Open Journal of Physical Chemistry
Vol.07 No.02(2017), Article ID:76164,22 pages
10.4236/ojpc.2017.72005
Simplified Coarse-Grained Dynamic Model for Real Gases
Panagis G. Papadopoulos1, Christopher G. Koutitas1, Yannis N. Dimitropoulos2, Elias C. Aifantis1
1Department of Civil Engineering, Aristotle University of Thessaloniki, Thessaloniki, Greece
2Department of Chemistry, University of Ioannina, Ioannina, Greece

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 13, 2017; Accepted: May 13, 2017; Published: May 16, 2017
ABSTRACT
A simplified model is proposed for an easy understanding of the coarse- grained technique and for achieving a first approximation to the behavior of gases. A mole of a gas substance, within a cubic container, is represented by six particles symmetrically moving. The impacts of particles on container walls, the inter-particle collisions, as well as the volume of particles and the inter-particle attractive forces, obeying a Lennard-Jones curve, are taken into account. Thanks to the symmetry, the problem is reduced to the nonlinear dynamic analysis of a SDOF oscillator, which is numerically solved by a step- by-step time integration algorithm. Five applications of proposed model, on Carbon Dioxide, are presented: 1) Ideal gas in STP conditions. 2) Real gas in STP conditions. 3) Condensation for small molar volume. 4) Critical point. 5) Iso-kinetic energy curves and iso-therms in the critical point region. Results of the proposed model are compared with test data and results of the Van der Waals model for real gases.
Keywords:
Real Gases, Coarse-Grained Molecular Dynamics, Particles Volume, Inter-Particle Attractive Forces, Lennard-Jones Curve, Step-by-Step Time Integration Algorithm, Condensation, Critical Point, Iso-Kinetic Energy Curves, Iso-Therms, Van der Waals Model

1. Introduction
Recently, in Computational Chemistry, the coarse-grained molecular dynamics technique is often used, by which millions of molecules are represented by a few hundred particles [1] - [13] . For example, if a mole of a gas substance is simulated by a thousand particles, by use of Avogadro number, it is noticed that every particle represents about 6 × 1020 molecules. By this technique, the computational handling of chemical problems becomes possible and usually a satisfactory approximation to observed behavior is achieved.
If we consider an amount of a gas substance, represented by a few particles, first in a large container (Figure 1(a)) and then in a small container (Figure 1(b)), the following two observations can be made [14] :
1. In the large container of Figure 1(a), the volume of particles is not significant, compared with the volume of the container. On the contrary, in the small container of Figure 1(b), the volume of particles is significant.
2. In the large container of Figure 1(a), the mean distance between a couple of particles is large, so, as well known from Physical Chemistry [14] and described by Lennard-Jones curve [15] , the inter-particle attractive forces result small up to negligible. On the contrary, in the small container, the mean distance, between a couple of particles, is small, so the inter-particle attractive forces exhibit significant values.
For the above two reasons, for a quite large molar volume, as in Figure 1(a), the volumes of particles and the inter-particle attractive forces can be ignored. So, the amount of gas substance under consideration obeys the ideal gas laws.
On the contrary, for a small molar volume, as in Figure 1(b), the particles volumes and the inter-particle attractive forces must be taken into account. That is, we have a real gas, which significantly deviates from the behavior of ideal gases.
J. D. van der Waals [16] , by taking into account the molecular volumes and the inter-molecular attractive forces, developed a semi-rational, semi-empirical model, which is simple and exhibits a satisfactory approximation to the observed behavior of real gases.
The kinetic behavior of gases, ideal and real ones, is often described by the Maxwell-Boltzmann stochastic model [17] . The stochastic models are accurate but complicated. On the other hand, they obey some required symmetries. And it is recognized [18] [19] that, alternatively to a stochastic model, a symmetric deterministic model can be used, which is much simpler, but usually exhibits satisfactory approximation to test data.
 (a) (b)
(a) (b)
Figure 1. An amount of a gas substance, represented by a few particles. (a) In a large container; (b) In a small container.
Also, a very coarse-grained model can be used, that is consisting of very few, very large particles. This is similar to the concept of fundamental vibration mode of structural dynamics. Where there exist a lot of high vibration modes (with small periods and usually small amplitudes, too), which are of negligible interest, but complicate the computation and require a very small time steplength. Whereas, the fundamental vibration mode is the simplest mode and, at the same time, the most representative of the dynamic behavior of the structure.
In the present work, such a symmetric deterministic model is proposed for real gases, which is very coarse-grained, that is, it consists of very few - very large particles, and is compared to corresponding test data [14] [20] , as well as to results of the Van der Waals model [14] [16] for real gases.
In the recent literature on the coarse-grained technique [1] - [13] , advanced models are proposed, for the accurate description of the behavior of real materials, which can be used in the Design. Whereas, the proposed here simplified model, aims to an easy understanding, of the coarse-grained technique, by researchers of other than Chemistry fields and to a first approximation to the behavior of gases.
2. Proposed Model
A mole of a gas substance is considered, within a cubic container of side L (Figure 2), represented by six equal spherical particles, each one with mass m = M/6, where M molar mass. A reference axes system Ixyz is considered, with origin I at the center of cube and the axes x, y, z parallel to the principal directions of cube. The centers of the six particles are located on the axes x, y, z, initially at the middles of distances of I from the centers of six faces of cube, that is they have initial coordinates 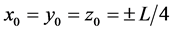 (Figure 2).
(Figure 2).
We assume that the six particles move symmetrically. So, by considering the plane Iyz (Figure 3(a)), what happens in this plane, the same happens in the planes Ixy, Ixz, too.

