World Journal of Neuroscience
Vol.2 No.1(2012), Article ID:17724,8 pages DOI:10.4236/wjns.2012.21003
Acetylcholine participates in pain modulation by influencing endogenous opiate peptides in rat spinal cord
![]()
1College of Pharmacy, Xinxiang Medical University, Xixiang, China
2Kamp Institute for Medical Research, Changsha, China
3Jiangsu Su Bei People’s Hospital, Yangzhou University, Yangzhou, China
Email: *bcd2009@126.com
Received 29 October 2011; revised 14 December 2011; accepted 9 January 2012
Keywords: Achetylcholine; Endogenous Opiate Peptide; Pain Modulation; Spinal Cord
ABSTRACT
The spinal cord is a necessary pathway that transfers the body nociceptive inputs to the brain, and endogenous opiate peptides (EOP) play an important role in pain modulation. Our previous work has proven that arginine vasopressin (AVP) antinociception in the caudate nucleus (CdN) relates with the acetylcholine (Ach) system mainly. The communication was designed to investigate the interrelations between Ach system and EOP system at the spinal level during pain process. The results showed that: 1) pain stimulation increased L-enkephalin (L-Ek), β-endorphin (β-Ep), dynorphin A1-13(DynA1-13), Ach and choline (Ch, an Ach metabolic product) concentrations in the spinal cord; 2) Ach increased L-Ek, β-Ep and DynA1-13 concentrations in the spinal cord; and 3) Atropine (M-receptor inhibitor) or hexahydric gallamine (Nreceptor inhibitor) decreased L-Ek, β-Ep and DynA1-13 concentrations in the spinal cord. The data suggested that Ach antinociception was involved in the EOP system at the spinal level.
1. INTRODUCTION
Hypothalamic paraventricular nucleus (PVN) is a complex neural structure, which has been implicated in a remarkable number of functions including control of pituitary-adrenocortical activity in response to stress [1], body fluid homeostasis [2], milk ejection reflex [3], hormone probation [4], circadian rhythm [5], food intake [6], gastrointestinal and cardiovascular functions [7], sexual activity [8], learning and memory [3]. Electrical stimulation of PVN increases pain threshold, whereas electrical cauterization of PVN decreases pain threshold; Microinjection of L-glutamate sodium into PVN, which only excites the PVN neurons [9], increases pain threshold; Removing the pituitary, which blocks the neurohypophysis effect, does not influence the role of PVN enhancing analgesia [10,11]. PVN can regulate pain process through the central nerve system rather than the peripheral organs.
Arginine vasopressin (AVP), a nonapeptide hormone, is an important bioactive substance that is synthesized in the PVN [3]. Pain stimulation increases AVP, not oxytocin (OXT), L-enkephalin (L-Ek), β-endorphin (β-Ep) and dynorphin A1-13(DynA1-13) concentrations in both the PVN tissue and PVN perfuse liquid; pain stimulation decreases the number of AVP-immunoreactive neurons and enhances AVP mRNA expression in the PVN; intraventricular injection of anti-AVP serum or V2 receptor antagonist, d(CH2)5[D-Ile2,Ile4,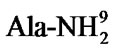 ]AVP completely reversed the antinociception induced by PVN stimulation [12]. AVP is the most important bioactive substance in PVN regulating pain process [13].
]AVP completely reversed the antinociception induced by PVN stimulation [12]. AVP is the most important bioactive substance in PVN regulating pain process [13].
The spinal cord is a necessary pathway that transfers the body nociceptive inputs to the brain. PVN stimulation increases and PVN cauterization decreases L-Ek, β- Ep, DynA1-13 concentrations in the spinal cord; the spinal cord pretreatment with naloxone, an opiate receptor antagonist partly reverses the analgesia induced by PVN stimulation [14]. PVN analgesia is involved in the endogenous opiate peptide (EOP) system in the spinal cord independently.
It is not clear how AVP in the PVN influences the EOP system at the spinal level. However, our previous study has pointed that AVP, which comes from the PVN, induces the caudate nucleus (CdN) releasing acetylcholine (Ach) during pain process [15,16]. Ach can influence the release of EOP in the brain including the hypothalamus [17-19]. The communication was designed to investigate the interaction between Ach system and EOP system at the spinal level during pain process.
2. MATERIALS AND METHODS
2.1. Animals
Adult male Sprague-Dawley rats weighing 180 - 220 g were used in all experiments (Animal Center of Yangzhou University, Yangzhou, Jiangsu, China). Animals were housed in a colony room under controlled temperature, humidity and a 12 hours light/dark cycle (light on at 6:00 AM), with food and water available ad libitum. All procedures were conducted according to the guidelines of the International Association for the Study of Pain [20] and approved by the Animal Care and Use Committee of Yangzhou University.
2.2 Materials
L-Ek, β-Ep and DynA1-13 were obtained from Peninsula Laboratories, San Carlos, CA, USA; 125Iodine was from Amersham Pharmacia, Buckinghamshire, UK; Ach, choline (Ch, an Ach metabolic product), atropine (M-receptor antagonist), hexahydric gallamine (N-receptor antagonist) and other chemical reagents were from Sigma Co., St. Louis, MO, USA.
Rabbit anti-rat L-Ek, β-Ep or DynA1-13 serum was made by Department of Neurobiology, Second Military Medical University, Shanghai, China. The specificity of each kind of antiserum was more than 99% reactivity with its corresponding antigen and less than 1% reactivity with other similar peptides. The effective dilution of the antiserum was 1:20,000 - 80,000 for radioimmunoassay.
