Open Journal of Genetics
Vol.4 No.4(2014), Article ID:47970,16 pages
DOI:10.4236/ojgen.2014.44028
Synthetic Lethality Induced by a Strong Drosophila Enhancer of Expanded Polyglutamine Tract
Ping Zhang, Qiming Wang, Hannah Hughes, Gino Intrieri
Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA
Email: ping.zhang@uconn.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 May 2014; revised 9 June 2014; accepted 8 July 2014
ABSTRACT
Proteins containing an expanded polyglutamine tract are neurotoxins. The expanded polyglutamine proteins influence a variety of cellular functions. In Drosophila the GMR-Gal4/UAS expression system has been widely used in an eye-based model to study human neurodegenerative diseases. This system has facilitated the isolation and characterization of abundant Drosophila genes that interact with the expanded polyglutamine proteins. We used the GMR-Gal4/UAS system to express three proteins containing an expanded polyglutamine tract, or an expanded polyglutamine tract alone. Doubling the dose of these proteins resulted in pupal lethality, indicating that these toxic proteins induced a sensitized condition that is prone to synthetic lethality. By using the GMRGal4/UAS system, we showed that a Drosophila gene interacts with three expanded polyglutamine proteins to induce a synthetic lethal phenotype. We further demonstrated that the synthetic lethality was mediated through the toxic expanded polyglutamine tract. Our study raises a possibility that conventional genetic screens may not recover synthetic lethal alleles, which are presumably stronger interacting alleles than the currently known modifiers of an expanded polyglutamine tract, due to synthetic lethality.
Keywords:Polyglutamine Diseases, Drosophila, Genetic Screen, GMR-Gal4/UAS System, Synthetic Lethal Mutations

1. Introduction
Nine dominant neurodegenerative disorders are caused by expansion of the unstable CAG trinucleotide repeats in gene coding regions, resulting in the production of expanded long glutamine proteins [1] . These diseases include Huntington’s disease (HD), dentatorubropallidoluysian atrophy (DRPLA), spinobulbar muscular atrophy (SBMA), and six types of spinocerebellar ataxias including Sca1, Sca2, Sca3, Sca6, Sca7 and Sca17. To study the expanded polyglutamine proteins (polyQ), the mouse model and small organism models including yeast, C. elegans and Drosophila have been employed [2] -[8] . Though the detailed mechanism of the expanded polyQ toxicity remains unclear, studies indicated that the mutated polyQ tract is intrinsically toxic in mice [9] , Drosophila [10] , and C. elegans [11] . Studies also demonstrated that the sequences surrounding an expanded polyQ tract influence the toxicity [12] -[16] .
Efforts to understand the neurotoxicity of the disease mutations include the identification of potential modifiers in the human genome from genome-wide linkage searches [17] . In the model organisms, large collections of modifiers, both loss-of-function and gain-of-function, have been identified through genetic screens [18] -[25] . The modifiers affect protein folding, protein quality control, transcriptional regulation, and mitochondrial function, among many others. From the diverse actions and interactions of the modifiers, the expanded polyQ proteins have emerged as pathogenic factors that affect cellular functions in a wide variety of pathways. However, it remains unresolved exactly how an expanded polyQ protein initiates the pathogenic pathways, or precisely, how an initial molecular interaction between the expanded polyQ protein and a cellular factor, or factors, triggers the development of the neurodegenerative diseases. It also remains unknown whether or not there is an early common pathogenic pathway for these diseases, since each of the disease proteins contains a toxic polyQ tract.
In Drosophila, a conditional binary gene expression system, the GMR-Gal4/UAS system, has been the choice of most genetic models to isolate and characterize Drosophila modifiers of neurodegenerative diseases [26] [27] . By using GMR (glass multiple reporter) that contains multiple copies of an eye enhancer element, this system targets gene activities in the Drosophila eyes that are largely dispensable for viability and fertility [28] -[30] . To model the expanded polyQ diseases in Drosophila, early investigations introduced the GMR-Gal4/UAS system to express human disease polyQ proteins in the Drosophila eyes, including HD, Sca1 and Sca3 [10] [18] [19] [31] . Expression of a toxic polyQ protein with GMR-Gal4/UAS induced retinal degeneration is often seen as a rough eye phenotype. This widely used gene expression system is sensitive and efficient in detecting modifiers that either enhance or suppress the eye defect. Here we showed that expression of a toxic polyQ protein in Drosophila with the GMR-Gal4/UAS gene expression system generates a condition that is not only sensitive for the eye morphology, but also is prone to lethality. We have also showed that, under a sensitized genetic background where a toxic polyQ tract is expressed, overexpressing a Drosophila gene, Dikar, with the GMR-Gal4/UAS system induced a synthetic lethal phenotype.
2. Materials and Methods
2.1. Drosophila Strains and Culture
Flies were raised on standard corn/agar media at 25˚C. Most of the alleles were obtained from the Bloomington Drosophila Stock Center. These included longGMR-Gal4 (No. 8605), shortGMR-Gal4 (No. 1104), UAS-Sca3- Q27 (No. 8149); UAS-Sca3-Q78 (No. 8150), UAS-41Q (No. 30540), P{SUPor-P}dikar (No. 13156), hs-FLP (No. 7), 2xEGFP (No. 6658), and Delta 2-3 (No. 1610). Three additional P{SUPor-P} insertions were also obtained from the center: KG09489 (63E1), KG02042 (67A7), and KG01319 (70C6). The d10135 chromosome was obtained from Berkeley Drosophila Genome Project. The d09108 allele was obtained from The Exelixis Collection at Harvard Medical School. An Httex1p-97QP allele on Chromosome 3 was obtained from Dr. Lawrence Marsh (University of California, Irvine).
2.2. Isolation of the d10135a Chromosome
We found that a Drosophila third chromosome caused a synthetic lethal phenotype in a genetic background where a transgene was expressed. The transgene, UAS-Sca3-Q78, encodes an expanded polyQ protein, which is a neural toxin [31] . The chromosome, d10135, carried two copies of a genetically engineer P element, XP, which could overexpress the adjacent endogenous Drosophila genes via the Gal4/UAS system [32] [33] . One of the XP elements on d10135 was located in 65C3 of the left arm, while the other was in 85A2 on the right arm. Flies homozygous for d10135 were viable and fertile, without obvious defects. When combined with a Gal4 driver, longGMR-Gal4, which expresses the Gal4 protein predominantly in the Drosophila eyes [34] , d10135 caused a rough eye surface defect.
To identify the XP element on the d10135 chromosome that is responsible for the rough eyes and the synthetic lethality, a combination of genetic and molecular approaches were taken. First, we used meiotic recombination to map the overexpressing mutation on d10135 that induced the eye defect. A series of 4 chromosomes were chosen and each of these chromosomes carried a P{SUPor-P} element on the left arm of Chromosome 3, in the locations of 63E1, 65C3, 67A7, and 70C6, respectively (Figure 1). P{SUPor-P} contains a yellow+ marker that gives rise to dark cuticle pigmentation in adults, in addition to a white+ marker. Females of the genotype y w/y w; +/+; d10135/P{SUPor-P} for each of the chromosomes were generated and crossed to males of genotype y w/Y; longGMR-Gal4/CyO; +/+. We asked if recombinant progeny, longGMR-Gal4/+; XP·P{SUPor-P}/+ were generated that displayed both the eye defect and yellow+ body color. All but one of the P{SUPor-P} elements produced abundant recombinants. The exception was the P{SUPor-P} element in 65C3, P{SUPor-P}dikar, that produced no recombinants (N > 1000). Since an XP element on d10135 was located in 65C3, the result showed that the overexpression mutation for the eye defect in the genotype containing d10135 and longGMR-Gal4 was caused by the XP element in 65C3.
In addition, recombination mapping was also used to locate the overexpression mutation on d10135 that interacted with UAS-Sca3-Q78 to generate the synthetic lethality. Females of the genotype y w/y w; +/+; d10135/ P{SUPor-P}dikar were generated and crossed to males of genotype y w/Y; longGMR-Gal4 UAS-Sca3-Q78/CyO; +/+. Because an interaction of d10135 with UAS-Sca3-Q78 caused lethality, the genotype of longGMR-Gal4 UAS-Sca3-Q78/+; d10135/+ would be lethal. If a recombination took place between an overexpression mutation on d10135 and P{SUPor-P}dikar, yellow-pigment progeny of longGMR-Gal4 UAS-Sca3-Q78/+ would be produced. However, progeny of this genotype were not recovered (N > 1000). Thus, the overexpression allele on d10135 failed to recombine with P{SUPor-P}dikar in 65C3, indicating that the XP in 65C3 on d10135 is responsible for both the eye defect and the interacting activity with UAS-Sca3-Q78.