Figure 2. Initial positions 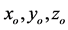 of the six particles of proposed model in the cubic container of side L.
of the six particles of proposed model in the cubic container of side L.
 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 3. The three successive characteristic states of the particles of proposed model, within the plane Ixy (a) of the container; (b) Initial state; (c) Impacts of particles on container walls; (d) Inter-particle collisions in the central region of the container. The arrows represent instantaneous velocities of the particles.
The particles are initially provided with equal speeds directed outwards. And they pass successively through three characteristic states: 1) Initial state (Figure 3(b)). 2) Impacts of particles on container walls (Figure 3(c)), where their speeds are inversed. 3) Inter-particle collisions, in the central region of the container, where again their speeds are inversed.
Obviously, all the six particles, as they move symmetrically, they pass simultaneously from the above three characteristic states.
The mutual repulsive forces F between a particle and a container wall (Figure 4(a)) are described by the repulsive part of a Lennard-Jones curve [14] [15] (Figure 4(b)). If the perpendicular distance 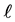 of the center of particle from container wall is quite large:
of the center of particle from container wall is quite large:
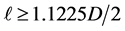
where D diameter of particle and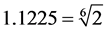 , then
, then

that is no force F is developed between the particle and the wall.
On the contrary, if 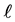 is quite small:
is quite small:
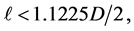
a mutual repulsive force F is developed between particle and wall, given by the equation (Figure 4(b)):
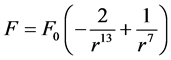 (1)
(1)
where 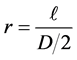 and the determination of force coefficient F0 of Lennard-Jones curve is described in the following Section 4.1.
and the determination of force coefficient F0 of Lennard-Jones curve is described in the following Section 4.1.
The mutual forces F, attractive or repulsive, between any couple of particles, with a distance 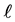 of their centers (Figure 5(a)), are described by the Lennard-Jones curve of Figure 5(b). In Figure 5(c) is shown enlarged the attractive part of this Lennard-Jones curve, because of the significance of attractive forces.
of their centers (Figure 5(a)), are described by the Lennard-Jones curve of Figure 5(b). In Figure 5(c) is shown enlarged the attractive part of this Lennard-Jones curve, because of the significance of attractive forces.
For any distance 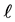 of the centers of two particles, their mutual force F is given by the following equation, corresponding to the Lennard-Jones curves of Figure 5(b) and Figure 5(c):
of the centers of two particles, their mutual force F is given by the following equation, corresponding to the Lennard-Jones curves of Figure 5(b) and Figure 5(c):
 (a) (b)
(a) (b)
Figure 4. (a) Perpendicular distance 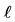 of a particle from container wall and mutual repulsive force F between particle and wall; (b) Repulsive part of a Lennard-Jones curve describing the function
of a particle from container wall and mutual repulsive force F between particle and wall; (b) Repulsive part of a Lennard-Jones curve describing the function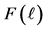 .
.

Figure 5. (a) Distance  and mutual inter-particle force F, attractive (+) or repulsive (−), between a couple of particles; (b) Lennard-Jones curve describing the function
and mutual inter-particle force F, attractive (+) or repulsive (−), between a couple of particles; (b) Lennard-Jones curve describing the function ; (c) The Lennard-Jones curve with enlarged its attractive part.
; (c) The Lennard-Jones curve with enlarged its attractive part.
 (2)
(2)
where 
Thanks to the symmetrical movement of all the six particles, it is enough to study the movement of only one particle, let choose that on right part of axis Iy of Figure 2 and name it R (right), as shown in Figure 6. If the distance of this particle from container wall at right is quite small , then the mutual particle-wall repulsive force is activated, according to Figure 4 and equation (1), and let call this force Fw.
, then the mutual particle-wall repulsive force is activated, according to Figure 4 and equation (1), and let call this force Fw.
At left of Figure 6, the particle R interacts with the other five particles of the model. The relative position of point R under consideration with respect to four of these particles, F, B, O, U (front, back, over, under) is symmetric. So, the horizontal resultant F4 of the four equal forces (attractive or repulsive), by which the points F, B, O, U act on point R is, according to Figure 5 and Figure 6 and Equation (2):

Figure 6. The particle R under consideration is moving on axis Iy. At right, it reaches up to impact with container wall. At left, it interacts with all the other five particles: (F, B, O, U) and L.
 (3)
(3)
where  and
and .
.
Finally, the left particle L acts on particle R, by an, always attractive, force , which is given by Figure 5 and Figure 6 and Equation (2), with
, which is given by Figure 5 and Figure 6 and Equation (2), with .
.
So, the horizontal force on particle R, due to inter-particle action, is

and the total horizontal force on particle R, due to inter-particle action and impact on wall is,

and the acceleration of particle R, under consideration, is, at any instant,
 .
.
3. Step-by-Step Algorithm
It has been described, in the previous Section 2, how the proposed model is reduced, thanks to the symmetric movement of its six particles, to the study of the movement of the single particle R (Figure 6). So, the problem is reduced to the nonlinear dynamic analysis of a SDOF (single degree of freedom) oscillator, which can be solved numerically by a step-by-step time integration algorithm.
For this purpose, the algorithm of trapezoidal rule (or Newmark algorithm of constant average acceleration) is chosen, combined with a predictor-corrector technique, with two corrections per step [21] , which has been proved simple and effective.
3.1. Flow-Chart
The flow-chart of the proposed algorithm is shown in Figure 7 and is briefly described below.
First, the constant input data are read: particle mass m and diameter D, force coefficient  of L-J (Lennard-Jones) curve, side L of cubic container, time step-length
of L-J (Lennard-Jones) curve, side L of cubic container, time step-length  of the algorithm.
of the algorithm.
The initial conditions are read: position y, temperature T and speed v of the particle R under consideration. The initial speed v results from initial temperature T, by a thermodynamic postulate, which will be described in following section 4.1.
The subroutine L-J (Lennard-Jones) is called, which, from the initial position y of the particle, determines the initial forces  and
and , acting on it, and its initial acceleration
, acting on it, and its initial acceleration .
.

