World Journal of Neuroscience
Vol.2 No.3(2012), Article ID:21850,4 pages DOI:10.4236/wjns.2012.23023
Tyrosine hydroxylase and Lewy body molecules immunoreactivity in the SNC neurons of an AS/AGU mutantrat
![]()
1College of Medicine, Umm Alqura University, Makkah, KSA
2School of Life Sciences, Glasgow University, Glasgow, UK
Email: *dr.alkushi@gmail.com
Received 11 March 2012; revised 10 April 2012; accepted 28 April 2012
Keywords: PKC-Gamma; Tyrosine Hydroxylase; Ubiquitin; Parkin
ABSTRACT
The AS/AGU rat has a recessive single point mutation in the gene coding for the gamma isoform of protein kinase C (PKC-γ) resulting in a failure to release dopamine in the striatum and impaired movement including a staggering gait, difficulty in initiating movement and a slight whole body tremor. This study examined the levels tyrosine hydroxylase, ubiquitin and parkin in individual SNC cell bodies. There was no evidence of a reduction in tyrosine hydroxylase levels although levels of ubiquitin and parkin were elevated in the cytoplasm. The findings support the hypothesis that the initial bar to dopamine availability in the striatum is reduced release, with substantia nigra cell death being a later phenomenon.
1. INTRODUCTION
The AS/AGU rat originated as a recessive mutation (agu) in a closed colony of Albino Swiss (AS) rats. The mutation is in the gene coding for the gamma isoform of protein kinase C [1]. The rats are characterized by a movement impairments including rigidity of the hind limbs, a staggering gait, a tendency to fall over every few steps, a slight whole body tremor and difficulty in initiating movements [2,3] by progressive dysfunction of the nigro-striatal dopaminergic (DA) and raphe-striatal serotonergic (5-HT) systems. The chief defect in both systems is a failure to release transmitter within the striatum under normal physiological conditions. Thus, extracellular dopamine levels in the mutant (measured using microdialysis with HPLC-ECD in conscious animals) are only 10% - 20% of control levels [4] and 5-HT is similarly reduced [5]. There is also a marked depletion in utilization of 2- deoxy-glucose in the substantia nigra pars compacta, subthalamic nucleus and ventrolateral thalamus [6]. At later ages, there is loss of aminergic cell bodies [7].
Clinical conditions such as Parkinson’s disease (PD) are characterized by widespread loss of dopaminergic cell bodies and terminals, but this can only be determined post mortem. Death is usually many years after the onset of symptoms. There is, therefore, little indication of the mechanism of cell loss or of the cellular changes taking place prior to that. In animal models of PD, the death of dopaminergic neurons following MPP+ or MPTP administration has been variously attributed to necrosis [8] to apoptosis [9-11] and to autophagy [12]. Furthermore, because the toxic agents used to produce models of PD act rapidly (e.g. MPTP/MPP+, 6-OH-DA), for reviews see [13] Schwarting and Huston, 1996; [14] Flint Beal, 2001, they are unlikely to give insight into any chronic stage during which function might potentially be retrievable.
Commonly used experimental procedures leading to death of SNC neurons (e.g. treatment with 6-hydroxydopamine or MPTP administration), [14] do not exhibit cell inclusions akin to Lewy bodies [15,16] although rotenone treatment is an exception [17]. Similarly, inclusions do not occur in the AS/AGU rat. However, it is unclear if molecules associated with Lewy bodies, such as ubiquitin, alpha-synuclein or parkin, are elevated.
The AS/AGU rat therefore presents an opportunity to examine dopaminergic cell bodies and terminals in a naturally occurring rat model which combines striatal dopamine dysfunction with motor disturbance. This study was undertaken to measure levels of the Lewy body associated proteins ubiquitin and parkin in the nigral cell bodies of the mutant and to compare them with control (unaffected) animals, individual cells of the two strains. Animals aged twelve months were used, as all mutants are reliably symptomatic at this age.
2. MATERIALS AND METHODS
A comparison of tyrosine hydroxylase, ubiquitin and parkin levels in individual midbrain cell bodies of AS and AS/AGU rats using a quantitative fluorescent ICC technique.
