Open Journal of Genetics
Vol.3 No.2A1(2013), Article ID:33679,11 pages DOI:10.4236/ojgen.2013.32A1003
Can population genomics guide future therapeutic gene transfer strategies for Parkinson’s disease?
![]()
1Department of Neurology, Georgetown University Medical Center, Washington DC, USA
2Department of Neuroscience, Georgetown University Medical Center, Washington DC, USA
Email: hjf8@georgetown.edu
Copyright © 2013 Massimo S. Fiandaca et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 April 2013; revised 1 May 2013; accepted 1 June 2013
Keywords: Biomarkers; Functional Imaging; Gene Therapy; Parkinson’s Disease; Population Genomics; Prodromal Phase
ABSTRACT
Medical and surgical therapies for patients with Parkinson’s disease (PD) are typically considered and initiated upon development of clinical signs, especially therapeutic gene transfer therapies. Early clinical trials delivering transgenes within the brains of PD patients have confirmed their safety and suggested mild to moderate efficacy. Confirmatory phase III trials have yet to be undertaken with any of the current treatment regimens. During the development of PD gene therapy, mapping of the human genome was finalized and provides major insights into the normal and pathogenic genetic variabilities of populations. Genome wide association studies (GWAS) have expanded the genetic defects and risk factors accompanying clinical PD. Advanced genomic investigations may allow asymptomatic individuals with a high risk of developing PD, and evident presymptomatic nigrostriatal deficiencies, to consider early treatment approaches. Herein we propose that certain genomically and clinically defined PD patients may provide unique opportunities for testing neuronotrophic gene therapy in a pathobiological environment that is antecedent to overt motoric dysfunction. Such an approach may finally allow testing of the disease-altering capabilities of therapeutic gene transfer in PD.
1. INTRODUCTION
While Parkinson’s disease (PD) afflicts only 1% of the world’s 55-year-old population, approximately 3% of those aged 75 years and older manifest this illness [1,2]. Characterized by levodopa-responsive, progressive motor deficits that feature resting tremor, rigidity, hypokinesia, and postural instability (cardinal signs), the afflicted PD population has been estimated to include 5 million people worldwide [3], and is expected to expand to nearly 10 million by the year 2030 [4]. The individual, familial, and societal costs associated with this disease on a worldwide scale are rising dramatically [5-10], primarily due to the increasing average age of the population in developed countries [3,4,11]. More than ever, clinicians and scientists seek elucidation regarding the complex pathophysiologic mechanisms leading to the reported “common cascade of events” that results in PD [12,13]. It is clear that both environmental and genetic factors are involved. Multidisciplinary approaches to comprehension of the mechanistic events (“cascades”) associated with PD will lead to improved therapeutic options.
A blossoming area of interest to investigators studying PD has been the prodromal phase of this disorder [14] (see Figure 1). While classical PD motor deterioration involves the gradual degeneration of the dopaminergic nigrostriatal pathways (DNPs) [15-17], prodromal signs are increasingly being documented in individuals that subsequently develop the more classic disease. The progressive clinical picture of PD was defined by Hoehn and Yahr in 1967 (stages I-IV, with stage V being bedridden and totally disabled) [18] and continues to be in use today. Such clinical/pathological observations have prompted Braak et al. [19] to postulate that PD develops through as series of progressive degenerative steps that initially do not involve nigrostriatal dopaminergic neurons (DANs). In this scheme [19], sentinel changes occur within the olfactory pathways, while concurrent brainstem participation in the disease process occurs in a caudal to rostral manner, culminating in the classical degeneration of DANs of the substantia nigra pars compacta (SNpc). Although the strongest risk factor continues to be a first degree relative having PD, common premotor signs correlated with subsequent degeneration of DNPs
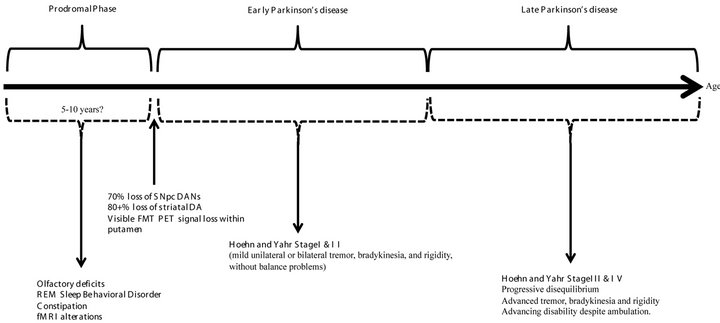
Figure 1. Putative time course associated with idiopathic Parkinson’s disease (PD). The prodromal phase is thought to last between 5 to 10 years, and features subtle changes, such as alterations in olfaction, sleep disturbances, altered gastrointestinal motility, and functional MRI (fMRI) changes. At onset of initial PD clinical signs, most patients have sustained significant loss in substantia nigra pars compacta dopaminergic neurons (SNpc DANs), major reduction in striatal dopamine (DA) levels, and noticeable alterations in striatal fluoromethyltyrosine positron emission tomography (FMT PET) signal within the dorsolateral putamen. The course of clinical PD is longer than the prodromal phase and is defined by the Hoehn and Yahr stages.
include hyposmia/anosmia, rapid eye movement (REM) sleep behavior disorder (RBD), and constipation [20,21], heralding olfactory and brainstem dysfunction.
Early development of gene transfer approaches for PD treatment [22] took advantage of the historical neurochemical and neurophysiological defects defined as hallmarks of this disorder [23-25]. Initial strategies (see Figure 2) featured enzyme replacement to ameliorate the distorted neurochemical balance, without the capability of affecting the intrinsic disease course [26]. Eventually, introduction of neuronotrophic factor genes, with the potential for rescue and restoration of the plagued DNP, have been posited to offer a disease-altering option [27- 30]. While all of the current PD gene transfer interventions appear safe, the attendant efficacy has been variable, but encouraging. Critique by many movement disorder specialists regarding this advanced treatment approach now relates to the timing of gene therapy in the clinical course of PD. Most believe that the window of opportunity for successful patient treatment may be before they develop motoric dysfunction, while the DNPs are not yet devastated. Neuronotrophic gene therapy, specifically delivered during this prodromal period, offers the potential to “rescue” the degenerating nigrostriatal DANs and thereby provides a disease-modifying approach [26]. Enzyme replacement gene therapy, along with deep brain stimulation (DBS) [31], could be reserved for treatment of more advanced PD stages.