Figure 7. Flow-chart of the proposed step-by-step time integration algorithm.
Within each step of the algorithm, first the steps counter n is increased by 1 and time t by .
.
Then, the prediction is performed, which determines the predicted values  of state variables and the subroutine L-J is called, which, from the given
of state variables and the subroutine L-J is called, which, from the given  determines the predicted acceleration
determines the predicted acceleration .
.
The first correction, by trapezoidal rule, determines the first corrections  of the state variables. The subroutine L-J, from
of the state variables. The subroutine L-J, from , finds the first correction of acceleration
, finds the first correction of acceleration .
.
The second and final correction finds the final values of , for present step, and the subroutine L-J, from y, determines the final forces
, for present step, and the subroutine L-J, from y, determines the final forces ,
,  and acceleration
and acceleration , for present step.
, for present step.
The output, of present step of algorithm, is printed: steps counter n, time t, position y and speed v of the particle, forces  due to inter-particle action and
due to inter-particle action and  due to impact on wall, instantaneous total kinetic energy, for all six particles
due to impact on wall, instantaneous total kinetic energy, for all six particles , where M = 6 m.
, where M = 6 m.
At the end of step of algorithm, three summations are made: The present force , due to inter-particle action, is summed to
, due to inter-particle action, is summed to . The present
. The present , due to impact on wall is summed to
, due to impact on wall is summed to . The present second power of speed
. The present second power of speed  is summed to
is summed to .
.
Then, if the first cycle of oscillation has not yet been completed, we continue with the next step of the algorithm.
When the first cycle of oscillation is completed, by returning to the initial state, if we continued the algorithm, everything would be repeated the same, with only a small algorithmic damping. So, the algorithm is interrupted and the global output data are printed, which are:
1) Mean inter-particle force .
.
2) Mean particle-wall impact force .
.
It results , as is due for global equilibrium.
, as is due for global equilibrium.
3) Pressure on wall , in Pascals = N/m2, which, divided by 101,325 N/m2, turns to atm units.
, in Pascals = N/m2, which, divided by 101,325 N/m2, turns to atm units.
4) Mean 2nd power of speed , and mean (rms) speed
, and mean (rms) speed  which, for small molar volumes, results significantly lower than the initial speed.
which, for small molar volumes, results significantly lower than the initial speed.
5) Mean total kinetic energy  in Joules = N×m.
in Joules = N×m.
3.2. Computer Program
Based on the step-by-step time integration algorithm, described in the previous section 3.1. and the flow-chart of Figure 7, a simple and short computer program has been developed, with only about 45 Fortran instructions for the main program of step-by-step algorithm and only about 25 Fortran instructions for the L-J (Lennard-Jones) subroutine, that is totally only about 70 Fortran instructions.
The program is written in the version Force 2.0 of Fortran, whose compiler is free available, even in Internet cafés.
4. Applications
The proposed simplified coarse-grained dynamic model for real gases is applied on Carbon Dioxide (CO2), which exhibits a particular behavior in Critical point region, as it condensates for rather high temperatures, slightly lower than .
.
From the next Section 4.1, it is apparent that, in order to calibrate the proposed model on other gases, the following data are required: molar mass, incompressibility limit of molar volume, as well as temperature, pressure and molar volume at the Critical Point.
4.1. Determination of Parameters
The numerical values of parameters of proposed model are determined below, which will be used in the following applications:
1) The mass of a particle is , where M = 0.044 kgr is the molar mass of Carbon Dioxide.
, where M = 0.044 kgr is the molar mass of Carbon Dioxide.
2) The diameter D of a particle is determined on the basis of criterion of in- compressibility of closely-packed equal spherical particles, as shown in Figure 8. According to experimental evidence [14] [16] [20] , the in-compressibility limit of molar volume, for Carbon Dioxide, is about V = 50 cm3, which corresponds to a cubic container with side L = 3.684 cm. In Figure 8, the spherical particles of proposed model are shown, closely -packed in such a small container. The inter-particle distances are 1.1225D and the particle-wall distances are 1.1225D/2, as, for smaller distances, mutual repulsive forces begin to develop (see Figure 4 and Figure 5). So, on the basis of configuration of Figure 8, the following inequality must be valid:
 from which
from which and a value D = 1.35 cm is chosen.
and a value D = 1.35 cm is chosen.

