Advances in Biological Chemistry
Vol.3 No.3A(2013), Article ID:33484,10 pages DOI:10.4236/abc.2013.33A003
Identification of the amino acid involved in the regulation of bacterial pyruvate, orthophosphate dikinase and phosphoenolpyruvate synthetase
![]()
1Biochemistry and Molecular Biology, James Cook University, Townsville, Australia
2Department of Biology, Minnesota State University, Moorhead, USA
Email: *james.burnell@jcu.edu.au
Copyright © 2013 Rebecca Tolentino et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 25 April 2013; revised 2 June 2013; accepted 12 June 2013
Keywords: Escherichia Coli; Listeria Monocytogenes; Enzyme Regulation; Phosphorylation/Dephosphorylation; Pyruvate Orthophosphate Dikinase; PEP Synthetase
ABSTRACT
Pyruvate, orthophosphate dikinase (PPDK) and phosphoenolpyruvate synthetase (PEPS) catalyze the conversion of pyruvate to phosphoenolpyruvate (PEP). Both are regulated by a phosphorylation-dephosphorylation mechanism involving a bifunctional serine/ threonine kinase and a pyrophosphorylase (PPDK regulatory protein, PDRP, and PEPS regulatory protein, PSRP, respectively). In plants the regulatory mechanism involves phosphorylation of a threonine residue that is separated by a single amino acid from the histidine residue that forms a phosphorylated intermediate during catalysis. Using antibodies, we demonstrated that the regulation of both Listeria monocytogenes PPDK and Escherichia coli PEP synthetase involves the phosphorylation of a threonine residue located close to the catalytic histidine residue. The amino acid located between the regulatory threonine and the catalytic histidine is highly conserved being serine in PPDK and cysteine in PEPS. Using site-directed mutagenesis we have shown that both PPDK and PEPS in which the serine and cysteine residues, respectively, were substituted with an alanine the enzymes could be regulated indicating that the serine and cysteine residues, respectively, are not essential for regulation. We also demonstrated that altering the intermediate amino acid did not alter the specificity of the regulatory proteins for their protein substrates.
1. INTRODUCTION
Pyruvate, orthophosphate dikinase (PPDK; EC 2.7.9.1) catalyzes the reversible conversion of pyruvate to phosphoenolpyruvate (PEP) and is important in C4 plants as it is generally recognized as catalyzing the rate-limiting step of C4 photosynthesis [1]. The enzyme is regulated by three separate mechanisms; by light-dependent gene expression [2,3], end-product inhibition [4] and by lightdependent post-translational modification [5]. The lightdependent post-translational regulation of PPDK is unusual in that it requires the protein to be catalytically phosphorylated prior to inactivation [6], involves an ADP-dependent phosphorylation [7], the activation involves a Pi-dependent dephosphorylation that results in the production of PPi [7] and, finally, a single enzyme catalyzes both the inactivation and the activation reactions [8].
Although details of the regulatory mechanism were elucidated in the middle 1980s the maize PPDK regulatory protein was not cloned until 2006 [9]. The cloning of maize PPDK regulatory protein (PDRP) revealed the regulatory protein had been sequenced from more than 200 bacterial species and had been identified as DUF299 (domain of unknown function). A ClustalW alignment of all known DUF299 amino acid sequences segregates them into two clearly separate clades; one clade is composed of species possessing PPDK and the other clade is composed of species possessing phosphoenolpyruvate synthetase (PEPS; EC 2.7.9.2). It should be pointed out that although the two proteins (PPDK and PEPS) share some homology, especially around a central catalytic site, they can be identified using the signature sequences identified by Tjaden and colleagues [10]. The PEP synthetase regulatory protein (PSRP) from E. coli has been cloned and expressed [11]. A study of the substrate specificity of the regulatory proteins from maize and Escherichia coli showed that the regulatory proteins were specific for their homologous protein substrate; that is, maize leaf PDRP only catalyzes the regulation of PPDK and E. coli PSRP only catalyzes the regulation of PEPS. This finding prompted a comparison of the active sites of PPDK and PEPS. A comparison of known PPDK and PEPS sequences revealed a high level of conservation of the amino acid sequence surrounding the catalytic histidine located within a central domain of the enzymes that was composed of GGXTS/CHAAI/VI/VA/SR with the regulatory threonine and catalytic histidine (shown in bold) conserved in all species.
To date, it has only been assumed that the regulatory mechanism of E. coli PEPS mimics the regulation of maize PPDK in that the threonine residue located in close proximity to the catalytic histidine is phosphorylated during inactivation. This assumption has now been confirmed using antibodies raised against a synthetic peptide containing a phosphothreonine residue. Moreover, this comparison revealed that the two enzymes can be identified by the amino acid located between the catalytic histidine and the regulatory threonine with a serine residue characteristic of all PPDKs and a cysteine residue present in all PEP synthetases (see Figure 1). The similarity of the two amino acids (serine and cysteine) and their ability to be phosphorylated raised two questions. Firstly, could the amino acid situated between the regulatory threonine and the catalytic histidine residue influence substrate specificity and secondly, could it be critical to the regulation of either or both proteins.It has been reported that both the threonine and the serine residues adjacent to the catalytic histidine of Arabidopsis PPDK can be phosphorylated [12]. The fact both cysteine and serine residues can be phosphorylated and that they lie between threonine and histidine residues, also capable of being phosphorylated, stimulated an investigation into the possible role of cysteine and serine in PEPS and PPDK, respectively. To date, reports of the regulation of E. coli PEP synthetase [11,13] have assumed that regulation involves the same mechanism found in plants that is, that the ADP-dependent inactivation of

Figure 1. A comparison of the amino acid sequences surrounding the regulatory threonine residue in maize, Arabidopsis and L. monocytogenes PPDK and E. coli PEP synthetase. The sequence of the phosphorylated peptide used to raise antibodies (A/B) is also provided.