The Human Genome Project (HGP) [32] has resulted in an explosion of information related to the genetic basis of diseases. Unfortunately this torrent of information has yet to be synthesized into a coherent, comprehensive understanding of the mechanistic foundation for many illnesses afflicting world populations. The enhanced appreciation for the genetic basis of many diseases is directly related to the completion of the HGP and the genesis of the burgeoning field of population genomics. Considered the comparative assessment of DNA (deoxyribonucleic acid) sequence variations occurring within differing populations [33], population genomic investigations are now commonly performed through genome wide association studies (GWAS) [34]. With constantly improving analytic techniques and throughput, these genetic surveys are yielding a tremendous amount of data regarding normal variations, as well as those associated with specific diseases, down to the level of single nucleotide polymorphisms (SNPs) and epigenomic variability [35]. To better differentiate disease-related DNA sequence variants, which comprise a small subset of all DNA sequence alternatives, it remains essential to gain confidence in what constitutes normal genome variability. The massive amount of nucleotide sequence data generated from each individual, through advanced genomic studies, has necessitated parallel innovations in large data management strategies, large data processing methods, and expanded large data storage technologies. Likewise, the validation of novel interpretive strategies for these immense data sets has elevated the field of bioinformatics

Figure 2. Timelines for therapeutic gene transfer strategies for Parkinson’s disease. Initial clinical trials in 2003 utilized the glutamic acid decarboxylase (GAD) gene to suppress excitatory output of the subthalamic nucleus. Financial difficulties of the corporate sponsor ended further investigations in 2012, despite significant clinical success in a Phase II trial. Aromatic amino acid decarboxylase (AADC) gene transfer was carried out in 2004 and carried through successful Phase I trials in centers in the United States and Japan. Future AADC trials are planned. Neurturin (NTRN) gene therapy trials, beginning in 2005, provided the first attempt at delivering a neuronotrophic factor to the degenerating nigrostriatal pathway. Both Phase II efforts have failed to show efficacy and future trials are in doubt. A tripartite gene transfer strategy (including the AADC gene) is being carried out through a collaborative effort in the European Union. So far, no clinical peer-reviewed publications are available but press releases and scientific presentations provide optimism utilizing this novel technique and delivered transgene set. Phase I/II results are anticipated in by 2014/2015. Finally, the anticipated trial delivering glial cell-derived neurotrophic factor (GDNF) to PD patients will begin at the National Institute of Health (NIH) in 2013. Preclinical investigations suggest a much more potent effect from GDNF compared to NTRN. The Phase I GDNF trial will utilize improved delivery methods, including real-time imaging, which will provide enhanced target distribution within the putamen and an extra measure of safety not previously available.
to greater prominence [36]. As we develop data-driven appreciation of certain disease mechanisms, founded on genomic interpretations, we approach a point of understanding the chromosomal basis for individual states of health and disease. Such keen knowledge may eventually allow the development of customized, or “personalized,” medical strategies tailored to the individual’s specific condition.
Population genomic investigations of normal individuals and those afflicted with PD have yielded important clues as to certain operative mechanisms associated with specific hereditary and non-hereditary pathologies. In this manuscript we consider how the improved understanding of the prodromal PD state, aided by genomic analyses and other interpretive clinical strategies, may specifically sanction future gene transfer therapeutics at a presymptomatic stage. If shown to be successful, such a sentinel treatment approach may prove beneficial in translational efforts with other human diseases, advancing the promise of genetic medicine [33].
2. GENOMIC STUDIES FOR PARKINSON’S DISEASE
Monogenic defects contribute to approximately 5% - 10% of the total PD population, while over 85% are considered idiopathic in nature. In the opinion of the learned majority, idiopathic PD involves at least a genetic predisposition as well as some environmental insult before clinical manifestation. Despite the low percentage of hereditary/familial cases, the specific alterations that have been genomically defined provide useful insights into the associated clinicopathology, and afford a rich environment for the development novel therapeutic approaches. While a direct link between a genetic abnormality and clinical PD had been previously associated only with small, infrequent familial cohorts, population genomic studies resulting from the HGP now allow relatively inexpensive screening for genetic mutations, without an antecedent family history. These genomic investigations expand our appreciation for the population at risk of developing genetically related forms of PD. In-depth investigations of the associated pathophysiology resulting from such defects promotes the mechanistic understanding of specific aspects of PD. An example of mechanistic information derived from a genetic form of PD has been associated with the leucine rich repeat kinase 2 (LRRK2) mutation. Asymptomatic carriers of the LRRK2 G2019S mutation, for example, were found to have quantifiably altered gait characteristics compared to control subjects [37], mirroring similar defects in symptomatic PD [38]. Similar prodromal motor characteristics may be useful in screening patients during PD evolution [39].
Genomic investigation of sporadic gene mutations within populations continue to delineate the monogenic defects that define or predispose an individual to familial PD (designated PARK1-PARK18, so far) [40]. Of these 18 DNA gene mutations, seven display autosomal dominant inheritance (PARK1, 3, 4, 5, 8, 11, and 13), six display autosomal recessive inheritance (PARK2, 6, 7, 9, 14, and 15), one is X-linked (PARK12), one is yet to be fully defined (PARK10), and the remaining three (PARK 16- 18) are genetic susceptibility loci [40,41]. Interestingly, these PARK genes are often downregulated in cases of idiopathic PD [42-44].