2.3. Surgery
Under pentobarbital sodium (35 mg/kg, intraperitoneal injection) anesthesia, the rat was implanted a chronic intrathecal catheter (PE-10, 12 cm in length, 0.6 cm outer diameter) extending into the lumbar enlargement of the spinal cord for intrathecal injection (ith).
All operations were carried out under aseptic conditions and the animals were allowed to recover for at least 14 days after the surgery.
2.4 Pain Stimulation
All animals were tested under the condition of free activity in the small cages (30 cm in diameter, 25 cm in height) from 8:00 to 10:00 am. We used the potassium iontophoresis inducing tail-flick served as pain stimulus. The small wet cotton with the potassium iontophoresis was set on the skin of the tail. The cotton was exposed to direct electrical current, and the anode led the potassium iontophoresis to permeate the skin of the tail. If the current was strong enough, the permeated potassium iontophoresis resulted in the animal feeling the pain stimulation. The intensity of current at the moment of the response was recorded as the pain threshold, which was expressed as mA (WQ-9E Pain Threshold Measurer, Shanghai, China).
Through the positive electrodes producing the direct electrical current was generated from Pain Threshold Measurer to induce the potassium iontophoresis into the animal tail skin and result in acute pain. The intensity was fixed to 1.2 - 1.4 × pain threshold (0.6 - 0.7 mA) for 1 min.
2.5. For Spinal Cord Administration
Ten μl of artificial cerebral spinal fluid (ACSF, containing 0.1 M NaCl, 1.0 mM KH2PO4, 4.0 mM KCl, 2.0 mM MgSO4, 2.0 mM CaCl2, 2.1 mM NaHCO3 and 8.0 mM Glucose), which the drug was dissolved, was gently injected into the lumbar enlargement of the spinal cord through the chronic intrathecal catheter over 10 min.
2.6. Prepare of Tissue Sample
After the decapitation, the lumber spinal cords were taken out and put into the liquid nitrogen quickly. After weighing, the tissues were homogenized in 1.0 ml of 0.1 M acetic acid at 4˚C. Two hours later, the same volume of 0.1 M sodium hydroxide was mixed in the homogenate. Using the centrifugation at 10,000 g at 4˚C for 20 min, the supernatants were withdrawn and stored at –80˚C for assay.
2.7. Radioimmunoassay (RIA)
The L-Ek, β-Ep and DynA1-13 concentrations were determined with specific rabbit antiserum. The peptides were labeled 125Iodine using the chloramines-T method and iodinated peptides werepurified by Sephadex G-50. The assay sensitivities for the L-Ek, β-Ep or DynA1-13 were 3.0, 1.2 and 6.3 pg/tube and intraand inter-assay coefficients of variation were less than 5.1% and 8.0%, respectively [21-23].
2.8. HPLC for Neurotransmitter Measurement
Ach concentration was measured by HPLC-ECD instrumentation, using a mobile phase, pH 8.5, containing 50 mM sodium phosphate buffer and 0.5% Kathon reagent (BAS) microbicide [24]. Samples were injected into a polymeric reversed phase column (BAS Ach-choline assay kit). Ach was then converted into hydrogen peroxide and betaine in a postcolumn enzyme reactor containing immobilizedacetylcholinesterase and choline oxidase (BAS). The hydrogen peroxide was detected electrochemically with a platinum electrode set at 50 mV (vs Ag/AgCl). The Ach standards was prepared weekly using ACSF and stored in –80˚C.
2.9. Statistical Analysis
Data were expressed as mean ± standard error of the mean (S.E.M.) and were analyzed between groups by the analysis of variance (ANOVA) and χ2 test. P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Pain Stimulation Increased the EOP and Ach Concentrations in the Spinal Cord
Giving the animal 1 min pain stimulation, the L-Ek concentration in the spinal cord was increased from 8.4 ± 2.1 pg/mg (tissue weight) to 35.5 ± 5.6 pg/mg in 5 min (P < 0.001) and 17.6 ± 4.3 pg/mg in 10 min (P < 0.05) after the stimulation (Figure 1(a)); the β-Ep concentration in the spinal cord was increased from 10.6 ± 2.7 pg/mg (tissue weight) to 48.9 ± 8.2 pg/mg in 5 min (P < 0.001) and 35.4 ± 6.5 pg/mg in 10 min (P < 0.01) after the stimulation (Figure 1(b)); the DynA1-13 concentration in the spinal cord was increased from 6.5 ± 1.8 pg/mg (tissue weight) to 18.7 ± 4.3 pg/mg in 5 min (P < 0.001) and 11.4 ± 2.1 pg/mg in 10 min (P < 0.01) after the stimulation (Figure 1(c)); the Ach concentration in the spinal cord was increased from 5.4 ± 1.6 pg/mg (tissue weight) to 21.2.5 ± 4.6 pg/mg in 5 min (P < 0.001) and 12.4 ± 3.7 pg/mg in 10 min (P < 0.001) after the stimulation (Figure 1(d)); and the Ch concentration in the spinal cord was increased from 4.7 ± 1.5 pg/mg (tissue weight) to 17.6 ± 5.3 pg/mg in 5 min (P < 0.001) and 9.8 ± 4.4 pg/mg in 10 min (P < 0.01) after the stimulation (Figure 1(e)). In control group, the L-Ek, β-Ep, DynA, 5-HT and 5-HIAA concentrations did not change.