PEPS involves the phosphorylation of a threonine located close to the catalytic histidine. Using antibodies raised against a polypeptide containing a phosphothreonine, the regulation of both E. coli PEP synthetase and L. monocytogenes PPDK has been shown to involve the phosphorylation of a threonine located in close proximity to the catalytic histidine.
To investigate the role of the specific amino acid located between the regulatory threonine and the catalytic histidine, the serine residue of PPDK was replaced with either a cysteine (similar size and capable of being phosphorylated and capable of forming hydrogen bonds), an alanine (similar size but incapable of being phosphorylated or forming hydrogen bonds), or a threonine (capable of being phosphorylated and forming hydrogen bonds but slightly larger than serine) and the specific activity and the ability of the mutated proteins to be regulated (inactivated and activated) were measured. Similarly, the cysteine residue of E. coli PEPS located between the regulatory threonine and the catalytic histidine was replaced with either a serine, a threonine or an alanine residue, and the specific activity and ability of the mutated enzymes to be regulated examined.
This report has confirmed the function of what has previously been identified as DUF299 as encoding a protein that catalyzes the reversible phosphorylation-dephosphorylation of either PPDK or PEPS in a mechanism that involves the phosphorylation of a threonine residue located close to the catalytic histidine. In addition, we have shown that the amino acid located between these two amino acids is not critical to either catalysis or regulation but replacing the amino acid with a similar but different amino acid decreases both the specific activity of the enzyme and decreases the rates at which the mutated enzymes are inactivated and activated. In addition, we have demonstrated that changing this amino acid does not alter the substrate specificity of the regulatory protein for its protein substrate.
Finally, this investigation of the regulation of PPDK by PDRP from Listeria monocytogenes represents the first report of the regulation of a bacterial PPDK by posttranslational modification.
2. MATERIALS AND METHODS
2.1. Cloning of L. monocytogenes PPDK and PDRP
For the cloning of PPDK, Listeria monocytogenes (ATCC19177) 0.5 μg genomic DNA was amplified by PCR using 20 pmol of forward and reverse primers (5’- TTTGAAGAGGTGGATCCTGTGAGAAAT-3’ and 5’- TGATTGTCATCGAGCTCTTTTTAGATAGT-3’, respectively), 20 nmol dNTPs, 10 μL Phusion buffer (HF) (5×), 1 unit Phusion High-Fidelity DNA polymerase
(Genesearch) and DNase-RNase free water in a total volume of 50 μL. A PCR protocol of heating at 98˚C for 30 sec, followed by 35 cycles of 98˚C for 15 sec, 55˚C for 30 sec and 72˚C for 4 min was employed. The product was digested with BamHI and SacI and the resulting fragment ligated into pROEXa digested with the same restriction enzymes. Plasmid DNA was amplified in NM522 cells and the DNA sequence determined (Macrogen, Korea). For PDRP, L. monocytogenes genomic DNA was amplified by PCR as described for L. monocytogenes PPDK with the L. monocytogenes PDRP forward and reverse primers (5’-GGAGGAGACCATGGAAAATCCGG-3’ and 5’-CTCACATTATTCACCTCTTC- 3’, respectively) except an annealing temperature of 45˚C was used and the extension time was reduced to 2 min. The product was subcloned into pGEMT, the DNA amplified in NM522 cells, plasmid DNA isolated and the DNA sequence determined. Following confirmation of the DNA sequence, the plasmid DNA was digested with NcoI and SpeI and the 825 bp insert ligated into NcoI and SpeI digested pROEXa. The DNA sequence of the pROEXa DNA was analyzed (Macrogen, Korea).
2.2. Cloning of E. coli PEPS and PSRP
PEPS and PSRP were cloned, expressed and purified as described previously [11].
2.3. Site-Directed Mutagenesis of L. monocytogenes PPDK and E. coli PEPS
Changes to the amino acid situated between the regulatory threonine and the catalytic histidine residues at the active site of both enzymes was achieved using paired mutagenic PCR primers as previously described [12] and the changes confirmed by DNA sequencing (Macrogen, Korea).
2.4. Protein Expression and Enzyme Purification
Recombinant enzymes were expressed as His-tagged fusion proteins and purified by Ni-affinity column chromatography. Cells containing the pROEX-PPDK and the pROEX-PDRP plasmids were cultured overnight in 5 mL LBA broth (LB media plus 100 μg·mL–1 ampicillin) that was used to inoculate 500 mL LBA in 2 L baffled flasks. Cultures were shaken at 37˚C for four hours, the cultures cooled to 16˚C prior to the addition of IPTG to a final concentration of 1 mM and shaken for 48 h. Cells were harvested by centrifugation (4000 × g, 10 min), resuspended in Column buffer (50 mM Hepes-NaOH, 300 mM NaCl, 5 mM MgCl2, 20% glycerol and 10 mM β-mercaptoethanol, pH 7.5), pelleted by centrifugation, resuspended in 10 mL of column buffer and frozen at –80˚C. Following thawing, the cells were broken using a pre-cooled French Press (2˚C) and the cell debris removed by centrifugation at 40,000 × g for 30 min at 2˚C. Imidazole (1 M at pH 7.5) was added to the supernatants to a final concentration of 5 mM before loading onto a 5 mL nickel-NTA column equilibrated with Column buffer at a flow rate of 1 mL·min–1. The column was washed with column buffer until the OD280nm decreased below 0.05, washed with column buffer containing 20 mM imidazole until the OD280nm decreased below 0.05 and protein eluted with Column buffer containing 200 mM imidazole.
