Open Journal of Genetics
Vol.4 No.1(2014), Article ID:43464,6 pages DOI:10.4236/ojgen.2014.41003
A Research of Pachyonychia Congenita Type 1 and Literature Analysis
Kun Guo, Shengxiang Xiao*, Songmei Geng*
Department of Dermatology, The Second Affiliated Hospital of Xi’an Jiaotong University, Xi'an, China
Email: *xiao_sx@163.com, *gsm312@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 3 December 2013; revised 1 January 2014; accepted 30 January 2014
ABSTRACT
Pachyonychia congenital (PC), consist of a group of rare autosomal-dominant ectodermal disorders. Symmetrically thickened, dystrophic fingernails and toenails are the defining characteristic of pachyonychia congenita. There are two main clinical subtypes of pachyonychia congenita: Pachyonychia congenita-1 and pachyonychia congenita-2. Pachyonychia congenita-U is another subtypes of pachyonychia congenita, where either a mutation has not been found or has not been investigated. Objectives: The present aim was to indentify the mutation of keratin 6a or keratin 16 gene in the pachyonychia congenita patient. Methods: The proband, her parents and 100 unrelated controls were subjected to mutation detection in keratin 6a or keratin 16 gene. Direct sequencing of all PCR products of the whole coding regions of keratin 6a or keratin 16 was performed to identify the mutation. Results: No mutation was found in keratin 6a or keratin 16 in the proband, her parents, and 100 unrelated and unaffected people. Conclusion: This study reported a Chinese female affected with pachyonychia congenita-1 without mutation in keratin gene.
Keywords: Pachyonychia Congenital; Pachyonychia Congenital-U; Mutation

1. Introduction
Pachyonychia congenital (PC) consists of a group of rare autosomal-dominant ectodermal disorders characterized predominantly by nail dystrophy and painful palmoplantar keratoderma. There are two main clinical subtypes of PC: PC-1, recognized as Jadassohn-Lewandowsky syndrome and PC-2, recognized as Jackson-Lawler syndrome. PC-1is caused by mutations in keratin 6a (K6a) gene or keratin 16 (K16) gene, accompanied by nail dystrophy, severe palmoplantar keratoderma, follicular keratoses and oral leukokeratosis. PC-2 is linked to mutations in keratin 6b (K6b) gene or keratin17 (K17) gene, associated with nail dystrophy, focal palmoplantar keratoderma, natal teeth and multiple steatocysts, which are the most predominant clinical findings distinguished from PC-1 [1] [2] . The yellow and thickened fingernails and/or toenails which caused by PC, affected patients’ appearance, social activities, even severe psychological illnesses. So do research on the gene mutation of PC to find the relationship between genotype and phenotype, would be useful for prenatal counseling patients, prenatal diagnosis and targeted therapy.
2. Clinical Report
The proband was a 16-year-old Chinese girl with thick nails who presented to the Second Affiliated Hospital of Xi’an Jiaotong University in January 2012. She was affected with the thickened fingernails and toenails at 6 month of age (Figure 1). Physical examination showed dystrophic and hyperkeratotic fingernails and toenails (Figure 1). Palmoplantar keratoderma, steatocystoma multiplex, cutaneous blisters, hair abnormalities and natal teeth were not found in this patient. Repeated fungal examination under microscope and culture excluded onychomychosis.
3. Material and Methods
The Human Medical and Ethical Committee of Xi’an Jiaotong University approved the investigation presented here and all study subjects gave informed consent. Five milliliters of peripheral blood was obtained from the proband, her parents, and 100 unrelated and unaffected people. Genomic DNAs were extracted from peripheral blood by a kit according to the instruction and used a template for PCR-mediated amplification of exon of the K6a, K17 gene, which no mutations were found. We also amplificated the exon of K6b and K16 gene with the same result. So did her parents and the 100 unrelated controls.