Figure 8. Closely-packed particles of proposed model, in a small container, in the limit of in-compressibility of Carbon Dioxide, according to experimental evidence [14] [16] [20] .
3) The force coefficient  of Lennard-Jones curve (Equations (1) and (2)) is determined by calibration of proposed model on the Critical point of Carbon Dioxide. For a cubic container with the critical volume
of Lennard-Jones curve (Equations (1) and (2)) is determined by calibration of proposed model on the Critical point of Carbon Dioxide. For a cubic container with the critical volume , that is
, that is  and for the critical temperature
and for the critical temperature , according to test data [14] [16] [20] , various values of
, according to test data [14] [16] [20] , various values of  are tried, until to achieve a value of pressure, with satisfactory approximation to the experimental critical pressure of CO2,
are tried, until to achieve a value of pressure, with satisfactory approximation to the experimental critical pressure of CO2, . In this way, a value
. In this way, a value  is obtained. Application on Critical point is described in Section 4.5.
is obtained. Application on Critical point is described in Section 4.5.
4) For the side L of cubic container, in STP (standard temperature-pressure) conditions, the value  is chosen, which corresponds to a volume V = 22.414 liters. And, in the Critical point region of CO2, values of L ranging from 4.0cm up to 9.0 cm are used, which correspond to container volumes V = L3 ranging from 64 cm3 up to 729 cm3.
is chosen, which corresponds to a volume V = 22.414 liters. And, in the Critical point region of CO2, values of L ranging from 4.0cm up to 9.0 cm are used, which correspond to container volumes V = L3 ranging from 64 cm3 up to 729 cm3.
5) The time step-length  of the proposed step-by-step integration algorithm can be determined on the basis of the accuracy criterion of the algorithm [21] ,
of the proposed step-by-step integration algorithm can be determined on the basis of the accuracy criterion of the algorithm [21] ,

where .
.
A maximum stiffness  appears in two cases: inter-particle collision (Figure 5 and Figure 6 and Equation (3)) and particle-wall impact (Figure 4 and Equation (1)). By linearization of branch AK in the Lennard-Jones curves in Figure 4(b) and Figure 5(b), the stiffness of the above two cases can be determined on the basis of Figure 9(a) and Figure 9(b), respectively. It is observed that both give the same value of stiffness, represented by Figure 9(c), which is
appears in two cases: inter-particle collision (Figure 5 and Figure 6 and Equation (3)) and particle-wall impact (Figure 4 and Equation (1)). By linearization of branch AK in the Lennard-Jones curves in Figure 4(b) and Figure 5(b), the stiffness of the above two cases can be determined on the basis of Figure 9(a) and Figure 9(b), respectively. It is observed that both give the same value of stiffness, represented by Figure 9(c), which is
 So,
So,
 and the time step-length of the algorithm must be, for accuracy [21] :
and the time step-length of the algorithm must be, for accuracy [21] :

 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 9. (a) Maximum stiffness at inter-particle collision; (b) Maximum stiffness at particle-wall impact; (c) Common maximum stiffness Kmax of both above cases.
However, the cost, from using a further shorter time step-length  of the algorithm, is negligible, as the computing time, for the first oscillation cycle of the model, is only a few seconds. So, a
of the algorithm, is negligible, as the computing time, for the first oscillation cycle of the model, is only a few seconds. So, a  is chosen, much shorter than that required by the above accuracy criterion of the algorithm, so that to achieve more accuracy.
is chosen, much shorter than that required by the above accuracy criterion of the algorithm, so that to achieve more accuracy.
6) The initial position of the particle R under consideration is , where L side of cubic container, as mentioned in Section 2 and according to Figure 2 and Figure 6.
, where L side of cubic container, as mentioned in Section 2 and according to Figure 2 and Figure 6.
7) Initial temperatures, ranging from  in liquid phase region up to
in liquid phase region up to  in gas phase region, are used.
in gas phase region, are used.
8) The initial speed , of the particle under consideration, is obtained from the initial temperature T0, by the thermodynamic postulate:
, of the particle under consideration, is obtained from the initial temperature T0, by the thermodynamic postulate:

where R = 8.3144 Joules−1∙K−1 is the value of gas constant for ideal gases. However, for small molar volumes, through the oscillation of the particle, the speed v is significantly reduced, which implies mean values of R much smaller than the initial one, as will be shown in the applications.
4.2. First Application. STP Conditions. Ideal Gas
A mole of Carbon Dioxide is considered, within a cubic container of side L = 28.195 cm, that is volume , with an initial temperature
, with an initial temperature
 , thus an initial speed of the particle
, thus an initial speed of the particle 
In this first application, point particles are assumed, that is with zero volume, and the inter-particle attractive forces are ignored. So, we have an ideal gas. This case is simple, so it will be solved by hand.
Within the first cycle of oscillation, the particle, starting from the position  (Figure 6), goes to impact on wall at right, where the speed is reversed. Then, an inter-particle collision occurs at left, where the speed is again reversed and the particle returns to the initial position. So, the particle runs twice the distance L/2 (Figure 6), with the constant speed v = 393.5 m/sec and the period of oscillation is
(Figure 6), goes to impact on wall at right, where the speed is reversed. Then, an inter-particle collision occurs at left, where the speed is again reversed and the particle returns to the initial position. So, the particle runs twice the distance L/2 (Figure 6), with the constant speed v = 393.5 m/sec and the period of oscillation is

In Figure 10(a), a sketch of the large container, with the point particles (of zero volume), in the initial state is shown, with the speeds directing outwards. In Figures 10(b)-(f), for the first oscillation cycle, the variations, with respect to time t, of five quantities, are presented: (b). Position y of the particle. (c). Speed v. (d). Inter-particle impulse . (e). Particle-wall impulse
. (e). Particle-wall impulse . (f). Total kinetic energy, for all six particles,
. (f). Total kinetic energy, for all six particles,
 where M = 6 m.
where M = 6 m.