Preclinical experimental genomic analyses investigating genetically based PD pathology were carried out in transgenic Drosophila that harbored the human mutated α-synuclein (mSYN) gene, using gene transcript microarrays [45]. This fly model clearly provided evidence for dysregulation of gene expression at all evaluated timepoints, even before clinical signs and development of significant neuropathology. Additional genomic microarray investigations have been used to compare expression of a select panel of genes in diverse experimental systems, including a genetic mouse model, a toxicant rat model, and human PD postmortem specimens [46], suggesting possible significance for the selected panel of genes. Unfortunately, other investigators using similar panels have not had comparable results [47]. Microarray analysis of human brain tissues has implicated the dysregulation of genes responsible for multiple cellular systems in the idiopathic PD state compared to controls, including those associated with the ubiquitin proteosomal system (UPS), endoplasmic reticulum (ER) stress response, mitochondrial function, synaptic transmission, and programmed cell death, among others [44,46-48]. Unfortunately, many of the transcript microarray studies provided discordant findings that may in part be due to differences in tissue analyzed (brain tissue vs. laser micro-dissected SNpc DANs) or the varying sensitivities of the assays used. Careful attention to sample comparisons and generally improved microarray methodologies and assay sensitivities will increase the concordance of future results. It appears, however, that current and future transcriptomic studies will increasingly utilize high throughput RNA (ribonucleic acid) sequencing technologies, rather than microarrays, and may provide even greater sensitivity and correlation between analyses of comparable specimens [49]. Massively parallel transcriptomic analyses have been shown to provide unbiased, objective gene identification and estimated alterations in gene expression [50,51], as well as the associated pathways that are causally, reactively, or independently connected to genetic, environmental, or complex disease pathogenesis [51,52]. Such methods have been successfully applied to the classification of individuals based on their molecular characteristics [53], and thereby, assist in the understanding of complex disease mechanisms [54], possibly featuring environmental and epigenetic influences that are not explained by the DNA sequence.
PD provides and excellent disease platform for studying these complex associations and better defining an approach toward individualized medicine. In a recent investigation [55], such methods determined novel alterations in gene sets associated with mitochondrial bioenergetics and glucose metabolism early in the time course of PD. From these investigations, it was noted that the peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) appears to be underexpressed in PD patients [55], thereby suggesting a novel therapeutic approach via methods that will increase expression of PGC-1α.
Biomarker development for PD and other neurological disorders currently consists of interrogation of peripheral blood leukocytes and plasma (or possibly cerebrospinal fluid, CSF) for distinct biological signatures. Currently, typical biomarker assessment strategies include acquisition of whole genome DNA sequencing, DNA epigenomic analyses, RNA sequencing, and plasma proteomics and metabolomics [56]. Such a comprehensive appraisal is necessary to develop a foundational understanding of states of health versus disease, as these analytic technologies become the state of the art. Once the actual biomarker signatures are defined and validated, a more specific panel of indicators may be interrogated and utilized to define the state of health (or disease). With advanced sequencing technologies, genomic investigations have effectively defined genetic variants occurring in both familial and sporadic PD cases [57], thereby closing the mechanistic gap between these entities. Further genomic definition of the variabilities between these PD subgroups may contribute to the validation of genetic susceptibility screening and therapeutic intervention in a prodromal stage of the disease.
3. DEFINING THE PRODROMAL STAGE OF PD
Although not yet precisely defined, the prodromal stage of PD is considered to be quite long [21,58]. During this phase of the disease, the cardinal PD signs are not present and patients rarely report any neurological issues to their physicians. As describe above [19], subtle changes in the olfactory or brainstem function occur during this period, which require specific testing to confirm. Defining genetically based biomarkers correlated to prodromal conditions such as loss of olfaction, RBD, or chronic constipation may further advance our definition of this pre-motor PD state.
Of additional importance is the recent coupling of genomically defined gene mutations to functional magnetic resonance imaging (fMRI) in humans. This non-invasive neuroimaging technology provides an excellent premortem clinical correlate of dysregulated gene function associated with PD [59]. With accumulating evidence for genomic and fMRI relationships in PD, one can postulate the eventual screening assessment of asymptomatic, at risk individuals for PD. Such fMRI determinations will further define the prodromal state and, together with other biomarkers of disease, allow discussion and consideration of future therapeutic interventions such as neuroprotective gene transfer.
4. THE CURRENT USE OF GENE TRANSFER TECHNOLOGIES IN HUMAN PARKINSON’S DISEASE
Over the last decade and prior to 2012, there have been five major clinical research centers in the world participating in clinical trials evaluating therapeutic gene transfer for the treatment of PD [60,61]. Thus far, clinical investigations have advanced through the Phase II trial process without indications of major safety concerns related to either the viral vectors involved or the therapeutic transgenes delivered. While a limited number of complications were noted with the stereotactic surgical procedures (hemorrhages, venous infarcts) [62], none were directly implicated to result from either the viral vectors or transgenes utilized. While hemorrhagic complications associated with functional neurosurgery are well known, and infrequent [63], additional reductions in frequency may be achieved through the use of imageguided stereotactic approaches in the future [64]. To date, all of these gene transfer treatments were carried out on patients significantly affected (moderate to severe, Hoehn and Yahr Stage III or IV off medication) by idiopathic PD. For some of these studies, the diagnosis of familial/genetic PD was a criterion for exclusion. Although initial selection of those afflicted with moderate or severe stages may be justified in the evaluation of a novel treatment modality, such as therapeutic gene transfer, the nigrostriatal dopaminergic substrate in advanced PD is typically devastated. While even newly diagnosed PD patients, showing minimal motor symptoms (Stage I) feature advanced loss of SNpc DANs (approximately 70%), and similar losses in striatal dopamine transporters, the putamenal DA levels are typically reduced to an even greater extent [65,66]. In the later stages of PD, these substrate levels are progressively compromised and, thereby, decrease the likelihood that treatment modalities, including gene therapy, can provide neural substrate stabilization or rescue.
Four different transgene constructs have been utilized by the five clinical research protocols prior to 2012 (Figure 2) [26]. Four of the clinical teams attempted to provide enzyme replacement gene therapy that would modulate the neurochemical alterations of afflicted individuals. These four approaches were designed to treat specific clinical signs associated with PD, but not alter disease course. The fifth group attempted treatment by providing a neuronotrophic factor gene, attempting not only to improve neurological function, but also to alter the course of the disease. The first successful therapeutic transgene delivery in PD conveyed the glutamic acid dehydroxylase (GAD) gene (enzyme replacement) to the subthalamic nucleus (STN) [27]. This novel approach attempted to improve the altered basal ganglia motor physiology of PD through production of the inhibitory neurotransmitter GABA (gamma amino butyric acid) within the STN; thereby reducing it’s excitatory tone. Utilization of electrical impulses to attenuate STN output is the basis for DBS within this subcortical target. Phase I and II trials using the GAD gene transfer paradigm have shown safety and efficacy [67,68]. Unfortunately, a Phase III trial did not materialize for this treatment method, despite promise and momentum. Bankruptcy of the corporate sponsor [69], unrelated to the specific gene transfer technology or clinical results, scuttled further development of this therapeutic.