4. Results
The proband diagnose as PC-1. But the direct sequencing of the PCR products which in the proband, her parents, and 100 unrelated and unaffected people revealed no mutation in K6a, K17, K6b and K16 gene. This proband always can be diagnosed as PC-U.
5. Discussion
The abnormal of keratins’ structure are closely with PC. Keratins are heterodimeric proteins responsed for forming the intermediate filament cytoskeleton of epithelial cells. Protein structure of keratins are similar consisting of a highly conserved central helical rod domain, non-helical linkers (L1, L12 and L2), variable domains V1 and V2. The conserved central helical rod domain flanked by variable domains V1 and V2 is divided into 1A, 1B, 2 A and 2B domains, which are connected by L1, L12 and L2. These highly conserved motifs are thought to be important in mediating end-to-end interactions during filament assembly. The helix boundary motifs are mutational hot spots for all keratin disorders, and most PC mutations reported to date have occurred in these regions [3] .
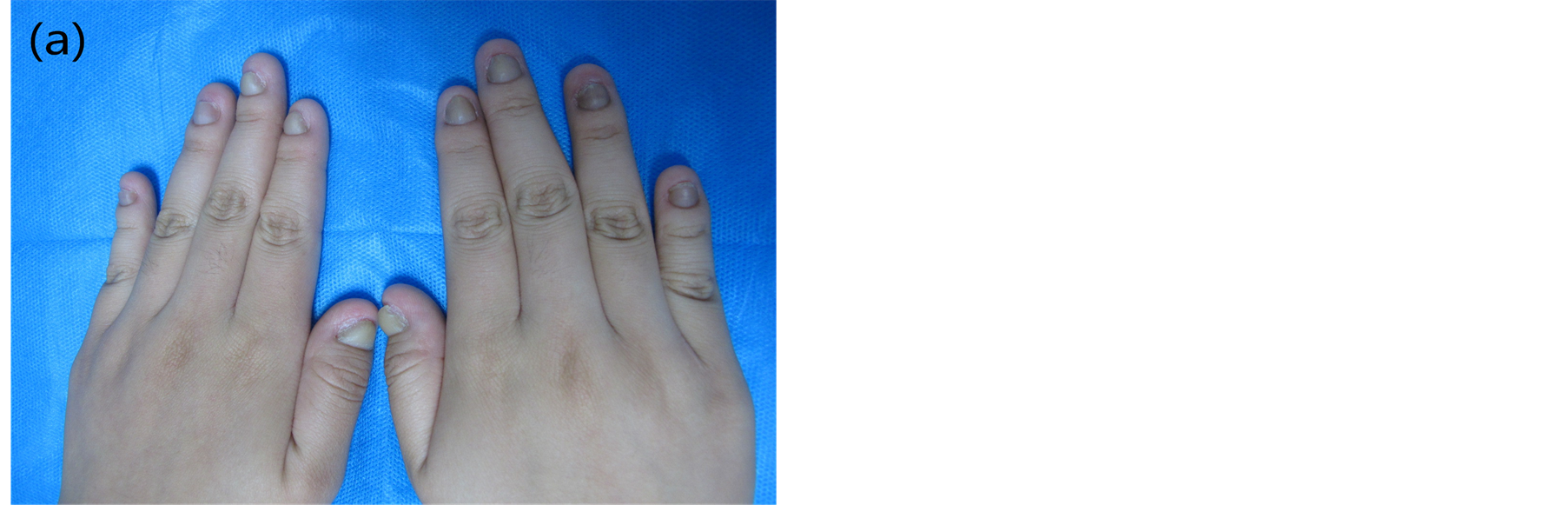
Figure 1. (a) Photograph of the thicken fingernails.