3.2. Ach Increased the EOP Concentrations in the Spinal Cord
Administration of 20 ng Ach into the spinal cord increased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 27.6 ± 5.2 pg/mg in 5 min (P < 0.001) and 16.7 ± 3.5 pg/mg in 10 min (P < 0.001); administration of 10 ng Ach into the spinal cord increased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 15.3 ± 3.2 pg/mg in 5 min (P < 0.01) and 9.5.7 ± 2.7 pg/mg in 10 min. In control group, the L-Ek concentration did not change (Figure 2(a)).
Administration of 20 ng Ach into the spinal cord increased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 31.2 ± 8.8 pg/mg in 5 min (P < 0.001) and 19.8 ± 6.1 pg/mg in 10 min (P < 0.05); administration of 10 ng Ach into the spinal cord increased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 19.3 ± 5.3 pg/mg in 5 min (P < 0.01) and 13.5 ± 4.4 pg/mg in 10 min. In control group, the β-Ep concentration did not change (Figure 2(b)).
Administration of 20 ng Ach into the spinal cord in-
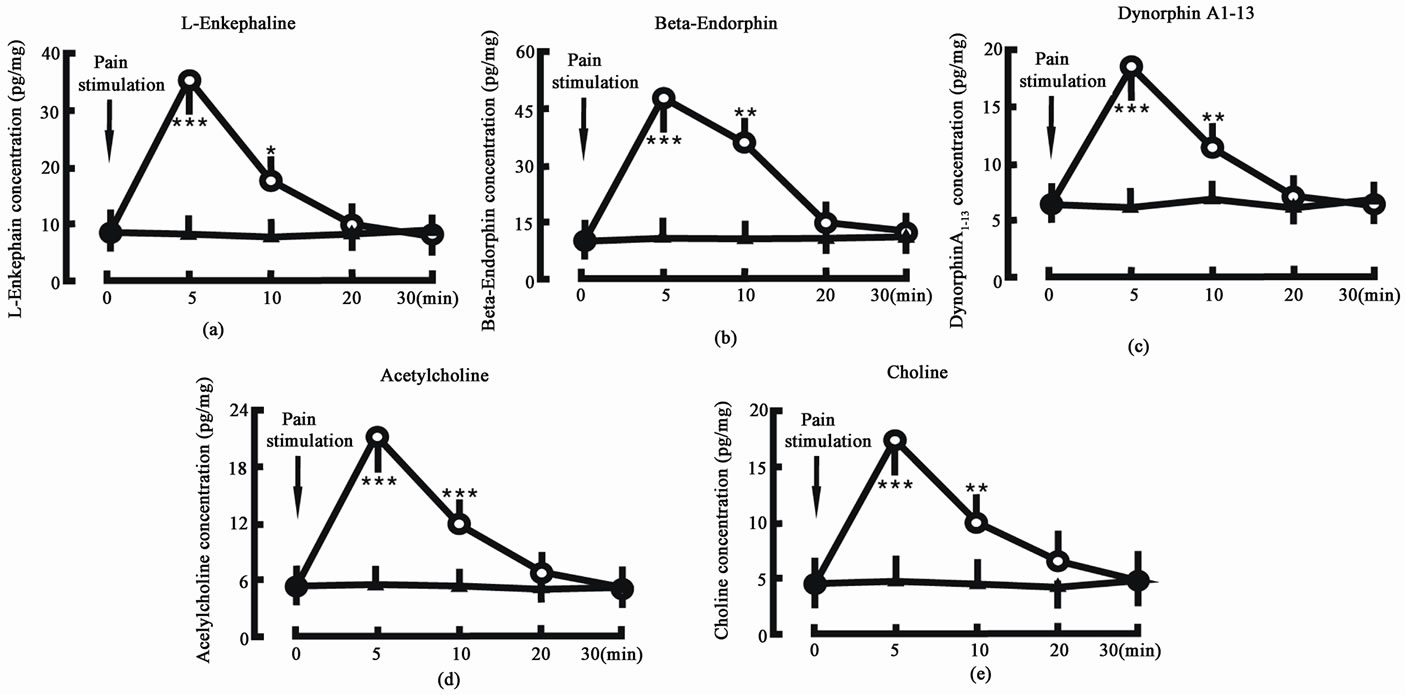
Figure 1. Effect of pain stimulation on endogenous opiate peptide and acetylcholine concentrations in the spinal cord. Pain stimulation denotes the beginning of pain stimulation. Pain stimulation group (○, n = 8): the animal was given 1 min pain stimulation; Control group (▲, n = 8): the animal was given the sham treatment. N indicates the animal number in each group. The data are expressed as mean ± standard error mean (SEM). *P < 0.05, **P < 0.01 and ***P < 0.001 are used for the comparison of the change of L-enkephalin, β-endorphin, dynorphin A1-13, acetylcholine or choline concentration from pain stimulation group and control group.
creased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 13.4 ± 3.5 pg/mg in 5 min (P < 0.01) and 10.7 ± 2.9 pg/mg in 10 min (P < 0.05); administration of 10 ng Ach into the spinal cord increased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 8.7 ± 2.5 pg/mg in 5 min and 7.2 ± 1.9 pg/mg in 10 min. In control group, the DynA1-13 concentration did not change (Figure 2(c)).
3.3. M-Receptor Inhibitor Decreased the EOP Concentrations in the Spinal Cord
Administration of 2 µg atropine into the spinal cord decreased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 2.9 ± 0.8 pg/mg in 5 min (P < 0.001), 4.2 ± 1.5 pg/mg in 10 min (P < 0.01) and 6.2 ± 1.8 pg/mg in 20 min; administration of 1 µg atropine into the spinal cord decreased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 5.1 ± 1.0 pg/mg in 5 min (P < 0.05), 6.2 ± 1.1 pg/mg in 10 min and 7.7 ± 1.9 pg/mg in 20 min. In control group, the L-Ek concentration did not change (Figure 3(a)).