Figure 10. First application. STP conditions. Ideal gas. (a) Large container of side L = 28.195 cm with point-particles in the initial state. In the following diagrams, variations of five quantities, with respect to time t; (b) Position y of the particle; (c) Speed v; (d) Inter-particle impulse ; (e) Particle-wall impulse
; (e) Particle-wall impulse ; (f) Total kinetic energy
; (f) Total kinetic energy .
.
At inter-particle collision and particle-wall impact, the impulse-momentum conservation equation can be written:

Here,  (tends to zero) and
(tends to zero) and  (tend to infinity). How- ever, as everyone, of the two above impulses, occurs once in an oscillation cycle, we can obtain the finite mean values of forces
(tend to infinity). How- ever, as everyone, of the two above impulses, occurs once in an oscillation cycle, we can obtain the finite mean values of forces ,
,  , by simply dividing the above impulses by the period τ:
, by simply dividing the above impulses by the period τ:

where ,
,  are opposite, as is due for equilibrium, and are noted in Figure 10(d) and Figure 10(e), respectively.
are opposite, as is due for equilibrium, and are noted in Figure 10(d) and Figure 10(e), respectively.
The pressure on the wall is

as was expected for an ideal gas in STP conditions.
As the speed is constant, the total kinetic energy is, at any instant,

The potential energy is

and the thermodynamic quantity is

It is observed that  as is due for an ideal gas. That is, the proposed model describes accurately the behavior of an ideal gas.
as is due for an ideal gas. That is, the proposed model describes accurately the behavior of an ideal gas.
4.3. Second Application. STP Conditions. Real Gas
The same input data, of the previous first application, in STP conditions, are again considered, that is a cubic container with side L = 28.195 cm, thus volume , and initial temperature
, and initial temperature , thus initial speed of the particle
, thus initial speed of the particle . However, now, the volume of particles, with diameter D = 1.35 cm, and the inter-particle attractive forces, described by a Lennard-Jones curve (Figure 5, Equation (2)), with a force coefficient
. However, now, the volume of particles, with diameter D = 1.35 cm, and the inter-particle attractive forces, described by a Lennard-Jones curve (Figure 5, Equation (2)), with a force coefficient  , are taken into account. So, we have a real gas, and the proposed step-by-step time integration algorithm is used, in order to follow the oscillation of the particle.
, are taken into account. So, we have a real gas, and the proposed step-by-step time integration algorithm is used, in order to follow the oscillation of the particle.
In Figure 11(a), is shown the large container of side L = 28.195 cm, with the particles of diameter D = 1.35 cm, in the initial conditions. In the Figures 11(b)-(f), are presented, within the first oscillation cycle, the variations, with respect to time t, of five quantities: b) position y of the particle. c) speed v. d) inter-particle force . e) particle-wall force
. e) particle-wall force . f) total kinetic energy
. f) total kinetic energy  , n = 6312 steps of the algorithm have been performed, within the first oscillation cycle, with a time-steplength
, n = 6312 steps of the algorithm have been performed, within the first oscillation cycle, with a time-steplength , thus the period is
, thus the period is .
.
The mean inter-particle force is
 and the mean particle-wall force
and the mean particle-wall force It is observed that
It is observed that , as is due for global equilibrium. The mean forces
, as is due for global equilibrium. The mean forces ,
,  are noted in the Figure 11(d) and Figure 11(e), respectively.The pressure on the wall is
are noted in the Figure 11(d) and Figure 11(e), respectively.The pressure on the wall is that is, it slightly deviates from the ideal gas value.The mean 2nd power of speed is
that is, it slightly deviates from the ideal gas value.The mean 2nd power of speed is

Figure 11. Second application. STP conditions. Real gas. (a) Large container of side L = 28.195 cm with particles of diameter D = 1.35 cm, in the initial state. In the following diagrams, variations of five quantities with respect to time t; (b) Position y of the particle; (c) Speed v; (d) Inter-particle force ; (e) Particle-wall force
; (e) Particle-wall force ; (f) Total kinetic energy
; (f) Total kinetic energy .
.
So, the mean (rms) speed is

that is, slightly larger than initial speed.
And the mean kinetic energy is

that is, it slightly deviates from the corresponding value of ideal gas. The mean kinetic energy is noted on the diagram K.E.-t of Figure 11(f), for comparison.
4.4. Third Application. Condensation for Small Molar Volume
A small cubic container with side L = 4.55 cm, thus molar volume  , is considered, the same as in the Critical point of Carbon Dioxide, according to test data [14] . And a low initial temperature
, is considered, the same as in the Critical point of Carbon Dioxide, according to test data [14] . And a low initial temperature , which implies a low initial speed of the particle
, which implies a low initial speed of the particle  A sketch of the small container, with the particles of diameter D=1.35cm, in the initial state, is shown in Figure 12(a).
A sketch of the small container, with the particles of diameter D=1.35cm, in the initial state, is shown in Figure 12(a).