Two groups of investigators have employed the delivery of the aromatic L-amino acid decarboxylase (AADC) gene (enzyme replacement) to the putamen (PUT) of PD patients in Phase I trials [61,70]. This enzyme is key to the production of DA within the striatum and is lost along with SNpc DANs. Replacement of this missing enzyme within the dopamine-depleted striatum of PD patients allows local DA restoration by providing an exogenous substrate, typically Sinemet® or levodopa. Data from the AADC trials disclosed robust evidence of transgene expression via postoperative positron emission tomography with 6-[18F]-fluoro-L-m-tyrosine (FMT-PET) within each transduced PUT, as well as a suggestion of clinical improvement [61,62], despite what may have been less than optimal targeting of the transgene [71]. The next series of investigations utilizing AADC gene transfer for PD are being designed, but similar strategies have shown potential in the treatment of pediatric AADC deficiency [72].
Phase I and II clinical trials have been completed utilizing the neurturin (NTRN) gene (neuronotrophic factor replacement) within the PUT of PD patients [30, 73]. Clinical safety was documented in both trials, but efficacy was not shown in the Phase II trial of NTRN gene delivery [30]. Investigators found no evidence of PET signal in targeted PUT post-op. Reasons considered for lack of efficacy in humans, using an approach that showed promise in the nonhuman primate [74], were postulated to include: 1) lack of significant putamenal coverage by the transgene; 2) suboptimal targeting in patients, due to lack of image guidance; 3) lack of adequate transport of transgene product from PUT to SNpc; and 4) reduced efficacy of NTRN in humans compared to glial cell-derived neurotrophic factor (GDNF). Postmortem assessment of transduction sites in several patients suggested at least evidence for postulates 1) and 3) [75]. The lack of clinical efficacy in the NTRN Phase II trial [66], and further consideration by the investigators [76], prompted a revised delivery strategy with targeting both the PUT and substantia nigra (SN) in a follow-up Phase IIb investigation (ClinicalTrials.gov identifier NCT00985517). A 2013 press release from the sponsoring company (Ceregene, Inc., San Diego, CA) regarding this recent Phase IIb study [77], however, indicated that the “trial did not demonstrate statistically significant efficacy on the primary endpoint (UPDRS-motor off).” The future of this specific commercial gene therapy product is unknown.
A fifth group of PD investigators delivered a tripartite transgene (enzyme replacement) to their patients [78]. The triple transgene codes for enzymes from the DA biosynthetic pathway and include AADC, tyrosine hydroxylase (TH), and guanosine triphosphate (GTP) cyclohydrolase 1 (GCH1), in Phase I/II clinical trials [78] (ClinicalTrials.gov identifier NCT00627588), due for completion in 2012 and with pending results. The basis for this approach is similar to that of transduction of AADC alone. According to preliminary unpublished re-
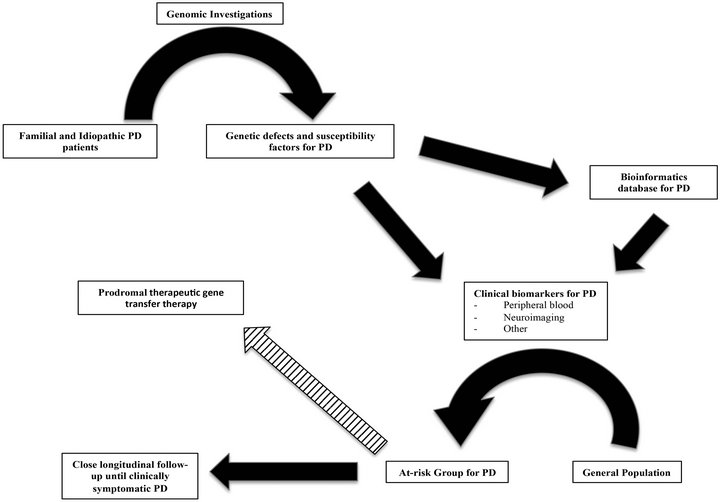
Figure 3. Schematic representation of how genomic investigations may impact Parkinson’s disease (PD) treatments, including future prodromal therapeutic gene transfer. Genomic analyses of familial and idiopathic PD expand our understanding of specific mechanisms underlying specific genetic defects and susceptibility factors. Gained knowledge from these investigations allows for the development of clinical biomarkers of PD and enhances the development of a vast bioinformatics database. Through the expanded use of clinical biomarkers of PD, drawing from rapidly increasing bioinformatic data, the general population will eventually be offered screening, to more precisely define the at-risk PD population during their disease prodrome. Such information may allow either social planning and close follow-up until clinical signs dictate treatment, or (striped arrow) lead to prodromal therapeutics that may alter disease course, such as gene transfer therapies.
ports [79], safety and prolonged clinical efficacy has been shown in the treated PD patients. A final analysis of the clinical data is pending.
As of 2013, a sixth clinical group is planning to deliver a fifth transgene, GDNF (neuronotrophic factor replacement) to the brain of PD patients in a Phase I trial (ClinicalTrials.gov identifier: NCT01621581). This group is currently recruiting PD patients and will deliver the more potent therapeutic neuronotrophic transgene using novel interventional MRI-related technologies that will address some of the concerns associated with the NTRN trials [27]. Putamenal coverage will be enhanced through the utilization of convection enhanced delivery (CED) of larger volumes of infusate, while targeting and coverage will be better documented via “real-time” MRI visualization of the infusion. The first patient in the planned safety study successfully underwent treatment in May of 2013 (K.S. Bankiewicz and J.D. Heiss, personal communications).
5. HOW GENOMICS MAY ALTER THE FUTURE OF THERAPEUTIC GENE TRANSFER FOR PARKINSON’S DISEASE
Genomic investigations have the potential to significantly impact not only the understanding of the mechanisms associated with both genetic and idiopathic PD, but may define the genetic basis for this variable disease entity through advancements in data analysis and bioinformatics (see Figure 3). The discovery that PGC-1α is underexpressed in PD via these methods provides a template for further mechanistic understanding and possible therapeutic approaches for this and other diseases. Additionally, through further discovery, validation, and correlation of genetic factors for PD, the use of genomics may eventually provide useful, specific, clinical biomarkers (e.g. blood-derived, or neuroimaging-defined). Such novel disease indicators, correlating closely with future disease presentation and progression, allow for therapeutic interventions (e.g. enhancers of PGC-1α expression, neuronotrophic factor gene transfer), within the prodromal phase of PD, where there is the greatest chance to impact the remaining pathobiologic substrate, heralding the era of disease-modifying treatments for neurodegenerative diseases.