Six male AS rats and six male AS/AGU mutants aged twelve months were killed by carbon dioxide euthanasia followed by intracardiac perfusions of 100 ml mammalian Ringer solution and 500 ml 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were removed and stored in 4% paraformaldehyde in phosphate buffer overnight before routine dehydration, embedding in paraffin wax at 57˚C and serial sectioning at 7 mm on a Spencer 820 microtome. Representative sections from the ribbons were collected and stained with 1% aqueous toluidine blue and examined so that matching pairs of unstained sections (AS and AS/AGU) could be taken from the central substantia nigra pars compacta of each animal. These corresponded approximately to coronal sections at –5.3 mm relative to bregma [18]. All sections were processed, examined and quantified in pairs from this stage onwards.
Sections were stretched in a mounting bath for 1 - 3 minutes at 40˚C and mounted on APES (3-aminopropyltriethoxysilane) coated slides, dried overnight at 37˚C and then at 56˚C for two hours. The sections were deparaffinized and rehydrated before undergoing a heat-mediated antigen retrieval technique [19] in boiling 0.01 M sodium citrate buffer (pH 6.0) and then at 120˚C for 1 min in a Prestige stainless steel pressure cooker [20]. After this, sections were rinsed in distilled water followed by 0.01 M phosphate buffered saline (PBS, 5 min) and treated with 1% normal goat serum (NGS, SigmaAldrich G9023, UK) in PBS with 3% Triton X-100 for 60 minutes to reduce non-specific background staining.
1) For fluorescent staining for tyrosine hydroxylase, sections were initially incubated for 24 h in a humidity chamber at 4˚C with the primary antibody (Monoclonal mouse anti-tyrosine hydroxylase, MAB 5280, Chemicon Europe Ltd.) at a concentration of 1:500, diluted in blocking serum (1% NGS in PBS with 0.3% Triton X-100). After 3 × 5 min washes in PBS, sections were incubated in a humidity chamber for 24 hours at 4˚C with a fluorescent antibody (Rhodamine Red-X goat-anti-mouse, 115-295-146, Jackson Immunoresearch) at a dilution of 1:100. Slides were covered during this (and all subsequent processes) to protect from bleaching. Sections were mounted with glass coverslips using Vectashield (H-1400, UK) after rinsing in distilled water (3 × 5 min), and fluorescent quantification was carried out the same day;
2) For fluorescent staining for ubiquitin (polyclonal rabbit anti-ubiquitin Z0458, DAKO, Cambridge, UK), sections were incubated in a humidity chamber for 24 h at 4˚C with a mixture of two primary antibodies (antityrosine hydroxylase and anti-ubiquitin) diluted at 1:500 in the blocking serum. After rinsing in PBS (3 × 5 min), the sections were incubated in a humidity chamber for 24 h at 4˚C with two fluorescent secondary antibodies with Fluorescein (goat-anti-mouse 115-095-116, Jackson Immunoresearch) for TH and Rhodamine (goat-anti-rabbit, Jackson Immunoresearch) for ubiquitin, at dilutions of 1: 100 in PBS for both. Slides were covered to give protection from bleaching in this step and the rest of the procedure. The sections were then mounted with glass coverslips using Vectashield (H-1400, UK) after rinsing in distilled water (3 × 5 min);
3) For fluorescent staining for parkin, the primary antiparkin antibody (anti-parkin I-126 raised in rabbit) was a gift from Professor Poul Henning Jensen, Institute of Medical Biochemistry, bldg. 170, University of Aarhus, DK-8000 Aarhus, Denmark [21]. Double staining procedures were identical to those for ubiquitin (see above).
Quantitative measurements of the immunofluorescence of individual cells was carried out on a Zeiss Axioskop using Zeiss Axiovision (version 4.8) software to perform densitometric analysis of fluorescence levels (Zeiss Axioskop with HBO 110 illuminator and Rhodamine filter set emission 575 - 640 nm, Germany). Image capture was carried out with a Zeiss AxiocamMRc.
For all animals, a minimum of 50 cells with visible nucleoli from each of three regions (rostral, central and caudal substantia nigra pars compacta) were highlighted from the captured file and the fluorescence measured densitometrically. In each case, readings were taken from unstained regions also to give a background fluorescence value, which was then subtracted from the display value. Values were compared between AS and AS/AGU pairs using a paired t-test.