Administration of 2 µg atropine into the spinal cord decreased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 4.8 ± 0.9 pg/mg in 5 min (P < 0.001), 6.1 ± 1.2 pg/mg in 10 min (P < 0.05) and 8.2 ± 2.2 pg/mg in 20 min; administration of 1 µg atropine into the spinal cord decreased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 7.1 ± 1.5 pg/mg in 5 min (P < 0.05), 8.6 ± 1.9 pg/mg in 10 min and 9.5 ± 2.4 pg/mg in 20 min. In control group, the β-Ep concentration did not change (Figure 3(b)).
Administration of 2 µg atropine into the spinal cord decreased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 2.8 ± 0.7 pg/mg in 5 min (P < 0.01), 3.7 ± 1.1 pg/mg in 10 min (P < 0.05) and 5.9 ± 1.5 pg/mg in 20 min; administration of 1 µg
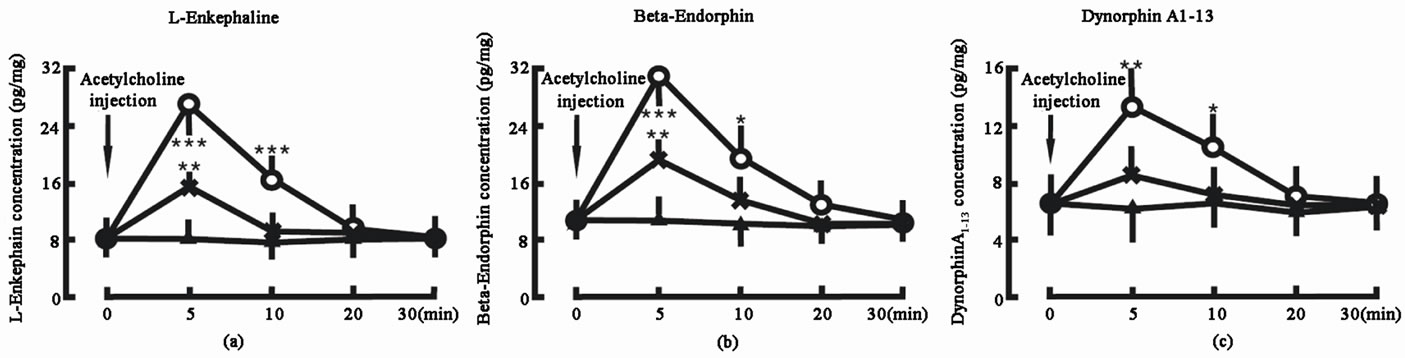
Figure 2. Effect of acetylcholine on the endogenous opiate peptide concentrations in the spinal cord. Acetylcholine injection denotes the beginning of acetylcholine administration. Acetylcholine 20 ng group (○, n = 8): the animal was given 20 ng acetylcholine into the spinal cord; Acetylcholine 10 ng group (Х, n = 8): the animal was given 10 ng acetylcholine into the spinal cord; Control group (▲, n = 8): the animal was not given any acetylcholine into the spinal cord. The data are expressed as mean ± standard error mean (SEM). *P < 0.05, **P < 0.01 and ***P < 0.001 are used for the comparison of the change of Lenkephalin, β-endorphin or dynorphin A1-13 concentration from acetylcholine 20 ng group or acetylcholine 10 ng group and control group.
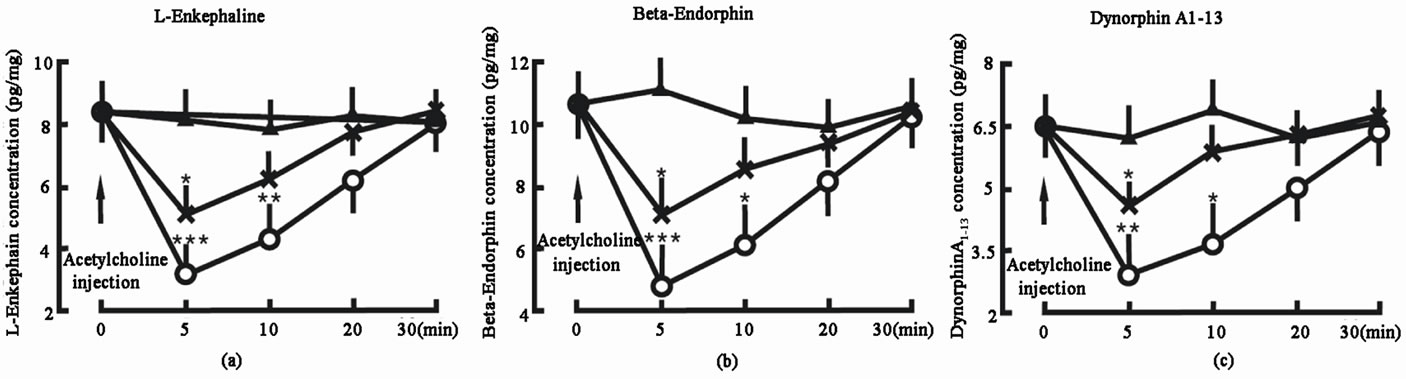
Figure 3. Effect of M-receptor inhibitor—atropine on the endogenous opiate peptide concentrations in the spinal cord. Atropine injection denotes the beginning of atropine administration. Atropine 2 μg group (○, n = 8): the animal was given 2 μg atropine into the spinal cord; Atropine 1 μg group (Х, n = 8): the animal was given 1 μg atropine into the spinal cord; Control group (▲, n = 8): the animal was not given any atropine into the spinal cord. The data are expressed as mean ± standard error mean (SEM). *P < 0.05, **P < 0.01 and ***P < 0.001 are used for the comparison of the hange of L-enkephalin, β-endorphin or dynorphin A1-13 concentration from atropine 2 μg group or atropine 1 μg group and control group.
atropine into the spinal cord decreased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 4.6 ± 1.0 pg/mg in 5 min (P < 0.05), 5.9 ± 1.6 pg/mg in 10 min and 6.3 ± 1.8 pg/mg in 20 min. In control group, the DynA1-13 concentration did not change (Figure 3(c)).