These results showed a second XP, which is located on the right arm of the d10135 chromosome, was involved neither in the rough eye defect, nor in the synthetic lethality. To remove the XP element in 85A2 on d10135, a chromosome that contained the XP in 65C3 and a P{SUPor-P} in 70C6 (Figure 1(b)) was retained from the previous recombination experiment (Figure 1(a)). To separate these two transposons, a second round of recombination was carried out. Progeny that displayed the mini-w+ expression, which was presumably from

Figure 1. Isolation of the d10135a chromosome. (a) Schematic drawings of the two XP elements on the parental d10135 chromosome and four SUPor-P elements on Chromosome 3. The centromere (a dot) of Chromosome 3 (a line) was shown. On d10135, one XP is in 65C3 on the left arm and the other is in 85A2 on the right arm (arrows). The SUPor-P element insertions (arrowheads) were used in the mapping, within the locations of 63E1, 65C3, 67A7, and 70C6, respectively. (b) Recombination to isolate the XP-65C3 element. A recombinant chromosome, which carried the XP-65C3 element (arrow) and a SUPor-P in 70C6 (arrowhead), gave rise to red-eyes and dark cuticle pigmentation (w+ y+). After another recombination between the XP and the SUPor-P (a cross symbol), a progeny that showed red eyes, but yellow cuticle, was selected. The reporter genes on XP and SUPor-P are w+ and w+ y+, respectively.
the XP in 65C3, and were yellow in body color, which indicated that the P{SUPor-P} in 70C6 was removed, were isolated. From one of these progeny, a strain was established to contain a d10135 derivative chromosome, d10135a, which carried only the XP element in 65C3. Further tests showed that this chromosome retained the characteristics of inducing the eye defect by GMR-Gal4, and interacting with UAS-Sca3-Q78.
2.3. UAS-Httex1p-97QP Alleles
UAS-Httex1p-97QP-w and UAS-Httex1p-97QP-s are two alleles on Chromosome 2. They were isolated by remobilizing a UAS-Httex1p-97QP insertion on Chromosome 3 by using a transposase source (Delta 2 - 3).
2.4. Primers for Cloning and Sequencing
a) Derived from genomic sequences P-Ge-hh: 5’TTGCGCTCCTGGTTAATTTT3’
P-Ge-1: 5’GCGCAGATAGCCAGATTAAC3’
P-Ge-2: 5’GCGCGAACTCTTTAACATAC3’
b) Derived from P element sequences P-XP-a: 5’AACGACGCATTTCGTACTCC3’ (1610 bp internal to the XP 5’-end with an outward orientation).
Pp31: 5’CGACGGGACCACCTTATGTTATTTCATCATG3’ (the 31-bp inverted terminal repeat of the P element and with an outward orientation).
PE5’: 5’AATTCGTCCGCACACAAC3’ (112 bp internal to the 5’ end of the P element and with an outward orientation).
PE3’: 5’TCGCACTTATTGCAAGCA3' (72 bp internal to 3’ end of the P element and with an outward orientation).
31A: 5’CGACACTCAGAATACTATTCC3’ (99 bp internal to 3’ end of the P element and with an outward orientation).
2.5. Immunostaining
Third instar larval imaginal eye discs were dissected and prepared for staining in a phosphate-buffered saline solution (PBS, 130 mM NaCl, 7mM Na2HPO4·2H2O, 3 mM NaH2PO4·2H2O, pH 7.0). The anti-HA staining procedures were adapted from Warrick et al. [31] . Prior to microscopic examination, DAPI (0.5 µg/mL) was added to the mounting solution (Prolong antifade, Molecular Probes, Inc.).
3. Results
3.1. A Drosophila Chromosome Interacted with a Transgene That Encodes an Expanded PolyQ Stretch
The d10135a chromosome was found to cause a synthetic lethal phenotype when combined with a transgene that expressed an expanded polyQ protein. It carried a genetically engineered XP element in 65C3. Because the XP element contains UAS constructs that are in an outward orientation at both ends of the transposon, it allows Gal4-driven overexpression of adjacent endogenous Drosophila genes [32] . When combined with the longGMR-Gal4 driver, d10135a caused eye defects, seen as a rough eye surface with conspicuous irregularly arranged bristles (Figure 2(a)). The rough eye defect suggests that a gene on d10135a was under the control of an UAS element within the XP insertion, and was overexpressed in the presence of longGMR-Gal4. The analysis also showed that overexpressing the gene on d10135a, UAS-d10135a, with longGMR-Gal4 did not induce other visible defects, though a slight reduction of pupal hatch rate was observed (~92% survival rate, N = 313, Figure 2(b)).
The UAS-Sca3-Q78 transgene encodes a protein with a 78 polyglutamine stretch that is a neurotoxin [31] . When driven by longGMR-Gal4, UAS-Sca3-Q78 induced damage to the adult eyes, seen as some loss of eye pigments (Figure 2). In addition, expressing UAS-Sca3-Q78 with longGMR-Gal4 was associated with a slightly reduced pupal hatching rate (~90% survival rate, N = 154, Figure 2). In the presence of longGMR-Gal4, a genotype containing UAS-Sca3-Q78 and the d10135a chromosome induced a lethal phenotype that was seen as

Figure 2. Eye defects and synthetic lethality associated with the d10135a chromosome. (a) Eye defects associated with three genotypes that expressed UAS-Sca3-Q78, UAS-d10135a, or coexpressed UAS-Sca3-Q78 and UAS-d10135a (rare escapers), by using the longGMR-Gal4 driver. The d10135a genotype without a driver showed wild type eyes. (b) Pupal survival rates of six genotypes that contained longGMR-Gal4, or shortGMR-Gal4. The pupal survival rate for each genotype was the percentage of pupae that emerged to adults: Pupal survival rate = No. of empty pupal cases (hatched adults)/No. of total pupal cases.
dead pupae, with only occasional adult escapers (~0.9% survival rate, N = 218, Figure 2). The rare escapers lost nearly all of the pigments in the eyes (Figure 2), and died within days after their hatch from the pupal cases. In conjunction with the eye defects, the lethal phenotype associated with the d10135a chromosome suggests that expression of a gene on the d10135a chromosome interacted with the UAS-Sca3-Q78 transgene, and significantly enhanced the toxicity of the expanded polyQ gene, resulting in a synthetic lethal phenotype.
There are two forms of the GMR-Gal4 drivers, longGMR-Gal4 and shortGMR-Gal4. longGMR-Gal4 carries pentamerized 39 bp glass-binding sites and it was used much less frequently than shortGMR-Gal4 that carries pentamerized 28 bp glass-binding sites [30] [34] . The unusual synthetic lethality associated with the longGMRGal4 driver could be attributed at least to three major factors. First, a chromosome position effect might have allowed the longGMR-Gal4 insertion to act in a non-eye tissue type that is essential for viability. Second, longGMR-Gal4 could hold an additional expression pattern that is drastically different from that of shortGMR-Gal4, inducing a non-eye expression. Third, the synthetic lethality could be caused by a general property of the GMRGal4 drivers that induce Gal4/UAS expression in a tissue type that is essential for viability. In a study that replaced the longGMR-Gal4 with a shortGMR-Gal4 driver, similar eye defects and synthetic lethality were both seen for the d10135a chromosome (Figure 2). Since the two independently generated GMR-Gal4 drivers induced the synthetic lethal phenotype, the results showed that it was a general property of the GMR-Gal4 drivers that induced the synthetic lethal phenotype.