2.5. Assay of Enzyme Activities
2.5.1. Spectrophotometric Assay
PPDK activity was measured spectrophotometrically in a coupled enzyme assay in which the PEP-dependent production of pyruvate was linked to NADH oxidation by lactate dehydrogenase. All assays were conducted at 25˚C in a Beckman DU650 spectrophotometer. Reaction mixtures contained 0.5 mM PEP, 0.5 mM AMP, 0.5 mM PPi, 1 mM MgSO4, 5 mM dithiothreitol, 0.2 mM NADH, 100 mM Hepes-KOH, pH7.5, 2.5 units lactate dehydrogenase in a total volume of 1 mL. Reactions were initiated by the addition of enzyme (PPDK or an aliquot of a PDRP reaction assay).
Inactivation reactions contained 0.5 to 1.0 units of PPDK, 50 mM Hepes-KOH, 5 mM MgSO4, 5 mM DTT, 0.1 mM ATP, 2 mM ADP and a variable amount of PDRP in a total volume of 0.22 mL. Inactivation was initiated by the addition of ADP/ATP (2 mM and 0.1 mM, respectively) and 20 mL aliquots removed at varying times intervals and the PPDK activity of these aliquots measured as described above.
2.5.2. Immunogenic Assay
PPDK and PEPS inactivation and activation was followed immunogenically using antibodies that had been specifically raised against a synthetic phosphopeptide corresponding to the threonine phosphorylated during inactivation of maize PPDK (residues 445 to 464 (AVGLILTERGGMpTSHAAVVAR) [14,15]. PDRP activetion activity was measured by initially running an inactivation reaction, removing the ADP and ATP by gel filtration, adding Pi to the eluate and measuring the PPDK activity at different time intervals as described above. An inactivation reaction (0.22 mL) was incubated for 10 min prior to loading onto a column of Sephadex G25 (medium) in a Pasteur pipette (9.0 × 0.5 cm) equilibrated with Column buffer minus NaCl. Column eluate was collected following elution of the void volume (determined using Blue Dextran) and activation initiated by the addition of Pi to a final concentration of 2 mM.
3. RESULTS
3.1. Cloning and Expression of L. monocytogenes PPDK and PDRP
Initial attempts to express active L. monocytogenes PPDK at 37˚C and 25˚C failed due to the expressed protein forming inclusion bodies, however active enzyme was obtained from cells initially grown at 37˚C but protein expression induced only once the cultures had been cooled to 16˚C. The protein expressed at 16˚C was soluble and was stable in storage at 0oC for several weeks. In addition, the protein was stable when stored at –80˚C. The enzyme was not affected by temperature; no changes in activity were detected on cooling or warming after storage at –80˚C or at 0˚C. The protein had an expected subunit size of 97 kDa and a band migrating with about the same mobility as a 100 kDa marker band was detected by Coomassie blue staining of an SDS-PAGE gel and western blotting using tetra-His antibodies. The optimum pH for the enzyme differed depending on the direction of the assay. In the pyruvate synthesizing direction the enzyme had a pH optimum of 7.5 and in the PEP forming direction had a pH optimum between 8.5 and 9.0. Under optimum pH conditions the PEP to pyruvate rate was about 2.5 times faster than the pyruvate to PEP rate. In the PEP synthesizing direction the enzyme was sensitive to MgSO4 concentrations with activity peaking at 1 mM and being inhibited at higher MgSO4 concentrations. The PDRP from L. monocytogenes was successfully expressed at 16˚C and was soluble and stable when stored on ice.
3.2. Inactivation and Activation Activities of L. monocytogenes PDRP
Purified expressed L. monocytogenes PDRP catalyzed both the ADP/ATP-dependent inactivation and the Pidependent activation of purified expressed L. monocytogenes PPDK (Table 1). The inactivation activity was dependent on the presence of both ATP and ADP and ATP could be replaced by PEP. Importantly there was no inactivation observed when PDRP was omitted from assays. Inclusion of pyruvate in inactivation reactions inhibited ADP/ATP-dependent inactivation completely. These results are consistent with the regulatory mechanism observed with plant PPDK suggesting that the posttranslational regulatory mechanism of bacterial PPDK is the same as that determined for plant PPDK. Specifically the mechanisms appear identical in their need of a catalytically phosphorylated form of PPDK as shown by the requirement for ATP or PEP together with ADP for inactivation and inhibition of ADP/ATP (PEP) dependent inhibition due to inclusion of pyruvate; pyruvate inhibits inactivation by dephosphorylating the catalytic histidine.
Activation activities were measured by monitoring inassay activation of inactivated PPDK. Activation assays were conducted by monitoring the increase of PPDK activity in cuvettes to which aliquots (0.02 mL) of inactivation reactions were added and Pi was added to a final concentration of 2 mM. PPDK activity increased in assays to which Pi had been added while PPDK activity remained essentially linear in assays not containing added Pi. This Pi-dependent activation mechanism appears consistent with activation activities previously reported for plant PPDK [16,17] and bacterial PEPS [11]. In plants, the regulatory mechanism of PPDK involves the reversible phosphorylation of a threonine residue located in close proximity to the catalytic histidine [18], however, the amino acids involved in the regulation of the bacterial form of the enzyme have not been investigated.