Amino acid substitution, deletion, or insert can change protein’ structure, which leading fracture or collapse of the cytoskeleton. The keratins associated with PC, K6a, K16, K6b and K17 are predominantly expressed in basal/suprabasal layers of palmoplantar skin, as well as in epidermal appendages and oral mucosa. Until November 2012, the number of new DNA Sequence Variant reported in K6a, K16, K17, K6b has been 35, 20, 23 and 3 respectively (http://www.interfil.org/). The most common mutations were substitution in PC compared with deletion mutation and insertion mutation, a mere 2 insertion mutations were reported all in K6a. 1A domain of keratin was far more frequent site than other domains, the second one being 2B. In PC-1, only 1 and 4 mutations were found in the tail and head domain of keratins respectely. However, no mutation was reported in these domains of keratin in PC-2.
The correlation between genotype and phenotype is more and more clearly due to the more reported numbers. The number of mutation reported association with PC-1, PC-2 is 167 and 66 respectively, include 57 mutations happened in K16 (Table 1), 105 mutations in K6a (Table 2).
Mutation in the helix boundary motifs produces more severe phenotype. The classical thicken nails usually begins after the first months of born in PC. More and more late onset PC has been found, which clinical observation of these patients with the onset of the characteristic nail changes of PC during the second and third decades of life for mutations in the non-helical domains or in the mid-region of the α-helical rod domain [4] [5] . Connors et al. detected the first case of late onset PC association with a mutation K354N in the central 2B domain of the K16 [6] . Terrinoni et al. reported a postzygotic mutation in the V1 domain of K16 with delayed onset PC-1 [7] . Cao et al. found a frame shift mutation in K16 causes the delayed onset PC-1 [8] . Our group has reported a mutation N109D in the second half of 1A domain of K17 with delayed onset PC-2

Table 1. Summary of reported mutations of K16 in pachyony congenita-1.
aPalmoplantar keratoderma, nonepidermolytic (focal); bEpidermolytic palmoplantar keratoderma; c mutantion.
Table 2. Summary of reported mutations of K6a in pachyony congenita-1.
dmutation.
[9] . These patients listed above have delayed onset of PC, the milder phenotypes. Their mutation located at the end of the helix 1A, the beginning of the helix 2B, V1 domain or V2 domain, which occurred in the less critical site of the keratins [6] [9] [10] . Mclean et al. advised cases of suspected PC, where either a mutation has not been found or has not been investigated use PC-U for short [3] [8] [11] .
The PC association with alopecia is very rare. Wilson et al. reported two PC-2 families with alopecia, and found homozygous dominant missense mutations in K17 [12] . K16, K17, K6a, K6b are specifically expressed in the hair follicle [13] , hereby we speculate the mutations of these genes would cause the abnormal hair growth influenced by the genetic and environmental factors.
6. Conclusion
In conclusion, we reported a Chinese female affected with PC-1or PC-U. Phenotype and severity depend on the genetic background and environmental factors as well as the underlying mutation [5] [14] . We did not find any mutation in this patient, but the report allow better prognostic predictions, patient counseling, and give some clues to the cause of the phenotypic variability [3] [15] .
Conflict of Interest
The authors state no conflict of interest.
Acknowledgements
We thank the patient and her family for their participation in this research.