3. RESULTS
Levels of Tyrosine Hydroxylase (TH), Ubiquitin and Parkin
Double labeling showed that 90% of cells in the area of the substantia nigra pars compacta stained positively for TH. This figure is made up of 89% which stained both for TH and ubiquitin and 1% which were ubiquitin-negative. A further 10% of cells were ubiquitin-positive and TH-negative. The frequencies obtained by double labeling for TH and parkin were identical to those of TH and ubiquitin.
Fluorescent measurements were not made on doublelabeled sections, but on adjacent sections where one was stained for TH and the adjacent one for either ubiquitin or parkin (see Figure 1). Table 1 shows that the fluorescence levels for all three molecules were at similar levels throughout the substantia nigra when this was divided into rostral, central and caudal thirds. However, TH levels were some 40% higher in AS/AGU mutants than in AS controls. Similarly, ubiquitin was 36% higher, and parkin 47% higher in the mutant rat SNC cells. These strain differences were highly significant.
 (a)
(a)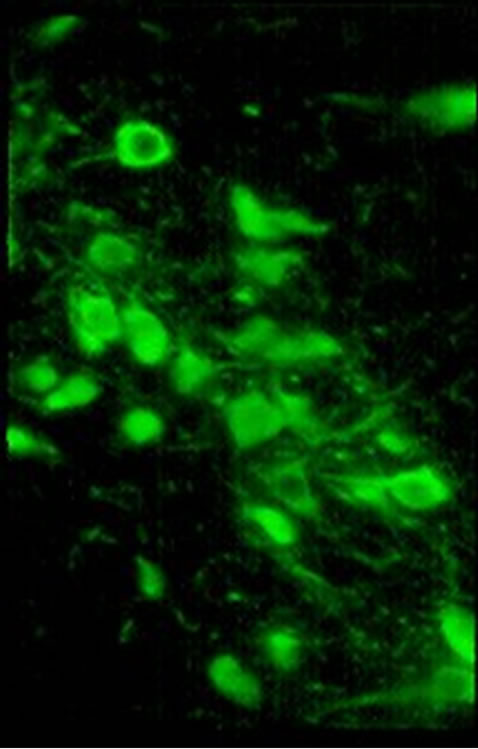 (b)
(b)
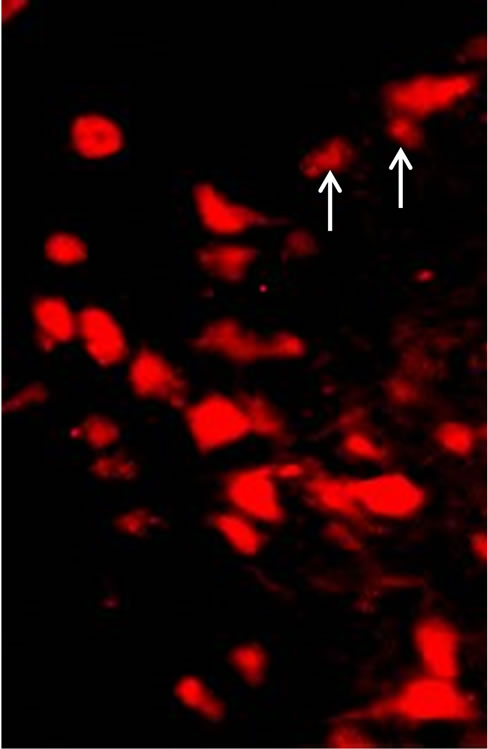
Figure 1. Ubiquitin (Ub) and TH immunostained cells of AS/AGU substantia nigra. (a) Ubcells (red) in (a), white arrows indicate examples of ubiquitin-positive cells which are non-dopaminergic and which were omitted from the count; (b) TH cells (green) (×500).