6. CONCLUSION
The field of genomics is rapidly gaining traction in helping define the basis for many human diseases, including PD. The definition and thorough investigation of specific genetic defects in familial PD allow the better comprehension of the mechanistic basis for particular pathobiology. In addition, genetic susceptibility factors discovered in those with idiopathic PD are also instrumental in defining an at-risk population. With the reduction in cost for genetic analyses and the expanded database of information related to health and disease spawned by the HGP, the era of molecular medicine has arrived. As such, a specific genetic profile for PD, found in asymptomatic individuals within the general population may eventually allow the optimal substrate for intervention via therapeutic gene transfer as well as other novel beneficial strategies.
7. ACKNOWLEDGEMENTS
Support for research on which this review is based comes from W81 XWH-09-1-0103 (H.J.F.). The authors wish to thank Drs. Tim Mhyre and Linda MacArthur for past discussions.
REFERENCES
- Lees, A.J., Hardy, J. and Revesz, T. (2006) Parkinson’s disease. Lancet, 373, 2055-2066. doi:10.1016/S0140-6736(09)60492-X
- de Lau, L.M. and Breteler, M.M. (2006) Epidemiology of Parkinson’s disease. Lancet Neurology, 5, 525-535. doi:10.1016/S1474-4422(06)70471-9
- Wirdefeldt, K., Adami, H.O., Cole, P., Trichopoulos, D. and Mandel, J. (2011) Epidemiology and etiology of Parkinson’s disease: A review of the evidence. European Journal of Epidemiology, 26, S1-S58. doi:10.1007/s10654-011-9581-6
- Dorsey, E.R., Constantinescu, R., Thompson, J.P., Biglan, K.M., Holloway, R.G., Kieburtz, K., Marshall, F.J., Ravina, B.M., Schifitto, G., Siderowf, A. and Tanner, C.M. (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology, 68, 384-386. doi:10.1212/01.wnl.0000247740.47667.03
- Chen, J.J. (2010) Parkinson’s disease: Health-related quality of life, economic cost, and implications of early treatment. The American Journal of Managed Care, 16, S87- S93.
- Winter, Y.M., Balzer-Geldsetzer, A., Spottke, J.P., Reese, E., Baum, J., Klotsche, J., Rieke, A., Simonow, K., Eggert, W.H., Oertel and Dodel, R. (2010) Longitudinal study of the socioeconomic burden of Parkinson’s disease in Germany. European Journal of Neurology: The Official Journal of the European Federation of Neurological Societies, 17, 1156-1163.
- Winter, Y., Balzer-Geldsetzer, M., von Campenhausen, S., Spottke, A., Eggert, K., Oertel, W.H. and Dodel, R. (2010) Trends in resource utilization for Parkinson’s disease in Germany. Journal of Neurological Science, 294, 18-22. doi:10.1016/j.jns.2010.04.011
- Winter, Y., von Campenhausen, S., Reese, J.P., BalzerGeldsetzer, M., Longo, K., Spiga, G., Boetzel, K., Eggert, K., Oertel, W.H., Dodel, R. and Barone, P. (2010) Costs of Parkinson’s disease and antiparkinsonian pharmacotherapy: An Italian cohort study. Neuro-Degenerative Diseases, 7, 365-372. doi:10.1159/000302644
- Vossius, C., Nilsen, O.B. and Larsen, J.P. (2010) Parkinson’s disease and hospital admissions: Frequencies, diagnoses and costs. Acta Neurologica Scandinavica, 121, 38-43. doi:10.1111/j.1600-0404.2009.01239.x
- Lokk, J., Borg, S., Svensson, J., Persson, U. and Ljunggren, G. (2012) Drug and treatment costs in Parkinson’s disease patients in Sweden. Acta Neurologica Scandinavica, 125, 142-147. doi:10.1111/j.1600-0404.2011.01517.x
- Olanow, C.W., Stern, M.B. and Sethi, K. (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology, 72, S1-S136. doi:10.1212/WNL.0b013e3181a1d44c
- Maguire-Zeiss, K.A. and Federoff, H.J. (2003) Convergent pathobiologic model of Parkinson’s disease. Annals of the New York Academy of Sciences, 991, 152-166. doi:10.1111/j.1749-6632.2003.tb07473.x
- Miller, R.M. and Federoff, H.J. (2005) Altered gene expression profiles reveal similarities and differences between Parkinson disease and model systems. The Neuroscientist: A Review Journal Bringing Neurobiology, Neu- rology and Psychiatry, 11, 539-549.
- Schapira, A.H., Agid, Y., Barone, P., Jenner, P., Lemke, M.R., Poewe, W., Rascol, O., Reichmann, H. and Tolosa, E. (2009) Perspectives on recent advances in the understanding and treatment of Parkinson’s disease. European Journal of Neurology: The Official Journal of the European Federation of Neurological Societies, 16, 1090- 1099.
- Hornykiewicz, O. (1975) Brain monoamines and parkinsonism. National Institute on Drug Abuse Research Monograph Series, 3, 13-21.
- Forno, L.S. (1996) Neuropathology of Parkinson’s disease. Journal of Neuropathology & Experimental Neurology, 55, 259-272. doi:10.1097/00005072-199603000-00001
- Wichmann, T. and DeLong, M.R. (1998) Models of basal ganglia function and pathophysiology of movement disorders. Neurosurgery Clinics of North America, 9, 223- 236.