Figure 12. Third application. Condensation for small molar volume. (a) Small container of side L = 4.55 cm with particles of diameter D = 1.35 cm, in the initial state. In the following diagrams, variations of four quantities, with respect to time t; (b) Position y of the particle; (c) Speed v; (d) Inter-particle force ; (e) Total kinetic energy
; (e) Total kinetic energy .
.
The application run by the proposed step-by-step algorithm, with  . The first oscillation cycle was completed in 475 steps, thus the period is
. The first oscillation cycle was completed in 475 steps, thus the period is 
In Figures 12(b)-(e), the variations, with respect to time t, of four quantities, are presented: b) position y of the particle. c) speed v. d) inter-particle force . e) total kinetic energy
. e) total kinetic energy 
In the present application, because of the low initial temperature, thus low initial speed and kinetic energy, too, the inter-particle attractive forces  overcome the kinetic energy of the particle, thus preventing it from reaching to impact on the wall. So zero particle-wall forces
overcome the kinetic energy of the particle, thus preventing it from reaching to impact on the wall. So zero particle-wall forces  and zero pressure P result, which mean that a liquid phase exists.
and zero pressure P result, which mean that a liquid phase exists.
Because of the zero particle-wall forces,  , the sum of inter-particle forces results zero,
, the sum of inter-particle forces results zero,  , for equilibrium, as noted in the Figure 12(d).
, for equilibrium, as noted in the Figure 12(d).
The mean 2nd power of speed results

thus the mean (rms) speed is  significantly smaller than the initial speed. Finally, the mean kinetic energy results
significantly smaller than the initial speed. Finally, the mean kinetic energy results

which is noted on the diagram K.E.-t of Figure 12(e), for comparison.
4.5. Fourth Application. Critical Point
The same small cubic container of side L = 4.55 cm of previous application is considered, which implies a volume  known, from experiments, as the critical molar volume of Carbon Dioxide [14] . And the initial critical temperature
known, from experiments, as the critical molar volume of Carbon Dioxide [14] . And the initial critical temperature  is provided, which implies an initial particle speed
is provided, which implies an initial particle speed
 In Figure 13(a), a sketch of the above small container, of side L = 4.55 cm, is shown, with the particles of diameter D = 1.35 cm, in the initial state, with the speeds directed outwards.
In Figure 13(a), a sketch of the above small container, of side L = 4.55 cm, is shown, with the particles of diameter D = 1.35 cm, in the initial state, with the speeds directed outwards.

Figure 13. Fourth application. Critical point. (a) The small container of side L = 4.55 cm with particles of diameter D = 1.35 cm, in the initial state. In the following diagrams, variations of five quantities with respect to time t; (b) Position y of the particle; (c) Speed v; (d) Inter-particle force ; (e) Particle-wall force
; (e) Particle-wall force ; (f) Total kinetic energy
; (f) Total kinetic energy  .
.
The application run by the proposed step-by-step algorithm, with  The first oscillation cycle was completed in 562 steps, so the period is
The first oscillation cycle was completed in 562 steps, so the period is 
In Figures 13(b)-(f), the variations, with respect to time t, of five quantities, are presented: b. position y of the particle. c. speed v. d. inter-particle force . e. particle-wall force
. e. particle-wall force . f. total kinetic energy
. f. total kinetic energy 
The mean inter-particle force results  The mean particle-wall force results
The mean particle-wall force results 
It is observed that  as is due for global equilibrium. The mean forces
as is due for global equilibrium. The mean forces ,
,  are noted on the diagrams Fi − t, Fw − t of Figure 13(d) and Figure 13(e), respectively.
are noted on the diagrams Fi − t, Fw − t of Figure 13(d) and Figure 13(e), respectively.
The pressure on the wall is

close to the experimental critical pressure  [14] .
[14] .
The mean 2nd power of speed is

Thus, the mean (rms) speed results

much smaller than the initial speed.
The mean kinetic energy results

which is noted on the diagram K.E.-t of Figure 13(f), for comparison.
The above mean kinetic energy  corresponds to a value of gas constant R = 2.856 Joules mole−1∙K−1, as obtained by equating
corresponds to a value of gas constant R = 2.856 Joules mole−1∙K−1, as obtained by equating  The present value of R is much smaller than the value R = 8.3144 of ideal gases. This will be discussed in the fifth application of next section 4.6.
The present value of R is much smaller than the value R = 8.3144 of ideal gases. This will be discussed in the fifth application of next section 4.6.
The present application is adapted to the Critical point by its initial temperature , which is the critical temperature of Carbon Dioxide, according to test data [14] . In the fifth application of next Section 4.6. The Critical point of CO2 will be determined in two different ways: By the group of iso- kinetic energy curves of Figure 14 and by the group of iso-therms of Figure 16. Both cases are close to the Critical point of present application.
, which is the critical temperature of Carbon Dioxide, according to test data [14] . In the fifth application of next Section 4.6. The Critical point of CO2 will be determined in two different ways: By the group of iso- kinetic energy curves of Figure 14 and by the group of iso-therms of Figure 16. Both cases are close to the Critical point of present application.
4.6. Fifth Application. Iso-Kinetic Energy Curves in the Critical Point Region
For side of cubic container ranging from L = 4.0 cm up to 9.0 cm, with a step , that is, volume V = L3 ranging from 64 cm3 up to 729 cm3. And for initial temperature ranging from
, that is, volume V = L3 ranging from 64 cm3 up to 729 cm3. And for initial temperature ranging from  up to
up to , with a step
, with a step , thus, for initial speed ranging from
, thus, for initial speed ranging from
 up to 459.8 m/sec, for every couple of L, T0, the corresponding pressure P and mean kinetic energy
up to 459.8 m/sec, for every couple of L, T0, the corresponding pressure P and mean kinetic energy  have been determined. Every point
have been determined. Every point