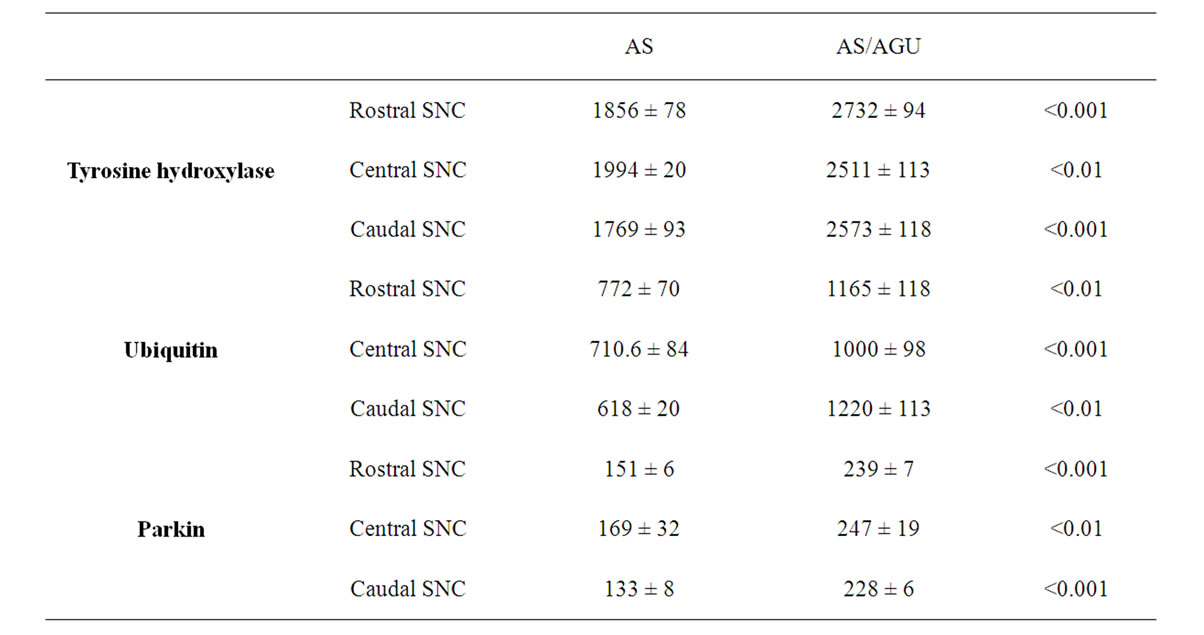
Table 1. Levels of tyrosine hydroxylase, ubiquitin and parkin in dopaminergic cells of the rostral, central and caudal substantia nigra pars compacta (SNC) of AS and AS/AGU rats aged 12 months (units are mean densitometric units ± SEM).
4. DISCUSSION
Nigral dopaminergic neurons in the AS/AGU rat are known to release very little dopamine in the striatum— with extracellular levels only 10% - 20% of normal [4]. However, there is no evidence that they lack the ability to synthesise dopamine—in fact whole tissue micropunches of the midbrain and striatum have shown that dopamine levels remain normal until six months or more [22]. The present experiment demonstrates that TH levels in the cell bodies of the SNC are actually elevated in the mutant (Table 1). One possible conclusion is that, whilst striatal release of dopamine is dysfunctional, the cell bodies of DA neurons are capable of normal physiological responses.
Whole tissue dopamine levels in micropunch samples from the dorsal and lateral caudate-putamen analysed by HPLC-ECD are known to be reduced by some 30% - 40% in the AS/AGU mutant rat compared to the AS control between 6 and 12 months of age [23]. Similar reductions of dopamine levels in the dorsal striatum have been seen in the weaver mouse [24], in post-mortem Parkinson’s disease patients [25] and in living patients with the disorder [26,27]. MPTP exposure can also greatly reduce striatal dopamine [28,29]. By contrast, extracellular levels of dopamine in the striatum as measured in microdialysis samples from conscious, freely-moving AS/AGU rats are reduced by 80% - 90% [4]. This leaves the possibility that dopamine is present in striatal terminals in reasonable amounts, but is not releasable under normal physiological conditions.
Levels of ubiquitin and parkin were elevated in nigral cell bodies of the AS/AGU mutant compared to the AS parent strain. In idiopathic Parkinson’s disease, levels of ubiquitin and parkin (which contains a ubiquitin-like homology domain at its N-terminus and may be involved in the recognition of the substrates and the subsequent degrading of mis-folded proteins) are elevated and the proteins incorporated into cell inclusions [30-39]. Such inclusions have not been found in laboratory models of Parkinson’s disease such as those produced through 6- OH-dopamine or MPTP toxicity [15,16], though they are present in rotenone-induced degeneration in rat [17].