3.2. Overexpression of the Dikar Gene by longGMR-Gal4 Caused Eye Defects and a Synthetic Lethal Phenotype When Coexpressed with UAS-Sca3-Q78
The XP element in 65C3 on d10135a was inserted into the promoter region of a computer-predicted gene, CG42799 or Dikar (Figure 3(a)) [32] [33] . The XP element has two UAS sites oriented outward at each end of the transposon, and one of them is flanked by a pair of FRT sites that are in a direct repeat orientation (Figure 3(b)). Upon exposure to the FLPase (Figure 4), the synthetic lethal genotype of longGMR-Gal4 UAS-Sca3-Q78/+; d10135a/+ was reverted frequently, suggesting that the UAS site flanked by the FRT pair in d10135a was responsible for inducing the synthetic lethality. Five independently isolated d10135a mutants, (-UAS)d10135a,

Figure 3. The Dikar gene and UAS-Dikar-a allele. (a) The genomic region of the Dikar gene (blue) and the predicted transcripts, with a gene span approximately from 6737k to 6721k of the genome coordinate, which was adapted from Flybase [33] . The P{XP}d10135a insertion was located at 6,736,213 in the Dikar’s first exon. ORFs are shown in orange. (b) The P{XP}d10135a insertion had a 3’ to 5’ P element orientation, as indicated by 3’-Pend and 5’-Pend. It contained two UAS elements in outward orientations, both of which could overexpress the flanking genomic sequences (dashed lines with arrows). The UAS elements nearby the 5’-Pend is flanked by two FRT sites in a direct repeat orientation (green arrowheads). FLPase treatment deleted the UAS element between the FRT sites (Del, deletion). Relative positions of a pair of PCR primers (arrows) are also shown. P-XP-a is derived from the XP sequence proximal to the FRT-UAS-FRT construct, while P-Ge-hh is derived from the Dikar’s genomic sequence (blue). (c) PCR products by using the P-XP-a and P-Ge-hh pair. Six genomic samples were used as templates for the PCR: d10135a from the parental d10135a strain; (-UAS)-1 through (-UAS)-5 from 5 independently generated mutants of the d10135a insertion.

Figure 4. Deletion of a UAS construct that is flanked by a pair of FRT sites in d10135a. To express FLPase, the genotype of hs-FLP y w/y w; +/+; d10135a/TM3, Sb was treated with heat shocks. longGMR-Gal4 UAS-Sca3-Q78/+; d10135a/+ was a synthetic lethal genotype with only rare escapers that showed severe eye defects. After treatment with FLPase, a large number of the genotype, longGMR-Gal4 UAS-Sca3-Q78/+; (-UAS)d10135a/+, were produced. In heat shock, larvae were treated to activate hs-FLP expression as described in [35] . (-UAS)d10135a, a d10135a mutant, in which one of the UAS sites was deleted; lGMR, longGMR-Gal4; Q78, UAS-Sca3-Q78.
were retained. Further tests showed that the (-UAS)d10135a mutants could no longer induce the eye defects in the presence of longGMR-Gal4, neither could they interact with UAS-Sca3-Q78. PCR experiment was used to confirm the loss of the UAS element flanked by the pair of the FRT sites on d10135a, by using a primer pair, P-XP-a and P-Ge-hh (Figure 3(b)). The predicted sizes of the PCR products for the d10135a chromosome and the mutant chromosome that lost the UAS, (-UAS)d10135a, were 1641 bp and 848 bp, respectively. As predicted, the (-UAS)d10135a mutants produced a PCR product of approximately 850 bp, which is smaller than that of the parental d10135a (approximately 1650 bp) (Figure 3(b) and Figure 3(c)). Thus, the results showed that overexpression of a Drosophila gene from the UAS element flanked by a pair of FRTs was the cause of both the eye defects and the interaction with UAS-Sca3-Q78. Several lines of evidence suggest that this Drosophila gene is a computer predicted gene with an annotation ID No. CG42799 or Dikar. First, the d10135a XP insertion is located in the promoter of the Dikar gene, generating a UAS-Dikar fusion gene (UAS-Dikar-a) with the UAS element franked by a pair of FRTs. Second, unlike enhancer elements in metazoans, UAS could only activate a promoter within a short range of a few hundred base pairs [36] -[38] . Third, since the nearest neighboring gene downstream of Dikar is over 23,000 base pairs away, it is unlikely that the UAS element in the UAS-Dikar-a allele could control endogenous Drosophila genes other than Dikar. Hereafter, we will assume that the Drosophila gene, which could interact with UAS-Sca3-Q78 when overexpressed, is the computer-generated Dikar gene.
3.3. UAS-Dikar-a Interacted with the Expanded PolyQ Tract
In addition to the expanded polyQ proteins, mRNAs that contain the CAG repeats encoding the toxic polyQ tracts could also be toxic [39] . Thus, the modifying effect of the Dikar gene on UAS-Sca3-Q78 might result from Dikar’s interaction with the transgene’s transcript, or its protein product, or both. To investigate how Dikar interacted with UAS-Sca3-Q78, another transgene, UAS-Httex1p-97QP was used. UAS-Httex1p-97QP expressed an expanded polyQ stretch, but its 97 glutamines were encoded by CAG/CAA alternate codons [40] . If Dikar’s interaction with UAS-Sca3-Q78 was mediated through the CAG repeats in the mRNA of the UAS-Sca3-Q78 transgene, a similar interaction with UAS-Httex1p-97QP then would not occur. However, if the interaction was mediated through the expanded polyQ tract, Dikar should likewise interact with the UAS-Httex1p-97QP transgene, because UAS-Httex1p-97QP also encodes a toxic polyQ tract.
As shown in Methods, two alleles of UAS-Httex1p-97QP on Chromosome 2 were generated by remobilizing a transposon carrying the transgene on Chromosome 3 into different chromosome locations, including a weak allele, UAS-Httex1p-97QP-w, and a strong allele, UAS-Httex1p-97QP-s. Expressing either of the UAS-Httex1p- 97QP alleles by longGMR-Gal4 caused eye defects, seen as slightly depigmented and glossy (Figure 5), but no significant viability problems were detected for genotypes expressing either allele (Figure 6). When UAS-Dikar-a was coexpressed with UAS-Httex1p-97QP-s, it interacted with UAS-Httex1p-97QP-s, as it did with UAS-Sca3-Q78.
As shown in Figure 6, a genotype coexpressing UAS-Dikar-a and UAS-Httex1p-97QP-s was synthetically le-
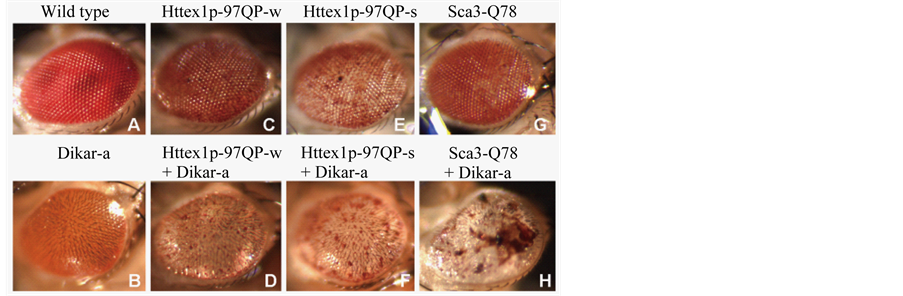
Figure 5. Eye defects induced by expressing UAS-Dikar-a, UAS-Httex1p-97QP-w, UAS-Httex1p-97QP-s, and UAS-Sca3-Q78 by longGMR-Gal4. Individually expressing the UAS-genes by longGMR-Gal4 induced loss of eye pigments (B, C, E, and G). Coexpressing UAS-Dikar-a with each of the transgenes that encoded a toxic polyQ stretch induced severe eye pigment loss (D, F, and H). Coexpressing Dikar-a with Sca3-Q78 also frequently induced grossly distorted eye morphology (H). Images in F and H were taken from rare escapers of the corresponding synthetic lethal genotypes.