References
- Leachman, S.A., Kaspar, R.L., Fleckman, P., Florell, S.R., Smith, F.J.D., McLean, W.H.I., et al. (2005) Clinical and Pathological Features of Pachyonychia Congenita. Journal of Investigative Dermatology Symposium Proceeding, 10, 3-17. http://dx.doi.org/10.1111/j.1087-0024.2005.10202.x
- Vishakha, M.S. and Sarah, L.S. (2007) A Novel Mutation in K6b in Pachyonychia Congenita Type 2. Journal of Investigative Dermatology, 127, 2060-2062. http://dx.doi.org/10.1038/sj.jid.5700814
- Wilson, N.J., Leachman, S.A., Hansen, C.D., McMullan, A.C., Milstone, L.M., Schwartz, M.E., et al. (2011)A Large Mutational Study in Pachyonychia Congenita. Journal of Investigative Dermatology, 131, 1018-1024. http://dx.doi.org/10.1038/jid.2011.20
- Paller, A.S., Moore, J.A. and Scher, R. (1991) Pachyonychia Congenital Tarda A Late-Onset Form of Pachyonychia Congenita. Archives of Dermatology, 12, 701-703. http://dx.doi.org/10.1001/archderm.127.5.701
- Zhou, H.L., Yang, S., Gao, M., Zhao, X.Y., Zhu, Y.G., Li, W., et al. (2007) A Novel Missense Mutation L468Q of Keratin 6a in Pachyonychia Congenita Type 1. Journal of the European Academy of Dermatology and Venereology, 21, 351-355. http://dx.doi.org/10.1111/j.1468-3083.2006.01930.x
- Connors, J.B., Rahil, A.K., Smith, F.J.D., McLean, W.H.I. and Milstone, L.M. (2001) Delayed Onset Pachyonychia Congenita Associated with a Novel Mutation in the Central 2B Domain of Keratin 16. British Journal of Dermatology, 144, 1058-1062. http://dx.doi.org/10.1046/j.1365-2133.2001.04199.x
- Terrinoni, A., Puddu, P., Didona, B., De Laurenzi, V., Candi, E., Smith, F.J.D., et al. (2000) A Mutation in the V1 Domain of K16 Is Responsible for Unilateral Palmoplantar Verrucous Nevus. Journal of Investigative Dermatology, 114, 1136-1140. http://dx.doi.org/10.1046/j.1523-1747.2000.00983.x
- Cao, L.H., Luo, Y., Wen, W., Liu, W.L., Jiang, L., Chen, C., et al. (2011) A Novel Frameshift Mutation in Keratin 16 Underlies Pachyonychia Congenita with Focal Palmoplantar Keratoderma. British Journal of Dermatology, 165, 1145- 1147. http://dx.doi.org/10.1111/j.1365-2133.2011.10450.x
- Xiao, S.X., Feng, Y.G., Ren, X.R., Tan, S.S., Li, L., Wang, J.M., et al. (2004) A Novel Mutation in the Second Half of the Keratin 17 1A Domain in a Large Pedigree with Delayed-Onset Pachyonychia Congenita Type 2. Journal of Investigative Dermatology, 122, 892-895. http://dx.doi.org/10.1111/j.0022-202X.2004.22408.x
- Steinert, P.M., Yang, J.M., Bale S.J. and Compton, J.G. (1993) Concurrence between the Molecular Overlap Regions in Keratin Intermediate Filaments and the Locations of Keratin Mutations in Genodermatoses. Biochemical and Biophysical Research Communications, 197, 840-848. http://dx.doi.org/10.1006/bbrc.1993.2555
- McLean, W.H., Hansen, C.D., Eliason, M.J. and Smith, F.J.D. (2011) The Phenotypic and Molecular Genetic Features of Pachyonychia Congenita. Journal of Investigative Dermatology, 131, 1015-1017. http://dx.doi.org/10.1038/jid.2011.59
- Wilson, N.J., Perez, M.L.C., Vahlquist, A., Schwartz, M.E., Hansens, C.D., McLean, W.H.I., et al. (2012) Homozygous Dominant Missense Mutation in Keratin 17 Leads to Alopecia in Addition to Severe Pachyonychia Congenita. Journal of Investigative Dermatology, 132, 1921-1924. http://dx.doi.org/10.1038/jid.2011.484
- Moll, R., Divo, M. and Langbein, L. (2008) The Human Keratins: Biology and Pathology. Histochemistry and Cell Biology, 129, 705-733. http://dx.doi.org/10.1007/s00418-008-0435-6
- Munro, C.S. (2001) Pachyonychia Congenita: Mutations and Clinical Presentations. British Journal of Dermatology, 144, 929-930. http://dx.doi.org/10.1046/j.1365-2133.2001.04216.x
NOTES

*Corresponding authors.