It is unclear what these elevations signify. Dysfunction of the ubiquitin-proteasome system (UPS) has been implicated in Parkinson’s disease and several other neurodegenerative disorders, and rats treated with proteasome inhibitors demonstrate neurodegeneration of midbrain cell groups [40,41]. Mutations in the gene responsible for parkin synthesis have been implicated in early onset forms of Parkinson’s disease [42,43].
REFERENCES
- Craig, N.J., Alonso, M.B.D., Hawker, K.L., Shiels, P., Glencorse, T.A., Campbell, J.M., Bennett, N.K., Canham, M., Donald, D., Gardiner, M., Gilmore, D.P., MacDonald, R.J., Maitland, K., McCallion, A.S., Russell, D., Payne, A.P., Sutcliffe, R.G. and Davies, R.W. (2001) A candidate gene for human neurodegenerative disorders: A rat PKC gamma mutation causes a Parkinsonian syndrome. Nature Neuroscience, 4, 1061-1062. doi:10.1038/nn740
- Clarke, D.J. and Payne, A.P. (1994) Neuroanatomical characterization of a new mutant rat with dopamine depletion in the substantia nigra. European Journal of Neuroscience, 6, 885-888. doi:10.1038/nn740
- Payne, A.P., Sutcliffe, R.G., Campbell, J.M., Favor, G., Russell, D., Bennett, N.K., Clarke, D.J., Branton, R., Davies, R.W., Simpson, E., Tsang, C. and Baxendale, R.H. (1998) Disordered locomotion in the AS/AGU mutant rat and the effects of L-DOPA or fetal midbrain grafts. Movement Disorders, 13, 832-834. doi:10.1002/mds.870130514
- Campbell, J.M., Gilmore, D.P., Russell, D., Growney, C.A., Favor, G., Weir, J., Stone, T.W. and Payne, A.P. (1998) Extracellular levels of dopamine and its metabolite 3,4-dihydroxy-phenylacetic acid measured by microdialysis in the corpus striatum of conscious AS/AGU mutant rats. Neuroscience, 85, 323-325. doi:10.1016/S0306-4522(98)00053-0
- Al-Fayez, M., Russell, D., Davies, R.W., Shiels, P., Baker, P.J. and Payne, A.P. (2005) Deficits in the midbrain raphe nuclei and striatum of the AS/AGU rat: A protein kinase C-g mutant. European Journal of Neuroscience, 22, 2792-2798. doi:10.1111/j.1460-9568.2005.04502.x
- Lam, A.G., Campbell, J.M., Bennett, N.K., Payne, A.P., Davies, R.W., Sutcliffe, R.G. and McCulloch, J. (1998) Local cerebral glucose utilization in the AS/AGU rat: A mutant with movement disorders. European Journal of Neuroscience, 10, 1963-1967. doi:10.1046/j.1460-9568.1998.00206.x
- Payne, A.P., Campbell, J.M., Russell, D., Favor, G., Sutcliffe, R.G., Bennett, N.K., Davies, R.W. and Stone, T.W. (2000) The AS/AGU rat: A spontaneous model of disruption and degeneration in the nigrostriatal dopaminergic system. Journal of Anatomy, 196, 629-633. doi:10.1046/j.1469-7580.2000.19640629.x
- Choi, W.S., Yoon, S.Y., Oh, T.H., Choi, E.J., O’Malley, K.L. and Oh, Y.J. (1999) Two distinct mechanisms are involved in 6-hydroxydopamineand MPP+-induced dopaminergic neuronal cell death: Role of caspases, ROS, and JNK. Journal of Neuroscience Research, 57, 86-94. doi:10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E
- Tatton, N.A. and Kish, S.J. (1997) In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyltransferase labelling and acridine orange staining. Neuroscience, 77, 1037-1048. doi:10.1016/S0306-4522(96)00545-3
- Spooren, W.P., Gentsch, C. and Wiessner, C. (1998) TUNEL-positive cells in the substantia nigra of C57BL/6 mice after a single bolus of 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Neuroscience, 85, 649-651.
- Serra, P.A., Sciola, L., Delogu, M.R., Spano, A., Monaco, G., Miele, E., Rocchitta, G., Miele, M., Migheli, R. and Desole, M.S. (2002) The neurotoxin 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine induces apoptosis in mouse nigrostriatal glia: Relevance to nigral neuronal death and striatal neurochemical changes. Journal of Biological Chemistry, 277, 34451-34461. doi:10.1074/jbc.M202099200
- Oztas, E. and Topal, T. (2003) A cell protective mechanism in a murine model of Parkinson’s disease. Turkish Journal of Medical Sciences, 33, 295-299.