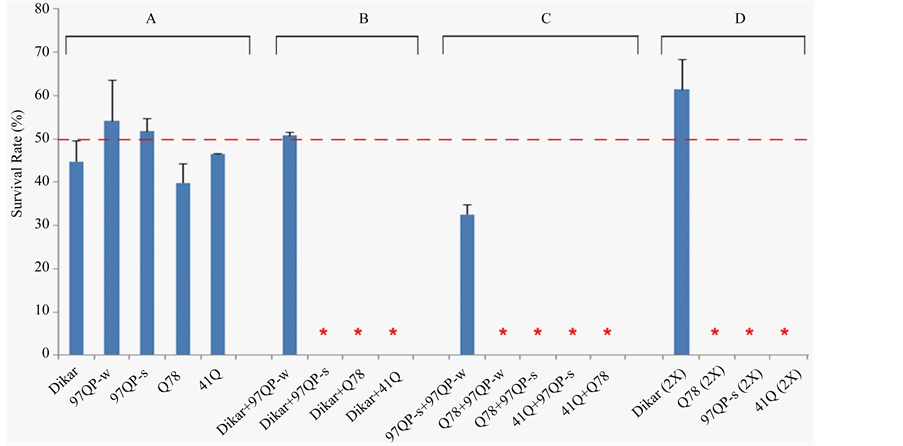
Figure 6. Synthetic lethality and synthetic-like lethality resulting from expressing transgenes by longGMR-Gal4. (A) Genotypes individually expressing UAS-Dikar-a, UAS-Httex1p-97QP-w, UAS-Httex1p-97QP-s, UAS-Sca3-Q78, or UAS-41Q. (B) Genotypes coexpressing UAS-Dikar-a and an expanded polyQ transgene. Synthetic lethality was shown for three genotypes. (C) Genotypes coexpressing two expanded polyQ transgenes. Synthetic lethality was shown for four genotypes. (D) Genotypes expressing two copies of each transgene (2X). Each genotype in the figure was generated from a cross, along with another genotype containing a CyO balancer. Thus, from each cross, the genotype that expressed a transgene, or coexpressed two transgenes, accounted for approximately 50% of the total progeny (red dashed line) when the overexpression did not cause a lethal effect. However, when a detrimental effect was induced by expressing the UAS-gene individually, or coexpressing the UAS-genes, the Cy+ progeny accounted for significantly less than 50% of the total progeny from the cross. Dikar, UAS-Dikar-a; 97QP-w, UAS-Httex1p-97QP-w; 97QP-s, UASHttex1p-97QP-s; Q78, UAS-Sca3-Q78; 41Q, UAS-41Q. These UAS-transgenes are all homozygous viable alleles. An asterisk indicates a synthetic lethal phenotype, or synthetic-lethal-like phenotype. The crosses to generate the indicated genotypes from left to right were carried out in vials (Table 1).
thal with rare escapers. In addition, the escapers showed severe eye defects, similar to that of the escapers of longGMR-Gal4 UAS-Sca3-Q78/+; UAS-Dikar-a/+ (Figure 5). Though a genotype coexpressing UAS-Dikar-a and UAS-Httex1p-97QP-w showed good viability, it was associated with severe eye damage (Figure 5, Figure 6).
The similarity between UAS-Dikar-a’s interactions with UAS-Sca3-Q78 and UAS-Httex1p-97QP-s suggests that the interactions are mediated through the toxic polyQ stretch, which is shared in the gene products of both UAS-Sca3-Q78 and UAS-Httex1p-97QP-s. If so, similar to these interactions with the Sca3-Q78 and Httex1p- 97QP-s proteins, interactions of the Dikar gene would occur with any protein containing an expanded polyQ tract. The UAS-41Q transgene encodes a protein that contains a 41 glutamine stretch but it contains no flanking domains [19] . As with UAS-Sca3-Q78 and UAS-Httex1p-97QP-s, a synthetic lethal interaction of UAS-Dikar-a with UAS-41Q was seen for the genotype of longGMR-Gal4/+; UAS-Dikar-a/UAS-41Q (Figure 6).
Taken together, the results showed that UAS-Dikar-a interacted with three transgenes, UAS-Sca3-Q78, UASHttex1p-97QP-s, and UAS-41Q. Because the products of these transgenes shared neither mRNA homology, nor a flanking domain, the Dikar’s interactions were mediated through the toxic polyQ stretches in each of the disease proteins, enhancing the neurodegenerative phenotypes in the eyes and possibly other tissue types that are essential for viability.
As a control, we examined the effect of coexpressing UAS-Dikar-a and a transgene, UAS-Sca3-Q27, which encodes a non-toxic 27 glutamine tract [31] , with the GMR-Gal4 drivers. The data showed that no significant interaction between UAS-Dikar-a and UAS-Sca3-Q27, on the eye defects, or viability.
3.4. Lethal Interaction between Two Toxic PolyQ Transgenes and Dose-Dependent Lethal Phenotypes
In addition to the interactions between Dikar and the toxic polyQ proteins, synthetic lethality resulted from ap

Table 1 .Crosses to generate the indicated genotypes from left to right in Figure 6.
lG, longGMR-Gal4; Dikar, UAS-Dikar-a; 97QP-w, UAS-Httex1p-97QP-w; 97QP-s, UAS-Httex1p-97QP-s; 41Q, UAS-41Q; T(2;3), T(2;3)CyO-TM6.
parent interactions between two expanded polyQ proteins were also seen. These included coexpressing UASSca3-Q78 and UAS-Httex1p-97QP-w, coexpressing UAS-Sca3-Q78 and UAS-Httex1p-97QP-s, coexpressing UAS-41Q and UAS-Httex1p-97QP-s, and coexpressing UAS-41Q and UAS-Sca3-Q78 (Figure 6(c)). Thus, similar to the synthetic lethality from Dikar’s interactions with the toxic polyQ stretch, synthetic lethality was also induced by coexpressing two expanded polyQ proteins.
Though all of these UAS-transgene alleles are homozygous viable, most of the genotypes expressing two copies of a disease transgene with longGMR-Gal4 were lethal, too. This dose-dependent lethal phenotype, or synthetic-like lethal phenotype, was seen for three genotypes, each of which expressed a single expanded polyQ protein at a higher level from two copies of a UAS-transgene, including UAS-Sca3-Q78, UAS-Httex1p-97QP-s, and UAS-41Q (Figure 6(d)).
However, unlike the transgenes that encode the toxic polyQ stretches, increasing the dose of UAS-Dikar-a appeared to have very little toxic effects, or no toxic effects at all, on viability, since a genotype expressing two copies of the UAS-Dikar-a allele with longGMR-Gal4 was viable (Figure 6(d)). In fact, the level of the Dikar’s expression could be further elevated without causing a serious problem for viability, because a stock containing two copies of the longGMR-Gal4 driver and two copies of the UAS-Dikar-a allele was successfully established.
3.5. Interaction of UAS-Dikar-a with the Expanded PolyQ Proteins Is a General Property of the Dikar Gene
Modifiers of the expanded polyQ proteins have been investigated intensively since these disease proteins were introduced into Drosophila in 1998 [31] [41] . Because UAS-Dikar-a was the only Drosophila allele that showed synthetic lethality with the expanded polyQ proteins, it was necessary to determine if this rarity was associated with an unusual UAS-Dikar allele that expressed its product at a higher level than the normal UAS-alleles. For example, at a low frequency, the newly generated XP insertion could be activated by the transposase, and its structure could be altered due to a local transposition onto the XP element, followed by more complex events including gap repair [42] -[45] . These secondary events, following the initial XP element insertion in Dikar, could give rise to a stronger UAS-expression structure, by adding additional GMR sequences on top of the 5 copies of the glass-binding sites present in the XP transposon.