- Hoehn, M.M. and Yahr, M.D. (1967) Parkinsonism: Onset, progression and mortality. Neurology, 17, 427-442. doi:10.1212/WNL.17.5.427
- Braak, H., Del Tredici, K., Rub, U., de Vos, R.A., Jansen Steur, E.N. and Braak, E. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24, 197-211. doi:10.1016/S0197-4580(02)00065-9
- Noyce, A.J., Bestwick, J.P., Silveira-Moriyama, L., Hawkes, C.H., Giovannoni, G., Lees, A.J. and Schrag, A. (2012) Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Annals of Neurology, 72, 893-901. doi:10.1002/ana.23687
- Postuma, R.B., Gagnon, J.F. and Montplaisir, J. (2010) Clinical prediction of Parkinson’s disease: Planning for the age of neuroprotection. Journal of Neurology, Neurosurgery, and Psychiatry, 81, 1008-1013. doi:10.1136/jnnp.2009.174748
- Fiandaca, M.S., Forsayeth, J. and Bankiewicz, K. (2008) Current status of gene therapy trials for Parkinson’s disease. Experimental Neurology, 209, 51-57. doi:10.1016/j.expneurol.2007.08.009
- Parkinson, J. (1817) An essay on the shaking palsy. Sherwood, Neely, and Jones, Paternoster Row, London.
- Bernheimer, H., Birkmayer, W., Hornykiewicz, O., Jellinger, K. and Seitelberger, F. (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Journal of Neurological Science, 20, 415-455. doi:10.1016/0022-510X(73)90175-5
- Lloyd, K.G., Davidson, L. and Hornykiewicz, O. (1975) The neurochemistry of Parkinson’s disease: Effect of Ldopa therapy. Journal of Pharmacological and Experimental Therapy, 195, 453-464.
- Fiandaca, M.S., Bankiewicz, K.S. and Federoff, H.J. (2012) Gene therapy for the treatment of Parkinson’s disease: The nature of the biologics expands the future indications. Pharmaceuticals, 5, 553-590. doi:10.3390/ph5060553
- Richardson, R.M., Kells, A.P., Rosenbluth, K.H., Salegio, E.A., Fiandaca, M.S., Larson, P.S., Starr, P.A., Martin, A.J., Lonser, R.R., Federoff, H.J., Forsayeth, J.R. and Bankiewicz, K.S. (2011) Interventional MRI-guided putaminal delivery of AAV2-GDNF for a planned clinical trial in Parkinson’s disease. Molecular Therapy, 19, 1048- 1057. doi:10.1038/mt.2011.11
- Su, X., Kells, A.P., Huang, E.J., Lee, H.S., Hadaczek, P., Beyer, J., Bringas, J., Pivirotto, P., Penticuff, J., Eberling, J., Federoff, H.J., Forsayeth, J. and Bankiewicz, K.S. (2009) Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Human Gene Therapy, 20, 1627-1640. doi:10.1089/hum.2009.103
- Kordower, J.H., Herzog, C.D., Dass, B., Bakay, R.A., Stansell, J., Gasmi, M. and Bartus, R.T. (2006) Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Annals of Neurology, 60, 706-715. doi:10.1002/ana.21032
- Marks, W.J., Bartus, R.T., Siffert, J., Davis, C.S., Lozano, A., Boulis, N., Vitek, J., Stacy, M., Turner, D., Verhagen, L., Bakay, R., Watts, R., Guthrie, B., Jankovic, J., Simpson, R., Tagliati, M., Alterman, R., Stern, M., Baltuch, G., Starr, P.A., Larson, P.S., Ostrem, J.L., Nutt, J., Kieburtz, K., Kordower, J.H. and Olanow, C.W. (2010) Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurology, 9, 1164-1172. doi:10.1016/S1474-4422(10)70254-4
- Benabid, A.L. (2003) Deep brain stimulation for Parkinson’s disease. Current Opinion in Neurobiology, 13, 696- 706. doi:10.1016/j.conb.2003.11.001
- Schmutz, J., Wheeler, J., Grimwood, J., Dickson, M., Yang, J., Caoile, C., Bajorek, E., Black, S., Chan, Y.M., Denys, M., Escobar, J., Flowers, D., Fotopulos, D., Garcia, C., Gomez, M., Gonzales, E., Haydu, L., Lopez, F., Ramirez, L., Retterer, J., Rodriguez, A., Rogers, S., Salazar, A., Tsai, M. and Myers, R.M. (2004) Quality assessment of the human genome sequence. Nature, 429, 365-368. doi:10.1038/nature02390
- Jorde, L.B., Watkins, W.S. and Bamshad, M.J. (2001) Population genomics: A bridge from evolutionary history to genetic medicine. Human Molecular Genetics, 10, 2199- 2207. doi:10.1093/hmg/10.20.2199
- Bush, W.S. and Moore, J.H. (2012) Chapter 11: Genomewide association studies. PLoS Computing Biology, 8, e1002822. doi:10.1371/journal.pcbi.1002822
- International HapMap (2003) The International HapMap Project. Nature, 426, 789-796. doi:10.1038/nature02168
- Moore, J.H., Asselbergs, F.W. and Williams, S.M. (2010) Bioinformatics challenges for genome-wide association studies. Bioinformatics, 26, 445-455. doi:10.1093/bioinformatics/btp713
- Mirelman, A., Gurevich, T., Giladi, N., Bar-Shira, A., Orr-Urtreger, A. and Hausdorff, J.M. (2011) Gait alterations in healthy carriers of the LRRK2 G2019S mutation. Annals of Neurology, 69, 193-197. doi:10.1002/ana.22165
- Hausdorff, J.M., Balash, J. and Giladi, N. (2003) Effects of cognitive challenge on gait variability in patients with Parkinson’s disease. Journal of Geriatric Psychiatry and Neurology, 16, 53-58.
- Maetzler, W. and Hausdorff, J.M. (2012) Motor signs in the prodromal phase of Parkinson’s disease. Movement Disorders, 27, 627-633.