3.3. Immunogenic Labeling of L. monocytogenes PPDK and E. coli PEPS
In an attempt to gain further evidence for the role of the threonine residue being involved in both the ADP/Pi regulation of L. monocytogenes PPDK and E. coli PEPS, antibodies raised against a peptide synthesized according to the amino acid sequence of maize PPDK, and con-
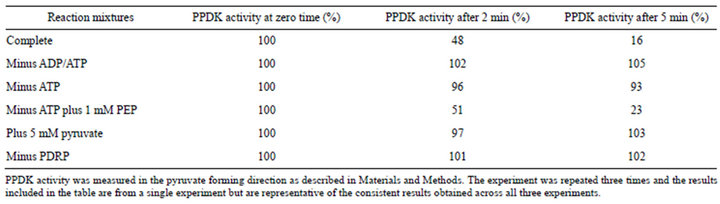
Table 1. Regulation of L. monocytogenes PPDK by L. monocytogenes PDRP.
taining a phosphorylated threonine residue, were used to test their reactivity against inactivated PPDK and PEPS. L. monocytogenes PPDK that had been inactivated in the presence of ADP, ATP and purified L. monocytogenes PDRP reacted immunogenically with the anti-phosphothreonine antibodies (Figure 2). The antibody labeling of PPDK in samples removed from an inactivation assay containing purified L. monocytogenes PPDK, PDRP, ADP and ATP was inversely proportional to the PPDK activity. In contrast, PPDK in samples taken from an activation assay indicated that as PPDK activity increased the level of antibody labeling decreased. And finally, preincubation of the antibody with the synthetic peptide used to raise the antibodies eliminated any labeling of inactivated L. monocytogenes PPDK. Collectively, these results indicate that the regulatory mechanism associated with the post-translational modification of L. monocytogenes PPDK is the same as that in C3 and C4 plants, that is, the mechanism involves the reversible phosphorylation of a threonine residue located in close proximity to the catalytic histidine residue. When the same experiments were conducted with purified expressed E. coli PEPS and PSRP, the same results were obtained. Inactivated PEPS reacted with the anti-phosphopeptide antibodies while activated PEPS did not (Figure 3). However, it should be noted that the intensity of labeling of inactivated E. coli PEPS was considerably lower compared to the labeling of inactivated L. monocytogenes PPDK when similar protein concentrations were probed in Western blots with the same concentrations of antibodies.
Aliquots of an inactivation reaction consisting of purified PPDK, purified PDRP, ADP and ATP were removed and the reactions stopped by adding to a solution containing SDS and EDTA. Equal volumes of the terminated reactions were loaded onto an SDS-PAGE gel, proteins separated and transferred onto PVDF membrane and probed with anti-phosphothreonine peptide antibodies. The positive control contained an aliquot of ADP-inactivated A. thaliana PPDK and the negative control an equivalent amount of A. thaliana PPDK that had not been inactivated.

Figure 2. L. monocytogenes PPDK is phosphorylated during ADP/ATP-dependent inactivation.
Aliquots of an inactivation reaction containing purified E. coli PEPS and PSRP were incubated with ADP and ATP and aliquots removed at different time intervals, the proteins separated and probed with anti-phosphothreonine phosphopeptide antibodies.

Figure 3. Time-course of the ADP/ATP-dependent inactivation of E. coli PEPS.
3.4. Substrate Specificity of PDRP and PSRP
The substrate specificity of the two bacterial regulatory proteins was investigated using both the spectrophotometric and the immunogenic assays. No ADP/ATP-dependent inactivation was detected when either L. monocytogenes PPDK was incubated with E. coli PSRP nor when E. coli PEPS was incubated with L. monocytogenes PDRP in either assay.
3.5. Further Identification of the Amino Acids Involved in Post-translational Modification of L. monocytogenes PPDK and E. coli PEPS
The amino acid sequence surrounding the catalytic sites of both L. monocytogenes PPDK and E. coli PEPS are very similar. In L. monocytogenes the sequence is TSH while in E. coli PEPS the sequence is TCH. Interestingly, all these amino acids can be phosphorylated and it has been reported that both the threonine and the serine residues adjacent to the catalytic histidine of Arabidopsis PPDK can be phosphorylated [12]. The fact both the cysteine and serine residues can be phosphorylated and that they lie between threonine and histidine residues, also capable of being phosphorylated, stimulated an investigation into the possible role of cysteine and serine in PEPS and PPDK, respectively.
3.5.1. Mutagenesis Studies with L. monocytogenes PPDK and E. coli PEPS
The amino acid located between the regulatory threonine and the catalytic histidine residues in both L. monocytogenes PPDK and E. coli PEPS sequences were altered by site-directed mutagenesis, expressed and purified by nickel affinity chromatography; the specific activities of the mutated PPDK and PEPS were determined and the regulation of the mutants by ADP-dependent inactivation and Pi-dependent activation were assessed. Results are summarized in Tables 2 and 3.
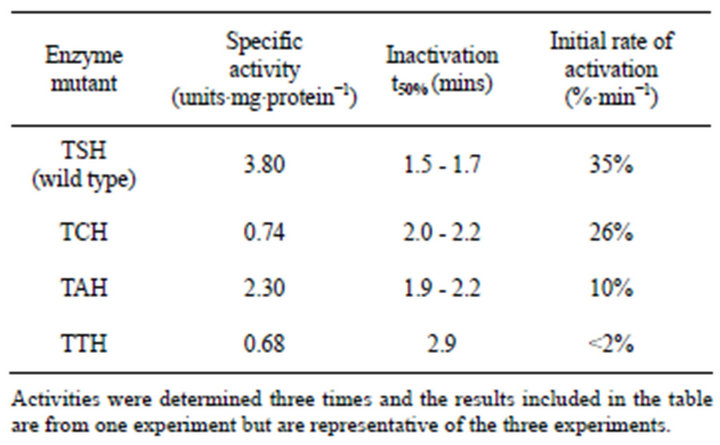
Table 2. Kinetic data for the TXH isozymes of L. monocytogenes PPDK.