To address a question of whether the strong interaction was unique to the UAS-Dikar-a allele, three additional UAS-Dikar alleles were generated independently from a local transposition experiment. Local transposition occurs nearby a starting P element at high frequency and the insertion sites are nearly entirely within the promoter regions of the targeted genes [44] [45] . Thus, the characteristics of local transposition, i.e., high frequency and promoter targeting, were ideal for isolating new alleles of UAS-Dikar. P{XP}d09108 is an XP insertion located nearby the Dikar promoter region and is only 635 bp upstream of the P{XP}d10135a insertion (Figure 7 and Figure 8). The d09108 insertion, which was reconfirmed by cloning and sequencing (Data not shown), did not show a rough eye defect when it was driven by GMR-Gal4. In a local transposition experiment, d09108 was used as a starting XP element to generate new alleles of UAS-Dikar alleles, in a two-step genetic screen (Figure 7). Briefly, in the first step of the genetic screen, the d09108 XP was activated by a transposase source to generate new insertions that showed the Dikar’s rough eye defect, as shown Figure 2, when combined with the shortGMR-Gal4 driver. In the second step of the genetic screen, the isolated insertions were investigated individually to determine if they interacted with the expanded polyQ proteins as UAS-Dikar-a did. The genetic
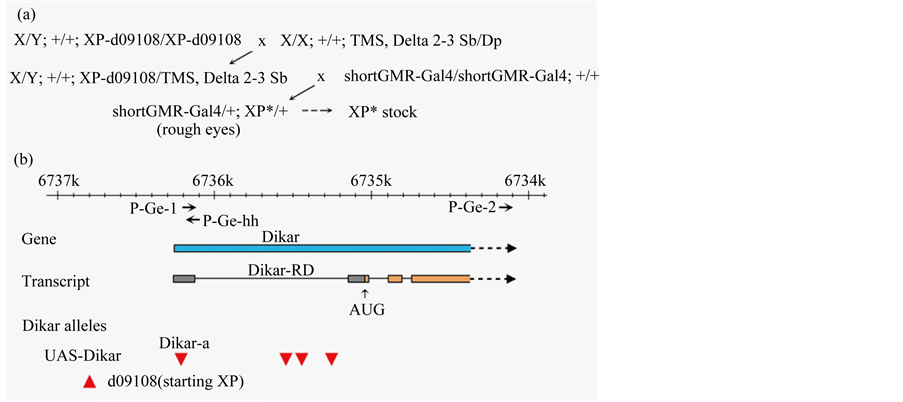
Figure 7. Additional UAS-Dikar alleles. (a) A local transposition from the nearby XP-d09108 element. XP* indicates a candidate for harboring a new XP insertions in Dikar. (b) Dikar al leles and the genomic sites of the corresponding P element insertions. An approximately 4 kb genomic region of the 5’ Dikar gene with a presumed transcription and translation initiation sites is shown [33] . Four UAS-Dikar alleles were produced by the insertions of the XP element (red triangles). UAS-Dikar-a on d10135a is at position {6,736,213}. Three additional UAS-Dikar alleles in the first intron, at positions of {5,735,543}, {5,735,442}, and {5,735,255}, were generated from remobilizing the XP-d09108 at {6,736,848}. Three PCR primers are indicated by arrows, P-Ge-1, P-Ge-2, and P-Ge-hh. A downward triangle indicates a P element insertion in a 3’ to 5’ orientation, while an upward triangle indicates a 5’ to 3’ orientation.
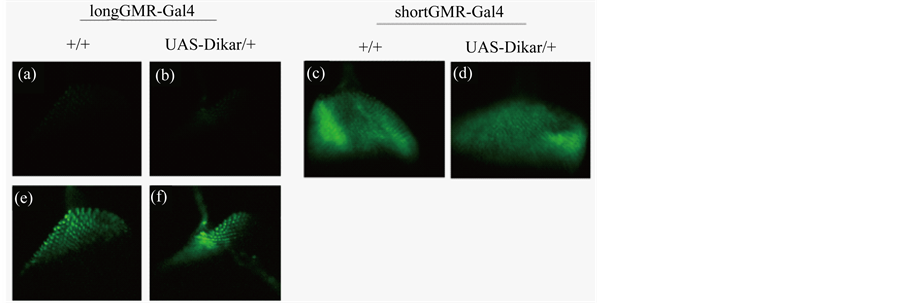
Figure 8. The effect of overexpressing Dikar on the Gal4/UAS system. A GFP reporter gene, UAS-2xEGFP, was used to examine the Gal4/UAS binary expression system. Representative images were taken for third instar larval imaginal eye discs of four genotypes. The signals from expressing the UAS-2xEGFP reporter gene were captured under identical exposure conditions (Panel (a) through (d)) to allow a direct comparison of the signal strengths between longGMR-Gal4 and shortGMRGal4. The images from Panel (e) and (f) were identical to those in Panel (a) and (b), except that they were digitally enhanced by increasing the brightness level.
screen was followed by molecular analyses, including cloning and sequencing with a set of primers derived from the genomic sequences and the P element sequences. The precise sites of the newly isolated XP insertions in the Dikar promoter region were then determined.
The molecular results showed that three newly isolated XP insertion sites are within the first intron of the Dikar gene, upstream of the translation initiation codon (Figure 7). We also determined that all these UAS-Dikar alleles induced synthetic lethality when coexpressed with an expanded polyQ tract by GMR-Gal4. Thus, a total of four UAS-Dikar alleles have been examined, which were independently isolated and dispersed within, or nearby, the promoter region of the Dikar gene. Since all four alleles showed identical phenotypes, including rough eyes when combined with a GMR-Gal4 driver, and synthetic lethality when coexpressed with the expanded polyQ proteins, the data indicate that the interaction between Dikar and an expanded polyQ stretch is a general property of the Dikar gene.
3.6. Effect of over Expressing Dikar on the Gal4/UAS Expression System
Dikar is a putative protein coding gene in Drosophila, though its function remains unknown. It is neither required for viability nor fertility [46] , while its mammalian homologue, the Cecr2 gene, plays an important role in neuronal development [47] [48] . The Dikar and Cecr2 proteins share a HAT domain (histone acetyltransferase) and they are components of ISWI complexes that covalently modify chromatin and regulate transcription [46] -[48] . Since Dikar is predicted to act in transcriptional regulation, overexpressing Dikar might enhance the Gal4/UAS gene expression system, which could lead to higher expression of the UAS-transgenes. It could subsequently give rise to higher toxicity, resembling the synthetic-like lethality from a double dose of a disease transgene (Figure 6). To ask if overexpressing Dikar had an effect on the Gal4/UAS expression system, an enhanced GFP report gene, UAS-2xEGFP [49] , was used. We constructed a pair of genotypes, longGMR-Gal4/+; UAS-2xEGFP/+ and longGMR-Gal4/+; UAS-2xEGFP/UAS-Dikar-a. The strength of the green fluorescence signals from these genotypes in the third instar larval eye discs was similar, suggesting that overexpressing Dikar had very little or no effect on the Gal4/UAS binary expression system (Figure 8). An additional pair of genotypes, shorGMR-Gal4/+; UAS-2xEGFP/+ and shortGMR-Gal4/+; UAS-2xEGFP/UAS-Dikar-a, was also constructed. Similar to that of the first pair, no significant difference of the GFP signals was seen between the second pair of genotypes (Figure 9), though the overall GFP signals from the shortGMR-Gal4 driver were much stronger than that of longGMR-Gal4, as expected [30] [34] . To further examine the effect of overexpressing Dikar on the Gal4/UAS expression system, expression of the UAS-Sca3-Q78 gene by using longGMR-Gal4 was also examined. This was facilitated by immunohistochemistry to detect the HA-tag that was fused to the Sca3- Q78 coding sequence in the UAS-Sca3-Q78 transgene [31] . The results showed that, in the presence or absence
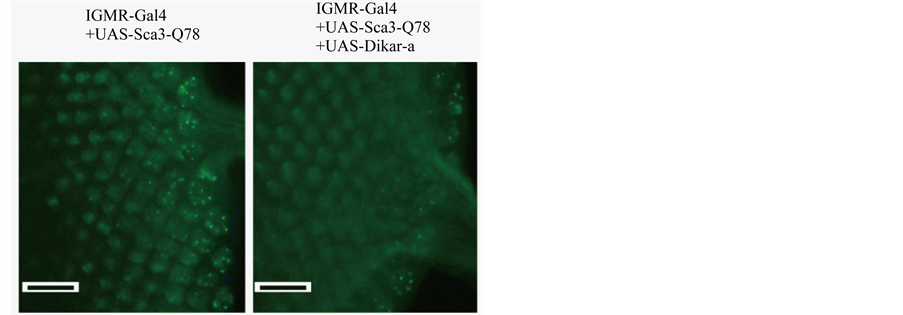
Figure 9. The effect of overexpressing Dikar on the Sca3-Q78 protein. A pair of the genotypes that expressed the UAS-Sca3-Q78 transgene with longGMR-Gal4 was examined. One of the genotypes expressed the HA-tagged Sca3-Q78 protein (green) in the presence of the UAS-Dikar-a allele. The protein was detected with anti-HA antibody. Accumulation of the protein was seen in the developing ommitidia (from left to right), while the fluorescent foci presumably resulted from nuclear localization of the protein [31] . Bar, 20 µM.
of the UAS-Dikar-a allele, the antibody staining against the HA-tag was similar in the third instar larval imaginal eye discs of a pair of genotypes, longGMR-Gal4 UAS-Sca3-Q78/+; +/+ and longGMR-Gal4 UAS-Sca3- Q78/+; UAS-Dikar-a/+ (Figure 9). Thus, the data from the GFP reporter and from the im munostaining suggest that overexpressing Dikar has no significant effect on the Gal4/UAS expression system, though Dikar is predicted to play a role in transcriptional regulation by selectively modifying chromatin.