3.4. N-Receptor Inhibitor Decreased the EOP Concentrations in the Spinal Cord
Administration of 2 µg hexahydric gallamine into the spinal cord decreased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 4.6 ± 1.4 pg/mg in 5 min (P < 0.01), 5.8 ± 1.8 pg/mg in 10 min and 7.3 ± 2.0 pg/mg in 20min; administration of 1 µg hexahydric gallamine into the spinal cord decreased the L-Ek concentration in the spinal cord from 8.4 ± 2.1 pg/mg (tissue weight) to 5.8 ± 1.3 pg/mg in 5 min, 6.8 ± 1.8 pg/mg in 10 min and 7.9 ± 2.1 pg/mg in 20 min. In control group, the L-Ek concentration did not change (Figure 4(a)).
Administration of 2 µg hexahydric gallamine into the spinal cord decreased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 5.8 ± 1.4 pg/mg in 5 min (P < 0.001), 7.2 ± 1.3 pg/mg in 10 min (P < 0.01) and 8.8 ± 2.2 pg/mg in 20 min; administration of 1 µg hexahydric gallamine into the spinal cord decreased the β-Ep concentration in the spinal cord from 10.6 ± 2.7 pg/mg (tissue weight) to 7.3 ± 1.6 pg/mg in 5 min (P < 0.05), 8.9 ± 1.9 pg/mg in 10 min and 9.5 ± 2.3 pg/mg in 20 min. In control group, the β-Ep concentration did not change (Figure 4(b)).
Administration of 2 µg hexahydric gallamine into the spinal cord decreased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 3.5 ± 0.7 pg/mg in 5 min (P < 0.01), 4.2 ± 1.1 pg/mg in 10 min and 5.8 ± 1.4 pg/mg in 20 min; administration of 1 µg hexahydric gallamine into the spinal cord decreased the DynA1-13 concentration in the spinal cord from 6.5 ± 1.8 pg/mg (tissue weight) to 4.7 ± 0.8 pg/mg in 5 min, 5.5 ± 1.1 pg/mg in 10 min and 6.8 ±1.2 pg/mg in 20 min. In control group, the DynA1-13 concentration did not change (Figure 4(c)).
4. DISCUSSION
PVN contains two types of functional cells, the largesized cells and small-sized cells. The large-sized cells, which are called magnocellular cells that synthesize either AVP or OXT and their respective neurophysins, areneuroendocrine cells, which project to the capillaries in the posterior pituitary lobe, where they secrete their contents as hormones into the systemic circulation. The small-sized cells, which are called parvocellular cells that synthesize these neuropeptides serve one of a number of specialized roles in the brain: some of these cells secrete releasing hormones into the portal circulation of the anterior pituitary gland; others activate, inhibit, or modulate activity in other neurons in the brain; and still other synapse on blood vessels in the brain, where they may influence vascular dynamics [3]. PVN has an analgesic role in the body nociceprion [10,11], which may be through EOP system in the spinal cord [14].
AVP has been identified as an important factor governing analgesia [6,25]. AVP intraventricular injection increases pain threshold and anti-AVP serum administration decreases pain threshold, whereas intrathecal or intravenous injection of AVP or anti-AVP serum does not influence pain threshold [26]. Our previous work has pointed that AVP is the most important bioactive substance in PVN antinociception [12,27,28]. AVP antinociception in PVN is through many brain areas including CdN [15]. Pain stimulation changes AVP concentration in CdN [28]; CdN treatment with AVP enhances analge-
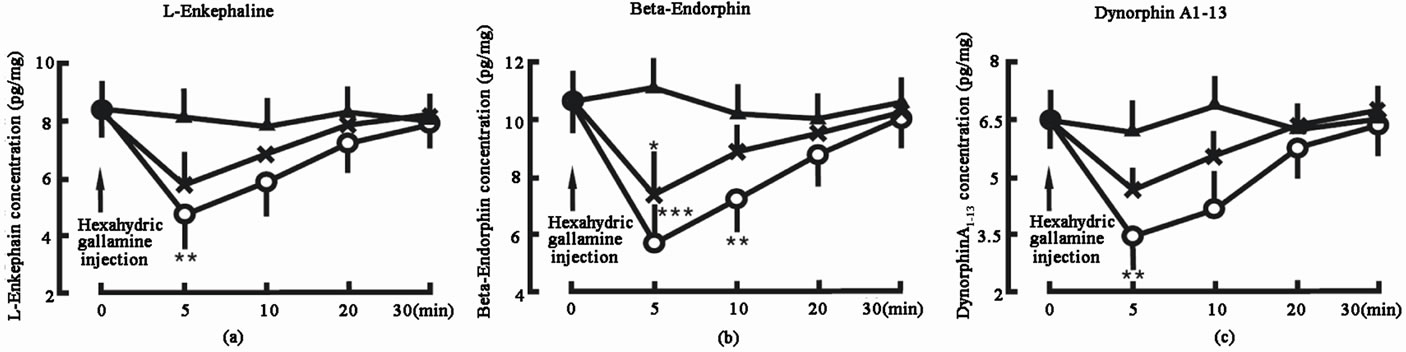
Figure 4. Effect of N-receptor inhibitor—hexahydric gallamine on the endogenous opiate peptide concentrations in the spinal cord. Hexahydric gallamine injection denotes the beginning of hexahydric gallamine administration. Hexahydric gallamine 2 μg group (○, n = 8): the animal was given 2 μg hexahydric gallamine into the spinal cord; Hexahydric gallamine 1 μg group (Х, n = 8): the animal was given 1 μg hexahydric gallamine into the spinal cord; Control group (▲, n = 8): the animal was not given any hexahydric gallamine into the spinal cord. The data are expressed as mean ± standard error mean (SEM). *P < 0.05, **P < 0.01 and ***P < 0.001 are used for the comparison of the change of L-enkephalin, β-endorphin or dynorphin A1-13 concentration from hexahydric gallamine 2 μg group or hexahydric gallamine 1 μg group and control group.