- Schwarting, R.K.W. and Huston, J.P. (1996) The unilateral 6-hydroxydopamine lesion model in behavioural brain research. Analysis of functional deficits, recovery and treatments. Progress in Neurobiology, 50, 275-331. doi:10.1016/S0301-0082(96)00040-8
- Flint, B.M. (2001) Experimental models of Parkinson’s disease. Nature Reviews Neuroscience, 2, 325-332.
- Forno, L.S., De Lanney, L.E., Irwin, I. and Langston, J.W. (1993) Similarities and differences between MPTP-induced parkinsonsim and Parkinson’s disease. Neuropathologic considerations. Advanced Neurology, 60, 600- 608.
- Dauer, W. and Przedborski, S. (2003) Parkinson’s disease: Mechanisms and models. Neuron, 39, 889-909. doi:10.1016/S0896-6273(03)00568-3
- Betarbet, R., Sherer, T.B., MacKenzie, G., Garcia-Osuna, M., Panov, A.V. and Greenamyre, J.T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature Neuroscience, 3, 1301-1306. doi:10.1038/81834
- Paxinos, G. and Watson, C. (1982) The rat brain in stereotaxic coordinates. Academic Press, Waltham.
- Shi, S.R., Key, M.E. and Kalra, K.L. (1991) Antigen retrieval in formalin-fixed, paraffin-embedded tissues: An enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. Journal of Histochemistry & Cytochemistry, 39, 741-748. doi:10.1177/39.6.1709656
- Norton, A.J., Jordan, S. and Yeomans, P. (1994) Brief, high-temperature heat denaturation (pressure cooking): A simple and effective method of antigen retrieval for routinely processed tissues. The Journal of Pathology, 173, 371-379. doi:10.1002/path.1711730413
- Muqit, M.M.K., Davidson, S.M., Smith, M.D.P., MacCormac, L.P., Kahns, S., Jensen, P.H., Wood, N.W. and Latchman, D.S. (2004) Parkin is recruited into aggresomes in a stress-specific manner: Over-expression of parkin reduces aggresome formation but can be dissociated from parkin’s effect on neuronal survival. Human Molecular Genetics, 13, 117-135. doi:10.1093/hmg/ddh012
- Campbell, J. M., Payne, A.P., Gilmore, D.P., Byrne, J.E., Russell, D., McGadey, J., Clarke, D.J., Branton, R., Davies, R.W. and Sutcliffe, R.G. (1997) Age changes in dopamine levels in the corpus striatum of Albino Swiss (AS) and AS/AGU mutant rats. Neuroscience Letters, 239, 54-56. doi:10.1016/S0304-3940(97)00871-9
- Campbell, J.M., Payne, A.P., Gilmore, D.P., Byrne, J.E., Russell, D., McGadey, J., Clarke, D.J., Davies, R.W. and Sutcliffe, R.G. (1996) Neostriatal dopamine depletion and locomotor abnormalities due to the Albino Swiss rat agu mutation. Neuroscience Letters, 213, 173-176.
- Roffler-Tarlov, S. and Graybiel, A.M. (1984) Weaver mutation has differential effects on the dopamine-containing innervation of the limbic and non-limbic striatum. Nature, 307, 62-66. doi:10.1038/307062a0
- Hornykiewicz, O. (1995) Striatal dopamine in dopa-responsive dystonia: Comparison with idiopathic Parkinson’s disease and other dopamine-dependent disorders. In: Segawa, M. and Nomura, Y., Eds., Age-Related Dopamine-Dependent Disorders, Karger, Basel, 101-108
- Leenders, K.L., Palmer, A.J., Quinn, N., Clark, J.C., Firnau, G., Garnett, E.S., Nahmias, C., Jones, T. and Marsden, C.D. (1986) Brain dopamine metabolism in patients with Parkinson’s disease measured with positron emission tomography. Journal of Neurology, Neurosurgery & Psychiatry, 49, 853-860. doi:10.1136/jnnp.49.8.853
- Leenders, K.L., Salmon, E.P., Tyrrell, P., Perani, D., Brooks, D.J., Sager, H., Jones, T., Marsden, C.D. and Frackowiak, R.S. (1990) The nigrostriatal dopaminergic system assessed in vivo by positron emission tomography in healthy volunteer subjects and patients with Parkinson’s disease. Archives of Neurology, 47, 1290-1298. doi:10.1001/archneur.1990.00530120034007
- Moratalla, R., Quinn, B., DeLanney, L.E., Irwin, I., Langston, J.W. and Graybiel, A.M. (1992) Differential vulnerability of primate caudate-putamen and striosome-matrix dopamine systems to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proceedings of the National Academy of Sciences of USA, 89, 3859-3863. doi:10.1073/pnas.89.9.3859
- Snow, B.J., Vingerhoets, F.J., Langston, J.W., Tetrud, J.W., Sossi, V. and Calne, D.B. (2000) Pattern of dopaminergic loss in the striatum of humans with MPTP induced parkinsonism. Journal of Neurology, Neurosurgery & Psychiatry, 68, 313-316.