4. Discussion
4.1. Expanded PolyQ and GMR-Gal4/UAS
The GMR-Gal4/UAS gene expression system was introduced into Drosophila, more than a decade ago, to study the expanded polyQ neurodegenerative diseases by expressing the neuronal toxic polyQ proteins in the eyes [31] [41] . The introduction has been followed by bursting investigations into all mechanistic aspects of the expanded polyQ neurodegenerative proteins by using the GMR-Gal4/UAS system. For example, to study Huntington’s disease, an array of transgenes was constructed to express various forms of the disease htt protein under the control of the GMR-Gal4/UAS system [24] . The GMR-Gal4/UAS system has also provided an efficient and sensitive tool for several genome-wide screens to identify modifiers that suppress or enhance the toxicity of the expanded polyQ proteins [18] [19] [23] -[25] . In addition to the loss-of-function modifiers, a large number of conditional gain-of-function (GOF) modifiers that could be overexpressed by using the GMR-Gal4 drive were also recovered from the screens. To isolate the GOF modifiers from the screens with the GMR-Gal4 driver, several collections of inducible UAS-GOF alleles were either obtained from an existing source [18] , or freshly isolated [19] [23] . These inducible GOF alleles were generated by the insertions of the EP element, which carried a UAS-expression construct in an outward orientation, similar to that of the XP element, to overexpress endogenous genes flanking the inserted EP elements in the presence of a GAL driver [50] .
Using the GMR-Gal4/UAS system we showed that synthetic lethality could be generated by coexpressing two expanded polyQ proteins while synthetic-like lethality could be produced by expressing a single expanded polyQ protein at a double dose. In addition, expressing two copies of the UAS-41Q with GMR-Gal4 induced a lethal phenotype, indicating that expressing a toxic polyQ tract alone with GMR-Gal4/UAS could lead to a synthetic-like lethality. These data suggest that expressing a toxic polyQ tract in Drosophila with the GMR-Gal4/ UAS system generates a sensitive, or even a critical, condition that is prone to lethality. The data also raise a possibility that, under this sensitized genetic background, overexpressing a Drosophila modifier might give rise to synthetic lethality. Nonetheless, no synthetic lethality was reported from coexpressing the GOF modifiers and the expanded polyQ proteins, despite of the large-scale genome-wide screens that contained more than 10,000 EP UAS-GOF alleles [18] [19] [23] . It is possible that only a small number of the Drosophila genes, when overexpressed by GMR-Gal4/UAS, could interact strongly with the expanded polyQ proteins to generate synthetic lethality, so that these rare alleles were not included in the genome-wide screens due to their sample sizes.
4.2. Synthetic Lethality and GMR-Gal4/UAS
Disruptions to the Drosophila eye development have practically no known effect on viability, or fertility. In an extreme case, the strong mutant alleles of the Drosophila eya gene (eyes absent) caused completely loss of adult eye facets [33] . Using GMR-Gal4/UAS in Drosophila, overexpressing a mouse gene (TIS11b), or a Drosophila gene (DTIS11), induced loss of eye facets, similar to that of the eya strong alleles [51] . Because the eyes are considered as nonessential to viability, it is generally believed that the widely used GMR-Gal4/UAS system is unbiased toward the recovery of modifiers. However, the results from our study have raised a concern about the use of the GMR-Gal4/UAS expression system to isolate expanded polyQ modifiers by monitoring the eye defects. A lethal-prone condition was implicated in our finding of the synthetic lethality, or synthetic-like lethality, as described above. We further showed that coexpressing Dikar, an endogenous gene, and the expanded polyQ proteins generated a synthetic lethal phenotype.
Though GMR-Gal4 operates primarily in the eyes, abundant evidence indicates that it is not restricted in the eyes. First, expression of the GMR-Gal4 driver outside of the eyes has been well documented. For example, early studies suggest that the progenitor of the GMR sequence is capable of inducing expressions in the larval brain and several other tissues, in addition to the developing eyes [28] . A recent study showed that GMR-Gal4 also operated in many other tissue types, including the wing discs, the trachea and the leg discs [52] . Second, GMR-Gal4/UAS frequently induced lethality at pupal stage [53] .
In our analysis, a “leaky” GMR-Gal4’s activity outside of the developing retina is likely the cause of the synthetic lethality. Since the ‘leaky’ gene activities are usually associated with the current gene expression technology, expressing a toxic polyQ protein could often lead to a lethality-prone condition outside of the primary target cell type. Studies with a series of mouse model systems showed that, in addition to the brain and the CNS, the vast majority of non-CNS organs and tissues were damaged by expressing expanded polyQ proteins [6] . Thus, expressing a protein containing an expanded polyQ tract is likely detrimental to a large number of cell types in all model organisms. It is also conceivable that the toxic polyQ expression generates lethal-prone conditions in many of these affected cell types, and strong modifiers of the expanded polyQ proteins induce synthetic lethality, similar to that of Dikar, in model organisms.
4.3. Dikar’s Synthetic Lethality with a Toxic PolyQ Tract
Examining the effects of coexpressing UAS-Dikar and UAS-Httex1p-97QP-s that encode a toxic polyQ tract with GAG/CAA alternate repeats, our data showed that Dikar’s synthetic lethality is not mediated through the CAG repeats in the mRNAs, though long CAG repeats in mRNAs are toxic in Drosophila [39] . It has also been well documented that flanking domains of the toxic polyQ tract have significant influence on the toxicity of the disease proteins [7] [16] . Our data showed that the flanking domains play no role in the synthetic lethality, since Dikar induced synthetic lethality when coexpressed with each of three disease proteins that share no homology in the flanking sequences. It is further supported by the data showing that Dikar also induced synthetic lethality when coexpressed with UAS-41Q, which encodes no flanking domains.
With respect to synthetic lethality, Dikar’s interactions with the expanded polyQ proteins appeared similar to the interactions between two expanded polyQ proteins (Figure 6(b) and Figure 6(c)). However, unlike a disease protein that can trigger a synthetic-like lethality (i.e., expressing a single toxic polyQ protein in a double dose), Dikar appeared unable to initiate, or trigger, a synthetic-like lethal pathway, since elevated UAS-Dikar expression in a genotype carrying two copies of UAS-Dikar-a did not induce lethal defects (Figure 6(d)).
The Dikar’s HAT domain belongs to the GCN5 subfamily, which mediates acetylation of histones at lysine residues. Acetylation by a GCN5 protein is generally correlated with the activation of transcription [54] . Though Dikar’s biological function remains unknown, mutant analysis showed that it is neither essential for viability, nor for fertility [46] . Its mouse homologue, Cecr2, on the other hand, is required for development, and a null allele of Cecr2 downregulated a number of transcription factors [47] [48] . Studies have shown that transcriptional deregulation resulted from impaired HAT activity is correlated with pathogenesis of the expanded polyQ disorders, but it remains unknown how overexpressing Dikar with GMR-Gal4/UAS impacts on transcription. Perhaps, not only Dikar’s synthetic lethality is caused by its interaction with the disease proteins, but also an amplifying effect resulting from Dikar’s chromatin remodeling activity that deregulates a series of the downstream genes.
Synthetic lethal alleles are likely stronger interacting alleles than the enhancers isolated from the conventional screens that relied on modifications to the eye defects. For example, synthetic lethal alleles may interact with expanded polyQ proteins early in the neurodegenerative pathways to deregulate a cascade of downstream gene activity, and thus induce more severe damages that lead to synthetic lethality. Currently, it is unclear whether there are more genes that could interact with the toxic polyQ proteins to induce synthetic lethal phenotypes. However, searching for additional synthetic lethal alleles will be of great importance, because understanding of their actions and interactions could shed light on one of the most intriguing yet unknown issues of the expanded polyQ diseases, i.e., the initiating poisonous event, or events of the expanded polyQ toxicity.
Acknowledgements
This work was supported by a grant from University of Connecticut. The authors would like to thank The Center for Applied Genetics and Technology at University of Connecticut for DNA sequence analysis.