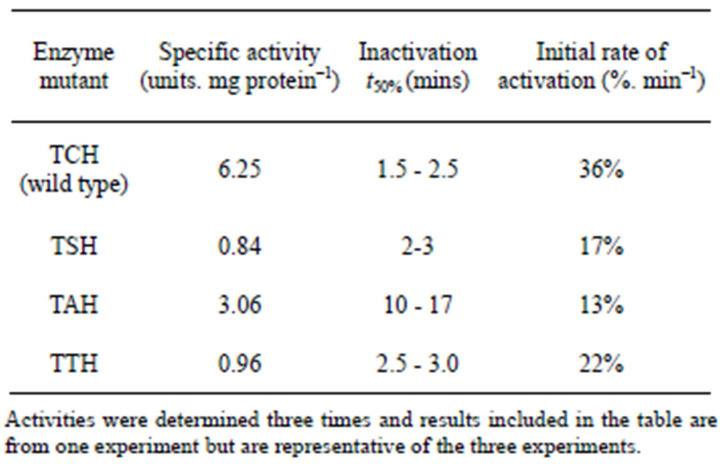
Table 3. Kinetic data for the TXH isozymes of L. monocytogenes PPDK.
1) The effect of Mutagenesis on L. monocytogenes PPDK and Its Regulation All four forms of L. monocytogenes PPDK (wild type, TAH, TCH and TTH) were catalytically active and capable of catalyzing the reaction in either direction, however, the specific activities of the four enzymes differed. Wild type PPDK had the highest specific activity while the TAH mutant was the next most active and the cysteine and threonine mutants similar exhibiting low specific activities. In addition, all four enzymes could be inactivated. The highest rate of inactivation was observed with the wild type enzyme while the cysteine mutant inactivated more rapidly compared to the alanine mutant with the threonine mutant inactivated at the slowest rate. The rates of Pi-dependent activation followed the same order with the wild type activating fastest followed by the cysteine mutant, the alanine mutant and the threonine mutant slowest of the four enzymes. So although the alanine mutant exhibited the second highest specific activity (about 60% of the wild type activity), it exhibited a slightly slower rate of inactivation but a significantly lower rate of Pi-dependent activation compared to the wild type. The TCH mutant had a significantly lower specific activity (about 20% of the wild type), inactivated at about the same rate as the TAH mutant and activated at about 75% the rate of the wild type. The TTH mutant exhibited the lowest specific activity, the slowest rates of inactivation and very low rates of Pidependent activation.
2) The Effect of Mutagenesis on the Specific Activity and Regulation E. coli PEPS The results for PEPS mutants were slightly different from those for the PPDK mutants. The wild type (TCH) PEPS had the highest specific activity, the greatest rate of inactivation, and the highest rate of activation. The TAH mutant had the second highest specific activity, however, of the four PEPS forms, the TAH mutant exhibited the slowest rate of inactivation and the slowest rate of activation Although the TSH and the TTH mutants were the least active PEPS mutants they both exhibited similar rates of inactivation and activation that were significantly greater than the rates observed for the TAH mutant.
These results clearly demonstrate the important role of the amino acid situated between the regulatory threonine and the catalytic histidine residues and, given the similarity of the amino acid sequence immediately downstream from the catalytic histidine that where the active site interacts with the rest of the protein must also differ due to the differences in the activities of the different mutant forms of the enzymes.
The results are consistent with the variable but conserved amino acid not only affecting the rate of catalysis but also affecting the binding (either directly or indirectly) of the regulatory protein to the enzyme substrate (PEPS or PPDK). In the case of alanine, replacement of either the cysteine in PEPS or the serine in PPDK resulted in a halving of the specific activity of the two enzymes. In addition, it indicated that the two amino acids influence the rate of phosphorylation or the rate of pyruvate-dependent dephosphorylation of catalytically phosphorylated histidine. So replacement of either serine or cysteine with an alanine decreased the catalytic rate. One common characteristic of serine and cysteine is that they are both capable of forming hydrogen bonds while alanine is not. One explanation for the decrease in catalytic activity observed with the alanine mutant may be that there is a requirement for the residue occupied by the alanine to form hydrogen bonds with a residue that is either upstream or downstream of the catalytic site. If this were the only effect that the substitution caused you would expect that the rate of inactivation would be decreased by about the same amount as the catalytic rate (due to the fact that ADP-dependent inactivation is dependent on the PPDK being catalytically phosphorylated). However, the rate of ADP-dependent inactivation of the TAH mutant is considerably slower for PEPS. So the effect of a substitution in PEPS has a greater effect on inactivation compared to the reduction in the catalytic reaction. Therefore the substituted residue must be affecting the binding of the regulatory protein to its protein substrate to a greater extent compared to its effect on the rate of ADP-dependent inactivation. Alternatively, the substitution may be affecting the pyruvate-dependent dephosphorylation of the catalytically phosphorylated histidine residue and this would be reflected by a difference in the rate of the reverse reaction compared to the forward reaction.
These results also clearly demonstrate that the amino acid situated between the regulatory threonine and the catalytic histidine is not involved in the ADP-dependent inactivation of either PPDK or PEPS as replacement of the serine and cysteine, respectively, with alanine, an amino acid that could not be phosphorylated, did not eliminate ADP-dependent inactivation. The fact that the alanine mutants did alter the specific activity and the rate of inactivation is evidence that the intervening amino acid plays a role at the active site of the enzyme and confirms the importance of the catalytic phosphorylation of the histidine for inactivation. The fact that the replacement of the serine (in PPDK) and cysteine (in PEPS) affected both the specific activity of the enzymes as well as the rates of inactivation is consistent with the suggestion that the amino acids play a role in the interaction of the catalytic histidine with the ATP-binding site of the enzymes; slower rates of catalytic phosphorylation will cause slower rates of ADP-dependent phosphorylation of the regulatory threonine residues of both enzymes.