sia, and CdN treatment with AVP receptor antagonist weakens analgesia [16]. Pain stimulation increases both AVP and Ach concentrations in the CdN perfusion liquid; AVP induces CdN releasing Ach, while AVP receptor antagonist including d(CH2)5Tyr(Me)AVP (V1 receptor antagonist) and d(CH2)5[D-Ile2, Ile4,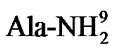 ]AVP (V2 receptor antagonist) inhibits this effect [15]. AVP analgesic effect in the CdN is involved in Ach system.
]AVP (V2 receptor antagonist) inhibits this effect [15]. AVP analgesic effect in the CdN is involved in Ach system.
Acetylcholine (ACh) is a neurotransmitter in both the peripheral nervous system and central nervous system in many organisms including humans. Acetylcholine is one of many neurotransmitters in the autonomic nervous system and the only neurotransmitter used in the motor division of the somatic nervous system.
Many experiments have reported that Ach in the spinal cord plays an important role in pan modulation [29,30]. Intrathecal injection (ith) of clonidine dose-dependently reduced tonic pain behaviors [31]. Subcutaneous injection of formalin into a paw of mice causes the licking and biting, which were inhibited by intrathecal injection of the muscarinic antagonist atropine or the M3 receptor antagonist 4-diphenylacetoxy-N-methylpiperidine methiodide [32].
Since Hughes et al. purified and identified L-Ek and M-Ek in 1975 [33], the endorphin had been confirmed in 1976 [34] and dynorphin in 1979 [35]. Endogenous opiate peptides include three series—enkephalin, endorphin and dynorphin [36], which have been proven to participate in the pain modulation in the spinal cord [37-40].
It has been proven that the interaction between Ach and endogenous opiate peptides in the brain relates with pain modulation [41]. The μ- and δ-opioids in the hypothalamic paraventricular nucleus (PVN) contribute to the control of ACh release [18]. The opioid peptide dynorphin A1-8 dynorphin B, methionine-enkephalin (M-Ek) or leucine-enkephalin (L-Ek) increased Ach release, which effect was reversed by the opiate antagonist naloxone [42]. The spinal cord is a necessary pathway that transfers the body nociceptive inputs to the brain. EOP system includes three series—enkephaline, endorphin and dynorphin, all of which are involved in analgesia [43]. PVN stimulation increases and PVN cauterization decreases the EOP concentrations in the spinal cord; administration of naloxone, an opiate receptor antagonist in the spinal cord partly attenuates the analgesia induced by PVN stimulation [14]. The date suggested that PVN regulating pain process may be through the EOP system at the spinal level. Ach system in the spinal cord is involved in the pain relieving effect [30]. Many studies have proven that there is interaction between Ach system and EOP system in central nerve system [17-19], which effects on the nociceptive suppression at the spinal level [44]. EOP induces Ach releasing evoked by K+ or glutamic acid [45], and Ach can cause the spinal cord releasing the neuropeptides [29]. Our present study showed that: 1) Pain stimulation increased L-Ek, β-Ep, DynA1-13, Ach and Ch concentrations in the spinal cord; 2) Ach increased L-Ek, β-Ep and DynA1-13 concentrations in the spinal cord; and 3) Atropine (M-receptor inhibitor) or hexahydric gallamine (N-receptor inhibitor) decreased LEk, β-Ep and DynA1-13 concentrations in the spinal cord. The data indicated that Ach antinociception at the spinal level was involved in the EOP system.
However, we have not investigated how Ach effects on the EOP system and where Ach comes from in the spinal cord during pain modulation.
5. ACKNOWLEDGEMENTS
This work was supported by Xinxiang Medical University, Kamp Institute for Medical Research, Jiangsu Su Bei People’s Hospital and grants from National Basic Research Program of China (2007CB936104) and the 863 National High Technology Research Development Program of China (2007AA021905).
REFERENCES
- Lightman, S.L. (1988) The neuroendocrine paraventricular hypothalamus: Receptors, signal transduction. mRNA and neurosecretion. Journal of Experimental Biology, 139, 31-49.
- Blair, M.L., Piekut, D., Want, A. and Olschowka, J.A. (1996) Role of the hypothalamic paraventricular nucleus in cardiovascular regulation. Clinical and Experimental Pharmacology and Physiology, 23, 161-165. doi:10.1111/j.1440-1681.1996.tb02590.x
- McEwen, B.B. (2004) The role of vasopressin and oxytocin in memory processing. Elsevier, Amsterdam.