Figure 14. Fifth application. Iso-kinetic energy curves  in the Critical Point region of Carbon Dioxide obtained by the proposed model. C.P. = Critical Point. In the drawning of successive curves, between 1100 and 1400 Joules the step is 50 Joules, between 1400 and 1800 Joules the step is 100 Joules, between 1800 and 3000 Joules the step is 200 Joules.
in the Critical Point region of Carbon Dioxide obtained by the proposed model. C.P. = Critical Point. In the drawning of successive curves, between 1100 and 1400 Joules the step is 50 Joules, between 1400 and 1800 Joules the step is 100 Joules, between 1800 and 3000 Joules the step is 200 Joules.
(V, P) was placed on the volume-pressure plane, with the corresponding  noted on it.
noted on it.
Then, by linear interpolation between successive values of , iso-kinetic energy curves, for rounded values of
, iso-kinetic energy curves, for rounded values of , were obtained, as shown in Figure 14, for
, were obtained, as shown in Figure 14, for  ranging from 1100 Joules up to 3000 Joules.
ranging from 1100 Joules up to 3000 Joules.
It is observed that, under the iso-kinetic energy curve of 1100 Joules, a Liquid phase exists, with zero pressures. Between the curve of 1100 Joules and the Critical curve of 1300 Joules, a Vapor phase exists with low pressures. And above the Critical curve, a Gas phase exists, with high pressures. That is, the Critical iso- curve of 1300 Joules is the boundary between the Vapor and Gas phases.
curve of 1300 Joules is the boundary between the Vapor and Gas phases.
By placing the above iso-kinetic energy curves of proposed model on the same P-V (pressure-molar volume) plane, together with the corresponding iso-therms of test data of Eastman-Rollefson [14] [20] and those of Van der Waals model [14] [16] and, by equating, at points of intersection of iso- curves with iso- therms,
curves with iso- therms,  values of gas constant R are obtained. And a variation of R values, in the Critical point region of Carbon Dioxide is revealed. Both, test data of Eastman-Rollefson and results of Van der Waals model exhibit similar trends, as regards this variation.
values of gas constant R are obtained. And a variation of R values, in the Critical point region of Carbon Dioxide is revealed. Both, test data of Eastman-Rollefson and results of Van der Waals model exhibit similar trends, as regards this variation.
It is observed that the gas constant R exhibits values, in Critical point region, ranging from 2.85 Joules mole−1∙K−1 up to 5.0, much smaller than the well- known value 8.3144, which is approximately valid for large molar volumes and accurately valid for ideal gases. The obtained variation of R values is described by the graph of Figure 15.

Figure 15. Variation of gas constant R values in the Critical Point region of Carbon Dioxide, obtained by comparison of iso- curves of proposed model of Figure 14 with corresponding iso-therms of Eastman-Rollefson tests [14] [20] and those of Van der Waals model [14] [16] .
curves of proposed model of Figure 14 with corresponding iso-therms of Eastman-Rollefson tests [14] [20] and those of Van der Waals model [14] [16] .
With the help of this graph, the iso-kinetic energy curves of proposed model, of Figure 14, have been transformed to the iso-therms shown in Figure 16, for temperatures ranging from T = 250 K up to 400 K with a step .
.
The above iso-therms of proposed model are compared with corresponding ones of the test data of Eastman-Rollefson [14] [20] , in Figure 17, as well as with those of Van der Waals model [14] [16] , in Figure 18.
It is observed, in the Figure 17 and Figure 18, that the proposed model better represents the wave-shaped isotherms of the Van der Waals model, in the Vapor region, than the horizontal linear isotherms of the test data by Eastman?Rollef- son. Also, the proposed model approximates better the larger incompressibility limit of the molar volume given by the Van der Waals model (Figure 18), than the smaller one of the test data by Eastman-Rollefson (Figure 17).
5. Conclusions
A simplified coarse-grained dynamic model, for real gases, is proposed. Five applications of this model, on Carbon Dioxide, are presented:
1) In STP conditions, by ignoring particle volume and inter-particle attractive forces, the proposed model accurately represents the behavior of an ideal gas.
2) Again in STP conditions, but taking into account the particles volume and inter-particle attractive forces, the proposed model slightly deviates from the behavior of an ideal gas, as was expected.
3) For a small molar volume and a low initial temperature, the inter-particle attractive forces overcome the initial kinetic energy of particles and prevent them from reaching at impact with container wall. So, a Liquid phase exists, with zero pressure.

Figure 16. Iso-therms of proposed model in Critical Point region of Carbon Dioxide, obtained from transformation of iso- curves of proposed model of Figure 14, by use of variation of gas constant R values of Figure 15. C.P.: Critical Point.
curves of proposed model of Figure 14, by use of variation of gas constant R values of Figure 15. C.P.: Critical Point.

Figure 17. Comparison of iso-therms of proposed model to corresponding ones of Eastman-Rollefson tests [14] [20] , in the Critical Point region of Carbon Dioxide.