Serine and cysteine have a number of properties in common. They are about the same size, are hydrophilic and both can form phosphorylated derivatives. In addition, they are both capable of forming hydrogen bonds with other amino acids. It may also be significant that the two amino acids are encoded by very similar codons. Serine can be encoded by six codons, (UCN, AGU and AGC) while cysteine is encoded by only two codons (UGU and UGC). Of the six serine codons, the serine located at the active site of almost all bacterial and plant PPDKs is encoded by UGU, and the cysteine located at the active site of almost all bacterial PEP synthetases is encoded by UCU indicating that only a single mutation was required to change a serine to a cysteine and vice versa.
Alanine differs from the wild type and other substituted intermediate amino acids in being incapable of being phosphorylated. Given that both alanine mutants are capable of being inactivated and activated indicates that the intermediate amino acid is not directly involved in the phosphorylation-dependent regulation but it may be indirectly involved by influencing the binding of the regulatory protein to its enzyme substrate.
3.6. Investigation of the Phosphorylation Status of the Catalytic Histidine during Pi-dependent Activation
It has been suggested that the activity of PPDK is regulated by the phosphate group attached to the regulatory threonine preventing pyruvate or PEP from binding to the catalytic histidine either sterically or due to the electronegativity of the negative charge [19]. The finding that the phosphorylation status of the regulatory threonine residue of both L. monocytogenes PPDK and E. coli PEPS could be determined using the anti-phosphothreonine peptide antibodies prompted a re-examination of the phosphorylation status of the catalytic histidine. In an attempt to investigate the effect of the phosphorylation status of the catalytic histidine on the rate of Pi-dependent activation the catalytic histidine of the L. monocytogenes was replaced with either an aspartate or a glutamate; these negatively charged amino acids were used to substitute for the catalytic histidine in an attempt to mimic the negative charge of catalytically-phosphorylated histidine. As expected, the mutated enzyme lacked catalytic activity. When the mutated enzyme was incubated with purified L. monocytogenes PPDK regulatory protein and ADP (in either the presence or absence of ATP), no phosphorylation of the regulatory threonine was detected using the anti-phosphothreonine antibodies that clearly reacted with inactivated wild type PPDK from L. monocytogenes. This inability to phosphorylate mutated PPDK precluded further study of the phosphorylation status of PPDK using the anti-phosphothreonine specific antibodies and indicated that an alternate strategy is required to investigate this characteristic further.
4. DISCUSSION
The results obtained in this study clearly demonstrate that the PPDK from bacteria (L. monocytogenes) is regulated by a mechanism that is identical with the mechanism described for plant PPDK. Using specific antibodies we have clearly demonstrated that the ADP-dependent inactivation of bacterial PPDK is due to phosphorylation of a threonine residue located very close to the catalytically important histidine residue. And using the same antibodies we have shown clearly that the mechanism of regulating E. coli PEP synthetase is very similar to the mechanism operating for L. monocytogenes PPDK.
The identification and the characterization of a bacterial PPDK regulatory system together with the characterization of a PEPS regulatory system has allowed a comparative study of homologous proteins.
The two systems are very similar sharing common characteristics including 1) a requirement for catalytic phosphorylation prior to inactivation 2) ADP being the phosphate donor in the inactivation reaction 3) a threonine residue close to the catalytic histidine being responsible for enzyme inactivation 4) inorganic phosphate being required for activation, and 5) both reactions being catalyzed by a single protein.
A close examination of the regulatory mechanisms involved in regulating PPDK and PEPS activity reveals that there is only one major difference between the two enzymes and that is the involvement of an extra phosphate in the reaction catalyzed by PPDK. However, this difference may affect the relationship between enzyme activity (PPDK versus PEPS) and rates of activation of PPDK and PEPS, respectively, due to the fact that the end product of both PPDK and PEPS activation is pyrophosphate that, in the case of PPDK, is also an end product and an inhibitor of PPDK activity. This is not the case for PEPS; pyrophosphate is not an end product of the reaction catalyzed by PEPS (see Figure 4). The similarity between the two regulatory systems has also been demonstrated in parallel mutagenesis studies [13, 20]. These studies revealed that two separate regions are involved in catalysis; a central P-loop required for the binding of the regulatory protein to its homologous protein substrate (PPDK for PDRP and PEPS for PSRP) and a C-terminal region that binds ADP and phosphate. Furthermore, these two sites are likely to be juxtaposed in the catalytically active regulatory proteins. Previous studies have been conducted to identify factors that affect the partitioning of pyruvate between anabolic and catabolic pathways. In E. coli cells grown on three-carbon sources such as lactate, pyruvate or alanine, pyruvate occupies the major metabolic branch point. The relative activities of PEP synthetase and pyruvate dehydrogenase are expected to control the relative rates of the anabolic and catabolic fates for pyruvate. Kinetic studies conducted using purified PEP synthetase and pyruvate dehydrogenase complex from E. coli were conducted in the early 1970s [21]. Significantly, these studies were performed in the absence of PEPS regulatory protein. In light of the discovery of the presence of PSRP in bacteria the effect of adenylate energy charge on the metabolic partitioning of pyruvate needs to be revisited using a system in which PSRP, PEPS and the pyruvate dehydrogenase complex are present.
With respect to the regulation of both PPDK and PEPS there has been some confusion as to the physiological origin of the catalytic phosphate required prior to ADPdependent inactivation. Since the role of both enzymes is anabolic and the level of activity of both PEPS and PPDK will be controlled by the intracellular ADP concentration, then it is logical to conclude that the phosphorylation of the catalytic histidine residue will be a
Since the catalytically-phosphorylated form of PPDK is the substrate for ADP-dependent inactivation, the rate of inactivation will be controlled by both the ratio of PEP and pyruvate as well as the concentration of ADP.