- Taylor, T., Gesundheit, N., Gyves, P.W., Jacobowitz, D.M. and Weintraub, B.D. (1988) Hypothalamic hypothyroidism caused by lesions in rat paraventricular nuclei alters the carbohydrate structure of secreted thyrotropin. Endocrinology, 122, 283-290. doi:10.1210/endo-122-1-283
- Pickard, G.F. and Turek, F.W. (1983) The hypothalamic paraventricular nucleus mediates the photoperiodic control of reproduction but not the effect of light on the circadian rhythm of activity. Neuroscience Letters, 43, 67-72. doi:10.1016/0304-3940(83)90130-1
- Leibowitz, S.F., Weiss, G.F. and Suh, J.S. (1990) Medial hypothalamic nuclei mediate serotonin’s inhibitory effect on feeding behavior. Pharmacology Biochemistry and Behavior, 37, 735-742.
- Ciriello, J., Kline, R.L., Zhang, T.X. and Caverson, M.M. (1984) Lesions of the paraventricular nucleus alter the development of spontaneous hypertension in the rat. Brain Research, 310, 355-359. doi:10.1016/0006-8993(84)90159-8
- Kiss, J.Z. (1988) Dynamism of chemoarchitecture in the hypothalamic paraventricular nucleus. Brain Research Bulletin, 20, 699-708. doi:10.1016/0361-9230(88)90080-9
- Robinson, M.B. (2006) Acute regulation of sodium-dependent glutamate transporters: A focus on constitutive and regulated trafficking. Handbook of Experimental Pharmacology, 175, 251-275. doi:10.1007/3-540-29784-7_13
- Yang, J., Chen, J.M., Yang, Y., Liu, W.Y., Song, C.Y. and Lin, B.C. (2008) Investigating the role of hypothalamic paraventricular nucleus in nociception of the rat. International Journal of Neuroscience, 118, 473-485. doi:10.1080/00207450601123563
- Yang, J. and Lin, B.C. (1992) Hypothalamic paraventricular nucleus plays a role in acupuncture analgesia through the central nervous system in the rat. Acupuncture electrotherapeutics research, 17, 209-220.
- Yang, J., Song, C.Y., Liu, W.Y., Wang, W. and Lin, B.C. (2006) Through the central V2, not V1 receptors influencing the endogenous opiate peptide system, arginine vasopressin, not oxytocin in the hypothalamic paraventricular nucleus involves in the antinociception in the rat. Brain Research, 1069, 127-138. doi:10.1016/j.brainres.2005.11.045
- Kordower, J.H. and Bodnar, R.J. (1984) Vasopressin analgesia: Specificity of action and non-opioid effects. Peptides, 5, 747-756. doi:10.1016/0196-9781(84)90017-2
- Yang, J., Yang, Y., Chu, J.G., Wang, G., Xu, H.T., Liu, W.Y., Wang, C.H. and Lin, B.C. (2009) Endogenous opiate peptides in the spinal cord are involved in the analgesia of hypothalamic paraventricular nucleus in the rat. Peptides, 30, 740-744. doi:10.1016/j.peptides.2009.01.004
- Wang, D.X., Yang, J., Gu, Z.X., Song, C.Y., Liu, W.Y., Zhang, J., Li, X.P., Li, H., Wang, G., Song, C. and Lin, B.C. (2010) Arginine vasopressin induces rat caudate nucleus releasing acetylcholine to participate in pain modulation. Peptides, 31, 701-705. doi:10.1016/j.peptides.2009.11.027
- Yang, J., Chen, J.M., Liu, W.Y., Song, C.Y. and Lin, B.C. (2006) Arginine vasopressin in the caudate nucleus plays an antinociceptive role in the rat. Life Science, 79, 2086-2090. doi:10.1016/j.lfs.2006.07.005
- Lendvai, B., Sándor, N.T. and Sándor, A. (1993) Influence of selective opiate antagonists on striatal acetylcholine and dopamine release. Acta Physiologica Hungarica, 81, 19-28.
- Rada, P., Barson, J.R., Leibowitz, S.F. and Hoebel, B.G. (2010) Opioids in the hypothalamus control dopamine and acetylcholine levels in the nucleus accumbens. Brain Research, 1312, 1-9. doi:10.1016/j.brainres.2009.11.055
- Sandor, N.T., Lendvai, B. and Vizi, E.S. (1992) Effect of selective opiate antagonists on striatal acetylcholine and dopamine release. Brain Research Bulletin, 29, 369-373. doi:10.1016/0361-9230(92)90070-E
- Zimmermann, M. (1983) Ethical guidelines for investigations of experimental pain in conscious animal. Pain, 16, 109-110. doi:10.1016/0304-3959(83)90201-4
- Wang, C.H., Zhu, Y.X., Liu, Z., Song, C.Y. and Zhu, Y.X. (1987) Radioimmunoassay for dynorphinA1-13. Acta Pharmacological Sinia, 8, 494-497.
- Zhu, Y.X., Guan, X.B., Cui, Y.Y., Liu, Z., Song, C.Y. amd Lin, B.C. (1986) Preparation for anti-β-endorphin serum and its radioimmuniassay. Academic Journal of Second Military Medical University, 7, 332-335.
- Zhu, Y.X., Liu, Z., Song, C.Y. and Lin, B.C. (1986) Preparation for L-enkephalin serum and its application. Acta Zoologica Sinia, 32, 213-219.