Figure 18. Comparison of iso-therms of proposed model with corresponding ones of Van der Waals model [14] [16] in the Critical Point region of Carbon Dioxide.
4) At the Critical point of Carbon Dioxide, the proposed model closely predicts the values of critical molar volume, temperature and pressure, known from experiments [14] .
5) Iso-kinetic energy curves have been determined, by the proposed model, in the Critical point region of Carbon Dioxide. By comparing these iso-kinetic energy curves to corresponding iso-therms of test data by Eastman-Rollefson [14] [20] and to those of Van der Waals model [14] [16] , a variation of values of gas constant R, in Critical point region, is revealed, ranging from 2.85 up to 5.0 Joules mole−1∙K−1. With the help of this variation of values of R, the iso-kinetic energy curves of proposed model are transformed to iso-therms, which are compared to corresponding ones of test data by Eastman-Rollefson, as well as to iso-therms of Van der Waals model. And a better agreement is achieved between the proposed model and the Van der Waals model, as shown in Figure 18, as regards a larger in-compressible molar volume and particularly the wave-shaped iso-therms in the Vapor region.
The above five numerical experiments show that the proposed simplified model can approximate the observed behavior of real gases.
In the present work, in order to achieve simplicity, the accuracy is reduced. However, if a refined version of the proposed model, with more particles, is developed, the accuracy can be improved.
Cite this paper
Papadopoulos, P.G., Koutitas, C.G., Dimitropoulos, Y.N. and Aifantis, E.C. (2017) Simplified Coarse- Grained Dynamic Model for Real Gases. Open Journal of Physical Chemistry, 7, 50-71. https://doi.org/10.4236/ojpc.2017.72005
References
- 1. Müller, E.A. and Jackson, G. (2014) Force-Field Parameters from the SAFT-γ Equation of State for Use in Coarse-Grained Molecular Simulation. Annual Review of Chemical and Biomolecular Engineering, 5, 405-427.
https://doi.org/10.1146/annurev-chembioeng-061312-103314 - 2. Herdes, C., Totton, T.S. and Müller, E.A. (2015) Coarse-Grained Force Field for the Molecular Simulation of Natural Gases and Condensates. Fluid Phase Equilibria 406, 91-100.
https://doi.org/10.1016/j.fluid.2015.07.014 - 3. Mejía, A., Herdes, C. and Müller, E.A. (2014) Force Fields for Coarse-Grained Molecular Simulations from a Corresponding States Correlation. Industrial & Engineering Chemistry Research, 53, 4131-4141.
https://doi.org/10.1021/ie404247e - 4. Avendano, C., Lafitte, T., Galindo, A., Adjiman, C.S., Jackson, G. and Müller, E.A. (2011) Saft-γ Force Field for the simulation of Molecular Fluids. 1. A Single-Site Coarse-Grained Model for Carbon Dioxide. The Journal of Physical Chemistry, 115, 11154-11169.
https://doi.org/10.1021/jp204908d - 5. Matteo, B., Oettel, M.M., Yelash, L. and Binder, K. (2009) (SI) Structure and Pair Correlations of a Simple Coarse-Grained Model for Super-Critical Carbon Dioxide. Molecular Physics, 107, 1-24.
- 6. Yelash, L., Müller, M., Paul, W. and Binder, K. (2006) How Well Can Coarse-Grained Models of Real Polymers Describe their Structure? The Case of Polybutadiene. Journal of Chemical Theory and Computation, 2, 588-597.
https://doi.org/10.1021/ct0502099 - 7. Theodorakis, P.E., Müller, E.A., Richard, V.K. and Matar. O.K. (2015) Superspreading: Mechanisms and Molecular Design. Langmuir, 31, 2304-2309.
https://doi.org/10.1021/la5044798 - 8. Adolfo, B.P., Cieplak, M. and Theodorakis, P.E. (2017) Combining the MARTINI and Structure-Based Coarse-Grained Approaches for the Molecular Dynamics Studies of Conformational Transitions in Proteins. Journal of Chemical Theory and Computations, 13, 1366-1374.
https://doi.org/10.1021/acs.jctc.6b00986 - 9. Kmiecik, S., Gront, D., Kolinski, M., Wieteska, L.A., Dawid, E. and Kolinski, A. (2016) Coarse-Grained Protein Models and Their Applications. Chemical Reviews, 116, 7896-7936.
https://doi.org/10.1021/acs.chemrev.6b00163 - 10. James, F., Dama, A.V., Sinitskiy, M.C., Weare, J., Roux, B., Aaron, R., Gregory, D. and Voth, A. (2013) The Theory of Ultra-Coarse-Graining. 1. General Principles. Journal of Chemical Theory and Computation, 9, 2466-2480.
- 11. Darlyan, A., James, F., Dama, A., Sinitskiy, G. and Voth, A. (2014) The Theory of Ultra-Coarse-Graining. 2. Numerical Implementation. Journal of Chemical Theory and Computation, 10, 5265-5275.
https://doi.org/10.1021/ct500834t - 12. James, F., Jaehyeok, D.J. and Voth, G.A. (2017) The Theory of Ultra-Coarse-Graining. 3. Coarse-Grained Sites with Rapid Local Equilibrium of Internal States. Journal of Chemical Theory and Computation, 13, 1010-1022.
- 13. Sanghi, T. And Aluru, N.R. (2012) Coarse-Grained Potential Models for Structural Prediction of Carbon Dioxide (CO2) in Confined Environments. The Journal of Chemical Physics, 136, 1-23.
https://doi.org/10.1063/1.3674979 - 14. Barrow, G.M. (1997) Physical Chemistry. McGraw-Hill, New York.
- 15. Wikipedia (2017) Lennard-Jones Potential.
- 16. Wikipedia (2017) Wan der Waals Equation.
- 17. Wikipedia (2017) Maxwell-Boltzmann Distribution.
- 18. Dougill, J.W. (1983) Path Dependence and General Theory for the Progressively Fracturing Solid. Proceedings of the Royal Society of London. Series A, Mathematical and Physical Sciences, 390, 341-351.
- 19. Papadopoulos, P.G. (1984) Biaxial Network Constitutive Model. Journal of Engineering Mechanics Division, 110, 449-464.
https://doi.org/10.1061/(ASCE)0733-9399(1984)110:3(449) - 20. Eastman, E.D. and Rollefson. G.K. (1947) Physical Chemistry. Mc Graw-Hill, New York.
- 21. Papadopoulos, P.G. (1984) A Simple Algorithm for Nonlinear Dynamic Analysis of Networks. Computers and Structures, 18, 1-8.