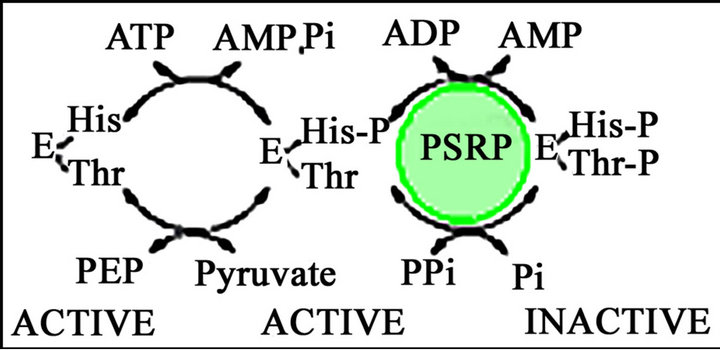
Figure 4. A scheme showing the regulation of E. coli PEPS.
function of the PEP to pyruvate ratio rather than a function of the ATP to AMP ratio. Assuming that this is the case any accumulation of PEP will increase the level of catalytic phosphorylation, and the level of enzyme activity will then be controlled by the intracellular ADP concentration. The requirement for PEPS (and PPDK) to be catalytically phosphorylated prior to activation allows an extra layer of complexity to the regulatory mechanism that permits the cell to more effectively partition pyruvate between energy use for anabolic reactions or energy production via the Krebs cycle and the electron transport chain.
It is significant that PPDK regulatory proteins are notpresent in either PPDK-possessing Archaea or unicellular eukaryotes and this may be an indication of their metabolic role in the conversion of PEP to pyruvate via glycolysis. This makes sense from an energetics viewpoint, as it would be counter productive to inactivate PPDK when ADP levels were high (ATP levels low) as the role of PPDK in these organisms is to increase the efficiency of ATP production during glycolysis. Given that the PEPS—catalyzed reaction is essentially irreversible, the presence of both PPDK and PDRP in bacteria may be an indication that the role of PPDK in bacteria is exclusively gluconeogenic.
In hindsight it makes sense that the Pi-dependent activation of PPDK results in the formation of PPi, a potent feedback inhibitor of PPDK activation and a product of the reaction catalyzed by PPDK. An understanding of the regulation of both PPDK and PEPS points to the fact that the role of PPDK in bacteria is to synthesize PEP from pyruvate rather than being involved in increasing the efficiency of ATP synthesis via glycolysis. Support for this hypothesis is two-fold. 1) ATP synthesis would tend to be stimulated by low adenylate energy charge (AMP concentrations high), conditions that would favour PPDK inactivation and inhibit PPDK activation). And 2) PDRP is absent from organisms (e.g., Entamoeba histolytica and Giardia lamblia) that use PPDK to catalyze the conversion of PEP to pyruvate [22,23]. Similarly, another oddity of the regulatory mechanism appears to be logical; that is the formation of pyrophosphate during phosphate-dependent activation as opposed to a simple phosphatase reaction; the release of inorganic phosphate would lead to the production of a substrate for activation and feed forward stimulation. The production of pyrophosphate, an end product of PPDK also provides a link to the feedback inhibition of PPDK.
The identification of a functional PPDK regulatory protein in bacteria coupled with the previous demonstration of an active PEPS regulatory protein may provide an opportunity for further comparative studies to expand our understanding of the regulatory proteins themselves.
5. SUMMARY
1) L. monocytogenes PPDK and PDRP have been cloned and expressed.
2) L. monocytogenes PPDK is regulated by a posttranslational modification mechanism involving an ADPdependent phosphorylation of a threonine residue located in close proximity to the active site histidine residue.
3) Both inactivated L. monocytogenes PPDK and E. coli PEPS cross-react with antibodies raised against a synthetic phosphopeptide corresponding to the threonine phosphorylated during inactivation of maize PPDK.
4) Both L. monocytogenes PPDK and E. coli PEPS could be inactivated and reactivated when the amino acid located between the regulatory threonine and the catalytic histidine residue was replaced with an amino acid that could not be phosphorylated (alanine).
5) The substrate specificity of the regulatory protein for its protein substrate was not altered by changing the amino acid separating the regulatory threonine or the catalytic histidine (serine in PPDK and cysteine in PEPS).
REFERENCES
- Edwards, G.E., Nakamoto, H., Burnell, J.N. and Hatch, M.D. (1985) Pyruvate, Pi dikinase and NADP-malate dehydrogenase in C4 photosynthesis: Properties and mechanism of light/dark regulation. Annual Reviews of Plant Physiology, 36, 255-286. doi:10.1146/annurev.pp.36.060185.001351
- Hudspeth, R.L., Glackin C.A., Bonner J. and Grula J.W. (1986) Genomic and cDNA clones for maize phosphoenolpyruvate carboxylase and pyruvate, orthophosphate dikinase: Expression of different gene-family members in leaves and roots. Proceedings of the National Academy of Science USA, 83, 2884-2888. doi:10.1073/pnas.83.9.2884
- Sheen, J.-Y. and Bogorad, L. (1987) Regulation of levels of nuclear transcripts for C4 photosynthesis in bundle sheath and mesophyll cells of maize leaves. Plant Molecular Biology, 8, 227-238. doi:10.1007/BF00015031
- Andrews T.J. and Hatch M.D. (1969) Properties and mechanism of action of pyruvate, phosphate dikinase from leaves. Biochemical Journal, 114, 117-125.