- Kumar, K.R., Djarmati-Westenberger, A. and Grunewald, A. (2011) Genetics of Parkinson’s disease. Seminars in Neurology, 31, 433-440. doi:10.1055/s-0031-1299782
- Tan, E.K., Kwok, H.H., Tan, L.C., Zhao, W.T., Prakash, K.M., Au, W.L., Pavanni, R., Ng, Y.Y., Satake, W., Zhao, Y., Toda, T. and Liu, J.J. (2010) Analysis of GWASlinked loci in Parkinson disease reaffirms PARK16 as a susceptibility locus. Neurology, 75, 508-512. doi:10.1212/WNL.0b013e3181eccfcd
- Hauser, M.A., Li, Y.J., Xu, H., Noureddine, M.A., Shao,Y.S., Gullans, S.R., Scherzer, C.R., Jensen, R.V., McLaurin, A.C., Gibson, J.R., Scott, B.L., Jewett, R.M., Stenger, J.E., Schmechel, D.E., Hulette, C.M. and Vance, J.M. (2005) Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with Parkinsonism. Archives of Neurology, 62, 917-921. doi:10.1001/archneur.62.6.917
- Moran, L.B., Duke, D.C., Deprez, M., Dexter, D.T., Pearce, R.K. and Graeber, M.B. (2006) Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics, 7, 1-11. doi:10.1007/s10048-005-0020-2
- Moran, L.B. and Graeber, M.B. (2008) Towards a pathway definition of Parkinson’s disease: A complex disorder with links to cancer, diabetes and inflammation. Neurogenetics, 9, 1-13. doi:10.1007/s10048-007-0116-y
- Scherzer, C.R., Jensen, R.V., Gullans, S.R. and Feany, M.B. (2003) Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Human Molecular Genetics, 12, 2457-2466. doi:10.1093/hmg/ddg265
- Miller, R.M. and Federoff, H.J. (2006) Microarrays in Parkinson’s disease: A systematic approach. NeuroRX, 3, 319-326. doi:10.1016/j.nurx.2006.05.008
- Simunovic, F., Yi, M., Wang, Y., Macey, L., Brown, L.T., Krichevsky, A.M., Andersen, S.L., Stephens, R.M., Benes, F.M. and Sonntag, K.C. (2009) Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain, 132, 1795-1809.
- Grunblatt, E., Mandel, S., Jacob-Hirsch, J., Zeligson, S., Amariglo, N., Rechavi, G., Li, J., Ravid, R., Roggendorf, W., Riederer, P. and Youdim, M. B. (2004) Gene expression profiling of Parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. Journal of Neural Transmission, 111, 1543-1573. doi:10.1007/s00702-004-0212-1
- Sirbu, A., Kerr, G., Crane, M. and Ruskin, H.J. (2012) RNA-Seq vs dualand single-channel microarray data: Sensitivity analysis for differential expression and clustering. PloS One, 7, e50986. doi:10.1371/journal.pone.0050986
- Scherzer, C.R., Offe, K., Gearing, M., Rees, H.D., Fang, G., Heilman, C.J., Schaller, C., Bujo, H., Levey, A.I. and Lah, J.J. (2004) Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Archives of Neurology, 61, 1200- 1205. doi:10.1001/archneur.61.8.1200
- Scherzer, C.R. (2009) Chipping away at diagnostics for neurodegenerative diseases. Neurobiology of Disease, 35, 148-156. doi:10.1016/j.nbd.2009.02.016
- Schadt, E.E., Lamb, J., Yang, X., Zhu, J., Edwards, S., Guhathakurta, D., Sieberts, S.K., Monks, S., Reitman, M., Zhang, C., Lum, P.Y., Leonardson, A., Thieringer, R., Metzger, J.M., Yang, L., Castle, J., Zhu, H., Kash, S.F., Drake, T.A., Sachs, A. and Lusis, A.J. (2005) An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genetics, 37, 710-717.
- Scherzer, C.R., Eklund, A.C., Morse, L.J., Liao, Z., Locascio, J.J., Fefer, D., Schwarzschild, M.A., Schlossmacher, M.G., Hauser, M.A., Vance, J.M., Sudarsky, L.R., Standaert, D.G., Growdon, J.H., Jensen R.V. and Gullans, S.R. (2007) Molecular markers of early Parkinson’s disease based on gene expression in blood. Proceedings of the National Academy of Sciences of the United States of America, 104, 955-960. doi:10.1073/pnas.0610204104
- Scherzer, C.R., Grass, J.A., Liao, Z., Pepivani, I., Zheng, B., Eklund, A.C., Ney, P.A., Ng, J., McGoldrick, M., Mollenhauer, B., Bresnick, E.H. and Schlossmacher, M.G. (2008) GATA transcription factors directly regulate the Parkinson's disease-linked gene alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America, 105, 10907-10912. doi:10.1073/pnas.0802437105
- Zheng, B., Liao, Z., Locascio, J.J., Lesniak, K.A., Roderick, S.S., Watt, M.L., Eklund, A.C., Zhang-James, Y., Kim, P.D., Hauser, M.A., Grunblatt, E., Moran, L. B., Mandel, S.A., Riederer, P., Miller, R.M., Federoff, H.J., Wullner, U., Papapetropoulos, S., Youdim, M.B., Cantuti-Castelvetri, I., Young, A.B., Vance, J.M., Davis, R.L., Hedreen, J.C., Adler, C.H., Beach, T.G., Graeber, M.B., Middleton, F.A., Rochet, J.C., Scherzer C.R. and Global, P.D.G.E.C. (2010) PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Science Translational Medicine, 2, 52ra73. doi:10.1126/scitranslmed.3001059
- Scholz, S.W., Mhyre, T., Ressom, H., Shah S. and Federoff, H.J. (2012) Genomics and bioinformatics of Parkinson’s disease. Cold Spring Harbor Perspectives in Medicine, 2, a009449.
- Peeraully T. and Tan, E.K. (2012) Genetic variants in sporadic Parkinson’s disease: East vs west. Parkinsonism & Related Disorders, 18, S63-S65. doi:10.1016/S1353-8020(11)70021-9
- Lang, A.E. (2011) A critical appraisal of the premotor symptoms of Parkinson’s disease: Potential usefulness in early diagnosis and design of neuroprotective trials. Movement Disorders, 26, 775-783.
- Van Nuenen, B.F., van Eimeren, T., van der Vegt, J.P., Buhmann, C., Klein, C., Bloem B.R. and Siebner, H.R. (2009) Mapping preclinical compensation in Parkinson’s disease: An imaging genomics approach. Movement Disorders, 24, S703-S710.
- Fiandaca, M.S. and Bankiewicz, K.S. (2010) Gene therapy for Parkinson’s disease: From nonhuman primates to humans. Current Opinion in Molecular Therapeutics, 12, 519-529.
- Muramatsu, S., Fujimoto, K., Kato, S., Mizukami, H., Asari, S., Ikeguchi, K., Kawakami, T., Urabe, M., Kume, A., Sato, T., Watanabe, E., Ozawa, K. and Nakano, I. (2010) A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Molecular Therapy, 18, 1731-1735.