References
- Orr, H.T. and Zoghbi, H.Y. (2007) Trinucleotide Repeat Disorders. Annual Review of Neuroscience, 30, 575-621.http://dx.doi.org/10.1146/annurev.neuro.29.051605.113042
- Winderickx, J., Delay, C., De Vos, A., Klinger, H., Pellens, K., Vanhelmont, T., Van Leuven, F. and Zabrocki, P. (2008) Protein Folding Diseases and Neurodegeneration: Lessons Learned from Yeast. Biochimica et Biophysica Acta, 1783, 1381-1395. http://dx.doi.org/10.1016/j.bbamcr.2008.01.020
- van Ham, T.J., Breitling, R., Swertz, M.A. and Nollen, E.A. (2009) Neurodegenerative Diseases: Lessons from Genome-Wide Screens in Small Model Organisms. EMBO Molecular Medicine, 1, 360-370. http://dx.doi.org/10.1002/emmm.200900051
- Yu, Z. and Bonini, N.M. (2011) Modeling Human Trinucleotide Repeat Diseases in Drosophila. International Review of Neurobiology, 99, 191-212. http://dx.doi.org/10.1016/B978-0-12-387003-2.00008-2
- Blum, E.S., Schwendeman, A.R. and Shaham, S. (2012) PolyQ Disease: Misfiring of a Developmental Cell Death Program? Trends in Cell Biology, 23, 168-174.
- Figiel, M., Szlachcic, W.J., Switonski, P.M., Gabka, A. and Krzyzosiak, W.J. (2012) Mouse Models of Polyglutamine Diseases: Review and Data Table. Part I. Molecular Neurobiology, 46, 393-429. http://dx.doi.org/10.1007/s12035-012-8315-4
- Ingram, M.A., Orr, H.T. and Clark, H.B. (2012) Genetically Engineered Mouse Models of the Trinucleotide-Repeat Spinocerebellar Ataxias. Brain Research Bulletin, 88, 33-42. http://dx.doi.org/10.1016/j.brainresbull.2011.07.016
- Switonski, P.M., Szlachcic, W.J., Gabka, A., Krzyzosiak, W.J. and Figiel, M. (2012) Mouse Models of Polyglutamine Diseases in Therapeutic Approaches: Review and Data Table. Part II. Molecular Neurobiology, 46, 430-466.http://dx.doi.org/10.1007/s12035-012-8316-3
- Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W., et al. (1996) Exon 1 of the HD Gene with an Expanded CAG Repeat Is Sufficient to Cause a Progressive Neurological Phenotype in Transgenic Mice. Cell, 87, 493-506. http://dx.doi.org/10.1016/S0092-8674(00)81369-0
- Marsh, J.L., Walker, H., Theisen, H., Zhu, Y.Z., Fielder, T., Purcell, J. and Thompson, L.M. (2000) Expanded Polyglutamine Peptides Alone Are Intrinsically Cytotoxic and Cause Neurodegeneration in Drosophila. Human Molecular Genetics, 9, 13-25. http://dx.doi.org/10.1093/hmg/9.1.13
- Brignull, H.R., Moore, F.E., Tang, S.J. and Morimoto, R.I. (2006) Polyglutamine Proteins at the Pathogenic Threshold Display Neuron-Specific Aggregation in a Pan-Neuronal Caenorhabditis Elegans Model. The Journal of Neuroscience, 26, 7597-7606. http://dx.doi.org/10.1523/JNEUROSCI.0990-06.2006
- Ellerby, L.M., Andrusiak, R.L., Wellington, C.L., Hackam, A.S., Propp, S.S., Wood, J.D., Sharp, A.H., Margolis, R.L., Ross, C.A., Salvesen, G.S., et al. (1999) Cleavage of Atrophin-1 at Caspase Site Aspartic Acid 109 Modulates Cytotoxicity. Journal of Biological Chemistry, 274, 8730-8736. http://dx.doi.org/10.1074/jbc.274.13.8730
- Goti, D., Katzen, S.M., Mez, J., Kurtis, N., Kiluk, J., Ben-Haiem, L., Jenkins, N.A., Copeland, N.G., Kakizuka, A., Sharp, A.H., et al. (2004) A Mutant Ataxin-3 Putative-Cleavage Fragment in Brains of Machado-Joseph Disease Patients and Transgenic Mice Is Cytotoxic above a Critical Concentration. The Journal of Neuroscience, 24, 10266-10279. http://dx.doi.org/10.1523/JNEUROSCI.2734-04.2004
- Jung, J., Xu, K., Lessing, D. and Bonini, N.M. (2009) Preventing Ataxin-3 Protein Cleavage Mitigates Degeneration in a Drosophila Model of SCA3. Human Molecular Genetics, 18, 4843-4852. http://dx.doi.org/10.1093/hmg/ddp456
- Suzuki, Y., Nakayama, K., Hashimoto, N. and Yazawa, I. (2010) Proteolytic Processing Regulates Pathological Accumulation in Dentatorubral-Pallidoluysian Atrophy. FEBS Journal, 277, 4873-4387. http://dx.doi.org/10.1111/j.1742-4658.2010.07893.x
- Zheng, Z. and Diamond, M.I. (2012) Huntington Disease and the Huntingtin Protein. Progress in Molecular Biology and Translational Science, 107, 189-214. http://dx.doi.org/10.1016/B978-0-12-385883-2.00010-2
- Zuccato, C., Valenza, M. and Cattaneo, E. (2010) Molecular Mechanisms and Potential Therapeutical Targets in Huntington’s Disease. Physiological Reviews, 90, 905-981. http://dx.doi.org/10.1152/physrev.00041.2009
- Fernandez-Funez, P., Nino-Rosales, M.L., de Gouyon, B., She, W.C., Luchak, J.M., Martinez, P., Turiegano, E., Benito, J., Capovilla, M., Skinner, P.J., et al. (2000) Identification of Genes that Modify Ataxin-1-Induced Neurodegeneration. Nature, 408, 101-106. http://dx.doi.org/10.1038/35040584
- Kazemi-Esfarjani, P. and Benzer, S. (2000) Genetic Suppression of Polyglutamine Toxicity in Drosophila. Science, 287, 1837-1840. http://dx.doi.org/10.1126/science.287.5459.1837
- Willingham, S., Outeiro, T.F., DeVit, M.J., Lindquist, S.L. and Muchowski, P.J. (2003) Yeast Genes that Enhance the Toxicity of a Mutant Huntingtin Fragment or Alpha-Synuclein. Science, 302, 1769-1772. http://dx.doi.org/10.1126/science.1090389
- Nollen, E.A., Garcia, S.M., van Haaften, G., Kim, S., Chavez, A., Morimoto, R.I. and Plasterk, R.H. (2004) Genome-Wide RNA Interference Screen Identifies Previously Undescribed Regulators of Polyglutamine Aggregation. Proceedings of the National Academy of Sciences of the United States of America, 101, 6403-6408. http://dx.doi.org/10.1073/pnas.0307697101
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S.C. and Muchowski, P.J. (2005) A Genomic Screen in Yeast Implicates Kynurenine 3-Monooxygenase as a Therapeutic Target for Huntington Disease. Nature Genetics, 37, 526-531.http://dx.doi.org/10.1038/ng1542
- Bilen, J. and Bonini, N.M. (2007) Genome-Wide Screen for Modifiers of Ataxin-3 Neurodegeneration in Drosophila. PLoS Genetics, 3, 1950-1964. http://dx.doi.org/10.1371/journal.pgen.0030177
- Zhang, S., Binari, R., Zhou, R. and Perrimon, N. (2010) A Genomewide RNA Interference Screen for Modifiers of Aggregates Formation by Mutant Huntingtin in Drosophila. Genetics, 184, 1165-1179. http://dx.doi.org/10.1534/genetics.109.112516
- Vossfeldt, H., Butzlaff, M., Prussing, K., Ni Charthaigh, R.A., Karsten, P., Lankes, A., Hamm, S., Simons, M., Adryan, B., Schulz, J.B., et al. (2012) Large-Scale Screen for Modifiers of Ataxin-3-Derived Polyglutamine-Induced Toxicity in Drosophila. PLoS One, 7, e47452. http://dx.doi.org/10.1371/journal.pone.0047452
- Brand, A.H. and Perrimon, N. (1993) Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development, 118, 401-415.