- Burnell, J.N. and Hatch, M.D. (1985) Light-dark modulation of leaf pyruvate, Pi dikinase. Trends in Biochemical Sciences, 111, 288-291. doi:10.1016/0968-0004(85)90090-8
- Burnell, J.N. and Hatch, M.D. (1984) Regulation of C4 photosynthesis: Catalytic phosphorylation as a prerequisite for ADP-mediated inactivation of pyruvate, Pi dikinase. Biochemical and Biophysical Research Communications, 118, 65-72. doi:10.1016/0006-291X(84)91068-4
- Ashton, A.R., Burnell, J.N. and Hatch, M.D. (1984) Regulation of C4 photosynthesis: Inactivation of pyruvate, Pidikinase by ADP-dependent phosphorylation and activation by phosphorolysis. Archives of Biochemistry and Biophysics, 230, 492-503. doi:10.1016/0003-9861(84)90429-6
- Burnell, J.N. and Hatch, M.D. (1983) Dark-light regulation of pyruvate, Pi dikinase in C4 plants: Evidence that the same protein catalyses activation and inactivation. Biochemical and Biophysical Research Communications, 111, 288-293. doi:10.1016/S0006-291X(83)80149-1
- Burnell, J.N. and Chastain, C.J. (2006) Cloning and expression of maize-leaf pyruvate, Pi dikinase regulatory protein gene. Biochemical and Biophysical Research Communications, 345, 675-680. doi:10.1016/j.bbrc.2006.04.150
- Tjaden, B., Plagens, A., Dorr, C., Siebers, B. and Hensel, R. (2006) Phosphoenolpyruvate synthetase and pyruvate, phosphate dikinase of Thermoproteus tenax: Key pieces in the puzzle of archaeal carbohydrate metabolism. Molecular Microbiology, 60, 287-298. doi:10.1111/j.1365-2958.2006.05098.x
- Burnell, J.N. (2010) Cloning and characterization of Escherichia coli DUF299; A bifunctional ADP-dependent kinase-Pi-dependent pyrophosphorylase from bacteria. BMC Biochemistry, 11, 1. doi:10.1186/1471-2091-11-1
- Reiland, S., Messerli, G., Baerenfaller, K., Gerrits, B., Endler, A., Grossmann, J., Gruissem, W. and Baginsky, S. (2009) Large-scale Arabidopsisphosphoproteome profiling reveals novel chloroplast kinase substrates and phosphorylation networks. Plant Physiology, 150, 889-903. doi:10.1104/pp.109.138677
- Bartlett, S., Seeliger, J. and Burnell, J.N. (2012) Identification of critical residues in the bifunctional phosphorenolpyruvate synthetase kinase/phosphotransferase of Escherichia coli. Current Topics in Biochemical Research, 14, 1.
- Chastain, C.J., Botschner, M., Harrington, G.S., Thompson, B.J., Mills, S.E., Sarath, G. and Chollet, R. (2000) Further analysis of maize C4-pyruvate, orthophosphate dikinase phosphorylation by its bifunctional regulatory protein using selective substitutions of the regulatory Thr- 456 and catalytic His-458 residues. Archives of Biochemistry and Biophysics, 375, 165-170. doi:10.1006/abbi.1999.1651
- Chastain, C.J., Fries, J.P., Vogel, J.A., Randklev, C.L., Vossen, A.P., Dittmer, S.K., Watkins, E.E., Fiedler, L.J., Wacker, S.A., Meinhover, K.C., Sarath, G. and Chollet, R. (2002) Pyruvate, orthophosphate dikinase in leaves and chloroplasts of C3 plants undergoes light-/dark-induced reversible phosphorylation. Plant Physiology, 128, 1368- 1378. doi:10.1104/pp.010806
- Burnell, J.N. (1984) Regulation of C4 photosynthesis: catalytic dephosphorylation and Pi-mediated activation of pyruvate, Pi dikinase. Biochemical and Biophysical Research Communications, 120, 559-565. doi:10.1016/0006-291X(84)91291-9
- Chastain, C.J. and Chollet, R. (2003) Regulation of pyruvate, orthophosphate dikinase by ADP-/Pi-dependent reversible phosphorylation in C3 and C4 plants. Plant Physiology and Biochemistry, 41, 523-532. doi:10.1016/S0981-9428(03)00065-2
- Burnell, J.N. (1984) Regulation of C4 photosynthesis: proximity of the catalytically important histidine and the regulatory important threonine residue of pyruvate, Pi dikinase. Biochemistry International, 9, 683-689.
- Burnell, J.N. and Hatch, M.D. (1985) Regulation of C4 photosynthesis: Purification and properties of the protein catalyzing ADP-mediated inactivation and Pi-mediated activation of pyruvate, Pi dikinase. Archives of Biochemistry Biophysics, 237, 490-503. doi:10.1016/0003-9861(85)90302-9
- Astley, H., Parsley, K., Aubry, S.,Chastain, C.J., Burnell, J.N., Webb, M.E. and Hibberd, J.M. (2011) The pyruvate, orthophosphate dikinase regulatory proteins of Arabidopsis are both functional and interact with the catalytic and nucleotide binding domains of pyruvate, orthophosphate dikinase. The Plant Journal, 68, 1070-1081. doi:10.1111/j.1365-313X.2011.04759.x
- Chulavatnatol, M. and Atkinson, D.E. (1973) Kinetic competition in vitro between phosphoenolpyruvate synthetase and the pyruvate dehydrogenase complex from Escherichia coli. Journal of Biological Chemistry, 248, 2716- 2721.
- Varela-Gómez, M., Moreno-Sánchez, R., Pardo, J.P. and Perez-Montfort, R. (2004) Kinetic mechanism and metabolic role of pyruvate phosphate dikinase from Entamoebahistolytica. Journal of Biological Chemistry, 279, 54124- 54130. doi:10.1074/jbc.M401697200
- Feng, X.M., Cao, L.J., Adam, R.D., Zhang, X.C. and Lu, S.Q. (2008) The catalyzing role of PPDK in Giardia lamblia. Biochemical and Biophysical Research Communications, 367, 394-398. doi:10.1016/j.bbrc.2007.12.139
NOTES
*Corresponding author.