- Lennox G., Lowe J., Morrell, K., Landon, M. and Mayer, R.J. (1989) Anti-ubiquitin immunocytochemistry is more sensitive than conventional techniques in the detection of diffuse Lewy body disease. Journal of Neurology, Neurosurgery & Psychiatry, 52, 67-71. doi:10.1136/jnnp.68.3.313
- Love, S. and Nicoll, J.A. (1992) Comparison of modified Bielschowsky silver impregnation and anti-ubiquitin immunostaining of cortical and nigral Lewy bodies. Neuropathology and Applied Neurobiology, 18, 585-592. doi:10.1111/j.1365-2990.1992.tb00830.x
- Spillantini, M.G., Crowther, R.A., Jakes, R., Cairns, N.J., Lantos, P.L. and Goedert, M. (1998) Filamentous alphasynuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neuroscience Letters, 251, 205-208. doi:10.1016/S0304-3940(98)00504-7
- Irizarry, M.C., Growdon, W., Gomez-Isla, T., Newell, K., George, J.M., Clayton, D.F. and Hyman, B.T. (1998) Nigral and cortical Lewy bodies and dystrophic nigralneurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. Journal of Neuropathology & Experimental Neurology, 57, 334- 337. doi:10.1097/00005072-199804000-00005
- Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Iwai, K., Chiba, T. and Tanaka, K. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature Genetics, 25, 302-305. doi:10.1038/77060
- Sherman, M.Y. and Goldberg, A.L. (2001) Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron, 29, 15-32. doi:10.1016/S0896-6273(01)00177-5
- Cookson, M.R. (2005) The biochemistry of Parkinson’s disease. Annual Review of Biochemistry, 74, 29-52. doi:10.1146/annurev.biochem.74.082803.133400
- Gai, W.P., Yuan, H.X., Li, X.Q., Power, J.T., Blumbergs, P.C. and Jensen, P.H. (2000) In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Experimental Neurology, 166, 324-333. doi:10.1006/exnr.2000.7527
- McNaught, K.S., Shashidharan, P., Perl, D.P., Jenner, P., and Olanow, C.W. (2002) Aggresome-related biogenesis of Lewy bodies. European Journal of Neuroscience, 16, 2136-2148. doi:10.1046/j.1460-9568.2002.02301.x
- Moreno-Gonzalez, I. and Soto, C. (2011) Misfolded protein aggregates: Mechanisms, structures and potential for disease transmission. Seminars in Cell & Developmental Biology, 22, 482-487. doi:10.1016/j.semcdb.2011.04.002
- McNaught, K.S., Perl, D.P., Brownell, A.L. and Olanow, C.W. (2004) Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Annals of Neurology, 56, 149-162. doi:10.1002/ana.20186
- Huang, Q. and Figueiredo-Pereira, M.E. (2010) Ubiquitin/ proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis, 15, 1292-1311. doi:10.1007/s10495-010-0466-z
- Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. and Shimizu, N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605-608. doi:10.1007/s10495-010-0466-z
- Kumar, K.R., Djarmati-Westenberger, A. and Grünewald, A. (2011) Genetics of Parkinson’s disease. Seminars in Neurology, 31, 433-440. doi:10.1055/s-0031-1299782
NOTES
*Corresponding author.