- Jackson, G.R. (2008) Guide to Understanding Drosophila Models of Neurodegenerative Diseases. PLoS Biology, 6, e53. http://dx.doi.org/10.1371/journal.pbio.0060053
- Moses, K. and Rubin, G.M. (1991) Glass Encodes a Site-Specific DNA-Binding Protein That Is Regulated in Response to Positional Signals in the Developing Drosophila Eye. Genes & Development, 5, 583-593. http://dx.doi.org/10.1101/gad.5.4.583
- Freeman, M. (1996) Reiterative Use of the EGF Receptor Triggers Differentiation of all Cell Types in the Drosophila Eye. Cell, 87, 651-660. http://dx.doi.org/10.1016/S0092-8674(00)81385-9
- Freeman, M. (1997) Personal Communication to FlyBase. http://flybase.org/reports/FBrf0091569.html
- Warrick, J.M., Paulson, H.L., Gray-Board, G.L., Bui, Q.T., Fischbeck, K.H., Pittman, R.N. and Bonini, N.M. (1998) Expanded Polyglutamine Protein Forms Nuclear Inclusions and Causes Neural Degeneration in Drosophila. Cell, 93, 939-949. http://dx.doi.org/10.1016/S0092-8674(00)81200-3
- Thibault, S.T., Singer, M.A., Miyazaki, W.Y., Milash, B., Dompe, N.A., Singh, C.M., Buchholz, R., Demsky, M., Fawcett, R., Francis-Lang, H.L., et al. (2004) A Complementary Transposon Tool Kit for Drosophila melanogaster Using P and Piggybac. Nature Genetics, 36, 283-287. http://dx.doi.org/10.1038/ng1314
- McQuilton, P., St Pierre, S.E. and Thurmond, J. (2012) FlyBase 101—The Basics of Navigating FlyBase. Nucleic Acids Research, 40, D706-D714. http://dx.doi.org/10.1093/nar/gkr1030
- Wernet, M.F., Labhart, T., Baumann, F., Mazzoni, E.O., Pichaud, F. and Desplan, C. (2003) Homothorax Switches Function of Drosophila Photoreceptors from Color to Polarized Light Sensors. Cell, 115, 267-279. http://dx.doi.org/10.1016/S0092-8674(03)00848-1
- Parks, A.L., Cook, K.R., Belvin, M., Dompe, N.A., Fawcett, R., Huppert, K., Tan, L.R., Winter, C.G., Bogart, K.P., Deal, J.E., et al. (2004) Systematic Generation of High-Resolution Deletion Coverage of the Drosophila melanogaster Genome. Nature Genetics, 36, 288-292. http://dx.doi.org/10.1038/ng1312
- Guarente, L. and Hoar, E. (1984) Upstream Activation Sites of the CYC1 Gene of Saccharomyces cerevisiae Are Active When Inverted but Not When Placed Downstream of the “TATA Box”. Proceedings of the National Academy of Sciences of the United States of America, 81, 7860-7864. http://dx.doi.org/10.1073/pnas.81.24.7860
- Struhl, K. (1984) Genetic Properties and Chromatin Structure of the Yeast Gal Regulatory Element: An Enhancer-Like Sequence. Proceedings of the National Academy of Sciences of the United States of America, 81, 7865-7869. http://dx.doi.org/10.1073/pnas.81.24.7865
- Dobi, K.C. and Winston, F. (2007) Analysis of Transcriptional Activation at a Distance in Saccharomyces cerevisiae. Molecular and Cellular Biology, 27, 5575-5586. http://dx.doi.org/10.1128/MCB.00459-07
- Li, L.B. and Bonini, N.M. (2010) Roles of Trinucleotide-Repeat RNA in Neurological Disease and Degeneration. Trends in Neurosciences, 33, 292-298. http://dx.doi.org/10.1016/j.tins.2010.03.004
- Steffan, J.S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L.C., Slepko, N., Illes, K., Lukacsovich, T., Zhu, Y.Z., Cattaneo, E., et al. (2004) SUMO Modification of Huntingtin and Huntington’s Disease Pathology. Science, 304, 100-104. http://dx.doi.org/10.1126/science.1092194
- Jackson, G.R., Salecker, I., Dong, X., Yao, X., Arnheim, N., Faber, P.W., MacDonald, M.E. and Zipursky, S.L. (1998) Polyglutamine-Expanded Human Huntingtin Transgenes Induce Degeneration of Drosophila Photoreceptor Neurons. Neuron, 21, 633-642. http://dx.doi.org/10.1016/S0896-6273(00)80573-5
- Engels, W. (1989) P elements in Drosophila melanogaster. In: Berg, D. and Home, M., Eds., Mobile DNA, American Society for Microbiology, Washington DC, 437-484.
- Zhang, P. and Spradling, A.C. (1993) Efficient and Dispersed Local P Element Transposition from Drosophila Females. Genetics, 133, 361-373.
- Timakov, B., Liu, X., Turgut, I. and Zhang, P. (2002) Timing and Targeting of P-Element Local Transposition in the Male Germline Cells of Drosophila melanogaster. Genetics, 160, 1011-1022.
- Sudi, J., Zhang, S., Intrieri, G., Hao, X. and Zhang, P. (2008) Coincidence of P-Insertion Sites and Breakpoints of Deletions Induced by Activating P Elements in Drosophila. Genetics, 179, 227-235. http://dx.doi.org/10.1534/genetics.107.085498
- Keuling, A., Yang, F., Hanna, S., Wang, H., Tully, T., Burnham, A., Locke, J. and McDermid, H.E. (2007) Mutation Analysis of Drosophila dikar/CG32394, Homologue of the Chromatin-Remodelling Gene CECR2. Genome, 50, 767-777. http://dx.doi.org/10.1139/G07-050
- Banting, G.S., Barak, O., Ames, T.M., Burnham, A.C., Kardel, M.D., Cooch, N.S., Davidson, C.E., Godbout, R., McDermid, H.E. and Shiekhattar, R. (2005) CECR2, a Protein Involved in Neurulation, Forms a Novel Chromatin Remodeling Complex with SNF2L. Human Molecular Genetics, 14, 513-524. http://dx.doi.org/10.1093/hmg/ddi048
- Fairbridge, N.A., Dawe, C.E., Niri, F.H., Kooistra, M.K., King-Jones, K. and McDermid, H.E. (2010) Cecr2 Mutations Causing Exencephaly Trigger Misregulation of Mesenchymal/Ectodermal Transcription Factors. Birth Defects Research Part A: Clinical and Molecular Teratology, 88, 619-625. http://dx.doi.org/10.1002/bdra.20695
- Halfon, M.S., Gisselbrecht, S., Lu, J., Estrada, B., Keshishian, H. and Michelson, A.M. (2002) New Fluorescent Protein Reporters for Use with the Drosophila Gal4 Expression System and for Vital Detection of Balancer Chromosomes. Genesis, 34, 135-138. http://dx.doi.org/10.1002/gene.10136
- Rorth, P., Szabo, K., Bailey, A., Laverty, T., Rehm, J., Rubin, G.M., Weigmann, K., Milan, M., Benes, V., Ansorge, W. and Cohen, S.M. (1998) Systematic Gain-of-Function Genetics in Drosophila. Development, 125, 1049-1057.
- Yeh, P.A., Yang, W.H., Chiang, P.Y., Wang, S.C., Chang, M.S. and Chang, C.J. (2012) Drosophila Eyes Absent Is a Novel mRNA Target of the Tristetraprolin (TTP) Protein DTIS11. International Journal of Biological Sciences, 8, 606-619. http://dx.doi.org/10.7150/ijbs.3782
- Li, W.Z., Li, S.L., Zheng, H.Y., Zhang, S.P. and Xue, L. (2012) A Broad Expression Profile of the GMR-GAL4 Driver in Drosophila melanogaster. Genetics and Molecular Research, 11, 1997-2002. http://dx.doi.org/10.4238/2012.August.6.4
- Flybase Recombinant Construct P{GAL4-ninaE.GMR}. http://flybase.org/reports/FBtp0001315.html
- Selvi, B.R., Cassel, J.C., Kundu, T.K. and Boutillier, A.L. (2010) Tuning Acetylation Levels with HAT Activators: Therapeutic Strategy in Neurodegenerative Diseases. Biochimica et Biophysica Acta, 1799, 840-853. http://dx.doi.org/10.1016/j.bbagrm.2010.08.012