- Christine, C.W., Starr, P.A., Larson, P.S., Eberling, J.L., Jagust, W.J., Hawkins, R.A., Van Brocklin, H.F., Wright, J.F., Bankiewicz, K.S. and Aminoff, M.J. (2009) Safety and tolerability of putaminal gene therapy for Parkinson’s disease. Neurology, 73, 1662-1669. doi:10.1212/WNL.0b013e3181c29356
- Zrinzo, L., Foltynie, T., Limousin, P. and Hariz, M.I. (2012) Reducing hemorrhagic complications in functional neurosurgery: A large case series and systematic literature review. Journal of Neurosurgery, 116, 84-94. doi:10.3171/2011.8.JNS101407
- Richardson, R.M., Kells, A.P., Martin, A.J., Larson, P.S., Starr, P.A., Piferi, P.G., Bates, G., Tansey, L., Rosenbluth, K.H., Bringas, J.R., Berger, M.S. and Bankiewicz, K.S. (2011) Novel platform for MRI-guided convection-enhanced delivery of therapeutics: Preclinical validation in nonhuman primate brain. Stereotactic and Functional Neurosurgery, 89, 141-151. doi:10.1159/000323544
- Wilson, J.M., Levey, A.I., Rajput, A., Ang, L., Guttman, M., Shannak, K., Niznik, H.B., Hornykiewicz, O., Pifl, C. and Kish, S.J. (1996) Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology, 47, 718-726. doi:10.1212/WNL.47.3.718
- Fearnley, J.M. and Lees, A.J. (1991) Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain, 114, 2283-2301.
- Kaplitt, M.G., Feigin, A., Tang, C., Fitzsimons, H.L., Mattis, P., Lawlor, P.A., Bland, R.J., Young, D., Strybing, K., Eidelberg, D. and During, M.J. (2007) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. The Lancet, 369, 2097-2105. doi:10.1016/S0140-6736(07)60982-9
- LeWitt, P.A., Rezai, A.R., Leehey, M.A., Ojemann, S.G., Flaherty, A.W., Eskandar, E.N., Kostyk, S.K., Thomas, K., Sarkar, A., Siddiqui, M.S., Tatter, S.B., Schwalb, J.M., Poston, K.L., Henderson, J.M., Kurlan, R.M., Richard, I.H., Van Meter, L., Sapan, C.V., During, M.J., Kaplitt, M.G. and Feigin, A. (2011) AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, shamsurgery controlled, randomised trial. The Lancet Neurology, 10, 309-319. doi:10.1016/S1474-4422(11)70039-4
- Annett, L.E., Dunnett, S.B., Martel, F.L., Rogers, D.C., Ridley, R.M., Baker, H.F. and Marsden, C.D. (1990) A functional assessment of embryonic dopaminergic grafts in the marmoset. Progress in Brain Research, 82, 535- 542. doi:10.1016/S0079-6123(08)62644-8
- Eberling, J.L., Jagust, W.J., Christine, C.W., Starr, P., Larson, P., Bankiewicz, K.S. and Aminoff, M.J. (2008) Results from a phase I safety trial of hAADC gene therapy for Parkinson’s disease. Neurology, 70, 1980-1983. doi:10.1212/01.wnl.0000312381.29287.ff
- Valles, F., Fiandaca, M.S., Eberling, J.L., Starr, P.A., Larson, P.S., Christine, C.W., Forsayeth, J., Richardson, R.M., Su, X., Aminoff, M.J. and Bankiewicz, K.S. (2010) Qualitative imaging of adeno-associated virus serotype 2-human aromatic L-amino acid decarboxylase gene therapy in a phase I study for the treatment of Parkinson disease. Neurosurgery, 67, 1377-1385.
- Hwu, W.L., Muramatsu, S., Tseng, S.H., Tzen, K.Y., Lee, N.C., Chien, Y.H., Snyder, R.O., Byrne, B.J., Tai, C.H. and Wu, R.M. (2012) Gene therapy for aromatic L-amino acid decarboxylase deficiency. Science Transnational Medicine, 4, 134ra161.
- Marks, W.J., Ostrem, J.L., Verhagen, L., Starr, P.A., Larson, P.S., Bakay, R.A.E., Taylor, R., Cahn-Weiner, D.A., Stoessl, A.J., Olanow, C.W. and Bartus, R.T. (2008) Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open label, phase I trial. Lancet Neurology, 7, 400-408.
- Herzog, C.D., Dass, B., Holden, J.E., Stansell, J. 3rd, Gasmi, M., Tuszynski, M.H., Bartus, R.T. and Kordower, J.H. (2007) Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Movement Disorders, 22, 1124-1132.
- Ceregene (2009) A phase I/II trial assessing the safety and efficacy of bilateral intraputaminal and intranigral administration of CERE-120 (Adeno-associated virus serotype 2 [AAV2]-neurturin [NTN]) in subjects with idiopathic Parkinson’s disease (06/17/2009). http://videocast.nih.gov/ram/rac061709.ram.
- Bartus, R.T., Herzog, C.D., Chu, Y., Wilson, A., Brown, L., Siffert, J., Johnson Jr., E.M., Olanow, C.W., Mufson E.J. and Kordower, J.H. (2011) Bioactivity of AAV2- neurturin gene therapy (CERE-120): Differences between Parkinson’s disease and nonhuman primate brains. Movement Disorders, 26, 27-36.
- Ceregene (2013) Press release-ceregene reports data from Parkinson’s disease phase 2b study. http://www.ceregene.com/press_041913.asp
- Jarraya, B., Boulet, S., Ralph, G.S., Jan, C., Bonvento, G., Azzouz, M., Miskin, J.E., Shin, M., Delzescaux, T., Drouot, X., Herard, A.-S., Day, D.M., Brouillet, E., Kingsman, S.M., Hantraye, P., Mitrophanous, K.A., Mazarakis N.D. and Palfi, S. (2009) Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Science Translational Medicine, 1, 1-10. doi:10.1126/scitranslmed.3000130
- Oxford Biomedica Press Release. (2011) Oxford biomedica announces interim update on prosavin® phase I/II study in Parkinson’s disease-15/12/2011. http://www.oxfordbiomedica.co.uk/press-releases/oxford-biomedica-announces-interim-update-on-prosavin-r-phase-i-ii-study-in-parkinson-s-disease