- Eva, C., Hadjiconstantinou, M., Neff, N.H. and Meek, J.L. (1984) Acetylcholine measurement by high-performance liquid chromatography using an enzyme-loaded postcolumn reactor. Analytical Biochemistry, 143, 320-324. doi:10.1016/0003-2697(84)90670-5
- Zajac, P. and Kasperska-Zajac, A. (1986) The role of arginine vasopressin (AVP) in pain transmission and perception. Postepy Higieny i Medycyny Doswiadczalnej, 55, 829-834.
- Yang, J., Song, C.Y., Liu, W.Y. and Lin, B.C. (2006) Only through the brain nuclei, arginine vasopressin regulates antinociception in the rat. Peptides, 27, 3341-3346. doi:10.1016/j.peptides.2006.08.019
- Yang, J., Liu, W.Y., Song, C.Y. and Lin, B.C. (2006) Only arginine vasopressin, not oxytocin and endogenous opiate peptides, in hypothalamic paraventricular nucleus play a role in acupuncture analgesia in the rat. Brain Research Bulletin, 68, 453-458. doi:10.1016/j.brainresbull.2005.10.002
- Yang, J., Liu, W.Y., Song, C.Y. and Lin, B.C. (2006) Through central arginine vasopressin, not oxytocin and endogenous opiate peptides, glutamate sodium induces hypothalamic paraventricular nucleus enhancing acupuncture analgesia in the rat. Neuroscience Research, 54, 49-56. doi:10.1016/j.neures.2005.10.006
- Dussor, G.O., Helesic, G., Hargreaves, K.M. and Flores, C.M. (2004) Cholinergic modulation of nociceptive responses in vivo and neuropeptide release in vitro at the level of the primary sensory neuron. Pain, 107, 22-32. doi:10.1016/j.pain.2003.09.022
- Schechtmann, G., Song, Z., Ultenius, C., Meyerson, B.A. and Linderoth, B. (2008) Cholinergic mechanisms involved in the pain relieving effect of spinal cord stimulation in a model of neuropathy. Pain, 139, 136-145. doi:10.1016/j.pain.2008.03.023
- Hama, A.T., Lloyd, G.K. and Menzaghi, F. (2001) The antinociceptive effect of intrathecal administration of epibatidine with clonidine or neostigmine in the formalin test in rats. Pain, 91, 131-138. doi:10.1016/S0304-3959(00)00425-5
- Honda, K., Harada, A., Takano, Y. and Kamiya, H. (2000) Involvement of M3 muscarinic receptors of the spinal cord in formalin-induced nociception in mice. Brain Research, 859, 38-44. doi:10.1016/S0006-8993(99)02456-7
- Hughes, J., Smith, T.W., Kosterlitz, H.W., Fothergill, L.A., Morgan, B. and Morris, H.R. (1975) Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature, 258, 577-580. doi:10.1038/258577a0
- Goldstenin, A., Tachibana, S., Lowney, L.I., Hunkapiller, M. and Hood, L. (1979) Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proceedings of the National Academy of Sciences of USA, 76, 6666-6670. doi:10.1073/pnas.76.12.6666
- Millan, M.J. and Herz, A. (1985) The endocrinology of the opioids. International Review of Neurobiology, 26, 1-83. doi:10.1016/S0074-7742(08)60072-0
- Bodnar, R.J. (2010) Endogenous opiates and behavior: 2009. Peptides, 31, 2325-2359. doi:10.1016/j.peptides.2010.09.016
- Herz, A. and Millan, M.J. (1990) Opioids and opioid receptors mediating antinociception at various levels of the neuraxis. Physiologia Bohemoslovaca, 39, 395-401.
- Rosenfeld, J.P. (1994) Interacting brain stem components of opiate-activated, descending, pain-inhibitory systems. Neuroscience & Biobehavioral Reviews, 18, 403-409. doi:10.1016/0149-7634(94)90053-1
- Stamford, J.A. (1995) Descending control of pain. British Journal of Anaesthesia, 75, 217-227.
- Zhao, Z.Q. (2008) Neural mechanism underlying acupuncture analgesia. Progress in Neurobiology, 85, 355-375. doi:10.1016/j.pneurobio.2008.05.004
- Lapchak, P.A., Araujo, D.M. and Collier, B. (1989) Regulation of endogenous acetylcholine release from mammalian brain slices by opiate receptors: Hippocampus, striatum and cerebral cortex of guinea-pig and rat. Neuroscience, 31, 313-325. doi:10.1016/0306-4522(89)90376-X
- Oron, L., Sarne, Y. and Michaelson, D.M. (1991) Effect of opioid peptides on electrically evoked acetylcholine release from Torpedo electromotor neurons. Neuroscience Letters, 125, 231-234. doi:10.1016/0304-3940(91)90036-S
- Toide, K. (2006) Basic research on analgesia: Basis of pain and novel pain targets. Nippon Yakurigaku Zasshi, 128, 321-325. doi:10.1254/fpj.128.321
- Li, Y.J., Zhang, Z.H., Chen, J.Y. and Qiao, J.T. (1994) Effects of intrathecal naloxone and atropine on the nociceptive suppression induced by norepinephrine and serotonin at the spinal level in rats. Brain Research, 666, 113-116. doi:10.1016/0006-8993(94)90290-9
- Arenas, E., Alberch, J., Sanchez Arroyos, R. and Marsal, J. (1990) Effect of opioids on acetylcholine release evoked by K+ or glutamic acid from rat neostriatal slices. Brain Research, 523, 51-56. doi:10.1016/0006-8993(90)91633-R
NOTES
*Corresponding author.