonik, B. (2001) Obstetric uses of intravenous immunoglobulin: Successes, failures, and promises. Journal of Allergy and Clinical Immunology, 108, S133-S138. doi:10.1067/mai.2001.117821
- Kaveri, S.V., Dietrich, G., Hurez, V. and Kazatchkine, M.D. (1992) Intravenous immunoglobulins (IVIg) in the treatment of autoimmune diseases. Clinical and Experimental Immunology, 86, 192-198. doi:10.1111/j.1365-2249.1991.tb05794.x
- Ehrenstein, M.R. and Notley, C.A. (2010) The importance of natural IgM: Scavenger, protector and regulator. Nature Reviews Immunology, 10, 778-786. doi:10.1038/nri2849
- Griffin, D.O. and Rothstein, T.L. (2012) Human b1 cell frequency: Isolation and analysis of human b1 cells. Frontiers in Immunology, 3, 122. doi:10.3389/fimmu.2012.00122
- Nimmerjahn, F. and Ravetch, J.V. (2006) Fc receptors: Old friends and new family members. Immunity, 24, 19- 28. doi:10.1016/j.immuni.2005.11.010
- Takai, T., Ono, M., Hikida, M., Ohmori, H. and Ravetch, J.V. (1996) Augmented humoral and anaphylactic responses in Fc RII-deficient mice. Nature, 379, 346-349. doi:10.1038/379346a0
- Jellusova, J. and Nitschke, L. (2011) Regulation of B cell functions by the sialic acid-binding receptors Siglec-G and CD22. Frontiers in Immunology, 2, 96. doi:10.3389/fimmu.2011.00096
- Hayami, K., Fukuta, D., Nishikawa, Y., Yamashita, Y., Inui, M., Ohyama, Y., Hikida, M., Ohmori, H. and Takai, T. (1997) Molecular cloning of a novel murine cell-surface glycoprotein homologous to killer cell inhibitory receptors. The Journal of Biological Chemistry, 272, 7320- 7327. doi:10.1074/jbc.272.11.7320
- Kubagawa, H., Burrows, P.D. and Cooper, M.D. (1997) A novel pair of immunoglobulin-like receptors expressed by B cells and myeloid cells. Proceedings of the National Academy of Sciences of the United States of America, 94, 5261-5266. doi:10.1073/pnas.94.10.5261
- Ujike, A., Takeda, K., Nakamura, A., Ebihara, S., Akiyama, K. and Takai, T. (2002) Impaired dendritic cell maturation and increased TH2 responses in PIR-B-/- mice. Nature Immunology, 3, 542-548. doi:10.1038/ni801
- Nitschke, L., Carsetti, R., Ocker, B., Köhler, G. and Lamers, M.C. (1997) CD22 is a negative regulator of B-cell receptor signalling. Current Biology, 7, 133-143. doi:10.1016/S0960-9822(06)00057-1
- Parr, D., Connell, G., Kells, D. and Hofmann, T. (1976) Fb’2, a new peptic fragment of human immunoglobulin G. Biochemical Journal, 155, 31-36.
- Snigurowicz, J. and Powiertowska, M.R. (1980) Papain hydrolysis products in four M-IgG subclass. Archivum Immunologiae, 28, 265-273.
- Virella, G. and Parkhouse, R. (1971) Papain sensitivity of heavy chain sub-classes in normal human IgG and localization of antigenic determinants for the sub-classes. Immunochemistry, 8, 243-250. doi:10.1016/0019-2791(71)90478-2
- Kubo, T., Uchida, Y., Watanabe, Y., Abe, M., Nakamura, A., Ono, M., Akira, S. and Takai, T. (2009) Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. The Journal of Experimental Medicine, 206, 1971-1982.doi:10.1084/jem.20082392
- Parnes, J.R. and Pan, C. (2000) CD72, a negative regulator of B-cell responsiveness. Immunological Reviews, 176, 75-85.doi:10.1034/j.1600-065X.2000.00608.x
- Li, D.H., Winslow, M.M., Cao, T.M., Chen, A.H., Davis, C.R., Mellins, E.D., Utz, P.J., Crabtree, G.R. and Parnes, J.R. (2008) Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis and Rheumatism, 58, 3192-3204.doi:10.1002/art.23812
- Hoffmann, A., Kerr, S., Jellusova, J., Zhang, J., Weisel, F., Wellmann, U., Winkler, T.H., Kneitz, B., Crocker, P.R. and Nitschke, L. (2007) Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nature Immunology, 8, 695-704. doi:10.1038/ni1480
- Ono, M., Bolland, S., Tempst, P. and Ravetch, J.V. (1996) Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc RIIB. Nature, 383, 263-266.doi:10.1038/383263a0
- Leucht, S., Uttenreuther-Fischer, M.M., Gaedicke, G. and Fischer, P. (2001) The B cell superantigen-like interaction of intravenous immunoglobulin (IVIG) with Fab fragments of V(H) 3-23 and 3-30/3-30.5 germline gene origin cloned from a patient with Kawasaki disease is enhanced after IVIG therapy. Clinical Immunology, 99, 18-29. doi:10.1006/clim.2001.5004
- Proulx, D.P., Aubin, E., Lemieux, R. and Bazin, R. (2009) Spontaneous internalization of IVIg in activated B cells. Immunology Letters, 124, 18-26. doi:10.1016/j.imlet.2009.03.012
- Séïté, J.F., Cornec, D., Renaudineau, Y., Youinou, P., Mageed, R.A. and Hillion, S. (2010) IVIg modulates BCR signaling through CD22 and promotes apoptosis in mature human B lymphocytes. Blood, 116, 1698-1704. doi:10.1182/blood-2009-12-261461
- Séité, J.F., Guerrier, T., Cornec, D., Jamin, C., Youinou, P. and Hillion, S. (2011) TLR9 responses of B cells are repressed by intravenous immunoglobulin through the recruitment of phosphatase. Journal of Autoimmunity, 37, 190-197.doi:10.1016/j.jaut.2011.05.014
- Saraiva, M. and O’Garra, A. (2010) The regulation of IL- 10 production by immune cells. Nature Reviews Immunology, 10, 170-181.doi:10.1038/nri2711
- O’Garra, A., Barret, F.J., Castro, A.G., Vicari, A. and Hawrylowicz, C. (2008) Strategies for use of IL-10 or its antagonists in human disease. Immunological Reviews, 223, 114-131.doi:10.1111/j.1600-065X.2008.00635.x
- Kessel, A., Peri, R., Haj, T., Snir, A., Slobodin, G., Sabo, E., Rosner, I., Shoenfeld, Y. and Toubi, E. (2011) IVIg attenuates TLR-9 activation in B cells from SLE patients. Journal of Clinical Immunology, 31, 30-38. doi:10.1007/s10875-010-9469-3