Open Journal of Physical Chemistry
Vol. 2 No. 2 (2012) , Article ID: 19162 , 6 pages DOI:10.4236/ojpc.2012.22013
Hydroxyalkylation of Cyclic Imides with Oxiranes. Part II. The Mechanism of Reaction in Presence of Triethylamine
Faculty of Chemistry, Rzeszów University of Technology, Rzeszów, Poland
Email: jml@prz.edu.pl
Received February 6, 2012; revised March 9, 2012; accepted April 12, 2012
Keywords: Imides; Oxiranes; Mechanism; Kinetics; Non-Kinetics Evidences
ABSTRACT
The mechanism of reaction of cyclic monoimides with oxiranes was established based upon kinetic studies and product analysis. It has been established that the reaction proceeds through initial formation of an adduct of imide and triethylamine. The crucial bond in adduct has ionic character; in non-aqueous solvents it is present as ion pair, while in water the adduct dissociate and free ions are present. The adduct enables the proton transfer from imide to oxirane. The rate determining step is reaction of imide and this adduct. Different values of entropy of transition states obtained from thermodynamic calculations suggest slightly different structure of transition state of rate determining step.
1. Introduction
The reaction of cyclic monoimides with oxiranes in presence of triethylamine (TEA) leads to N-(hydroxy-alkyl) imides of the general formula (I):
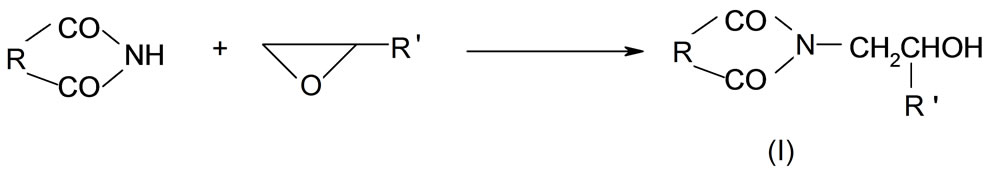
where R = (-CH2-)2, (-CH2-)3, -Ph R' = -H, -CH3
As it was presented in [1] the reaction follows the rate law (2):
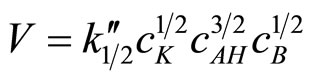 (2)
(2)
where cK, cAH, and cB mean the catalyst, imide groups, and oxirane concentrations, respectively.
The process can be considered to have the competitive and consecutive character consistent with reaction of N-(hydroxyalkyl)imides, which are the primary products, with oxirane. However the consecutive product is formed with low yield, ca 5 - 10 mol%. The rate law describing the reaction of N-(2-hydroxyalkyl)imides with oxiranes obeys another kinetics (first order related to oxirane concentration vs 1/2 for the first step), indicating that the reactions of oxiranes with N-(hydroxyalkyl)imides, which reveals the alcohol character, occur via different mechanism [2-6]. Based upon the rate constants the monoimides were ordered according to lowering reactivity as: PI ≥ SI > GI >HMSI. It has been also found that ethylene oxide (EO) was more reactive than propylene oxide (PO). The free energy of activation (ΔG≠) was also obtained for those processes. The temperature dependence was linear which provided further evidence that the same mechanism operates for all studied systems.
Here we propose the mechanism of those reactions based on product analysis, intermediate identification and kinetic measurements.
2. Experimental
2.1. Substrates
The substrates were the same as in Part I.
2.2. Conductometric Measurements
The conductivity of imides, TEA, and their mixtures was studied in aqueous solutions at variable molar ratio in thermostated conductometric cells of 20 cm3 capacity at 20˚C (±0.1). The solutions were then diluted twice and conductivity was measured again. No PI solutions were measured due to its low solubility in water.
2.3. Cryoscopic Measurements
The cryoscpic measurements were done for 0.12-0.13- molal solutions of imide and TEA at 1:1 molar ratio (the total concentration of both was 0.25 mol/kg solvent in dioxane (Kcryos. = 4.64). The method was standardized by determination of molar mass of imide and TEA. The presence of of imide-TEA adduct was deduced based on following approach. The total concentration of both reagents ct was the sum of imide (cAH) and catalyst (cK) concentrations. The actual molal concentration of all components (ce) present in solution (imide, TEA, and adduct) were found from the measurements of decrease of the freezing point solution related to solvent from (3):
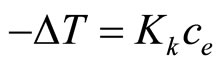 (3)
(3)
If the adduct is present in solution of mixture the ce < ct is expected. If x is the concentration of adduct, the equilibrium:
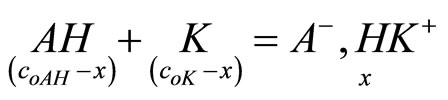 (4)
(4)
takes place and hence the association constant Ka can be obtained from (5):
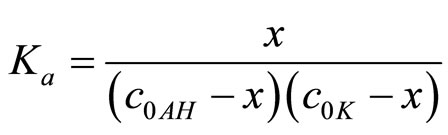 (5)
(5)
where the association degree β is defined as the ratio of adduct concentration (x) to component concentration (co) which is present in lower concentration:
 (6)
(6)
3. Results and Discussion
3.1. Mechanism of Reaction
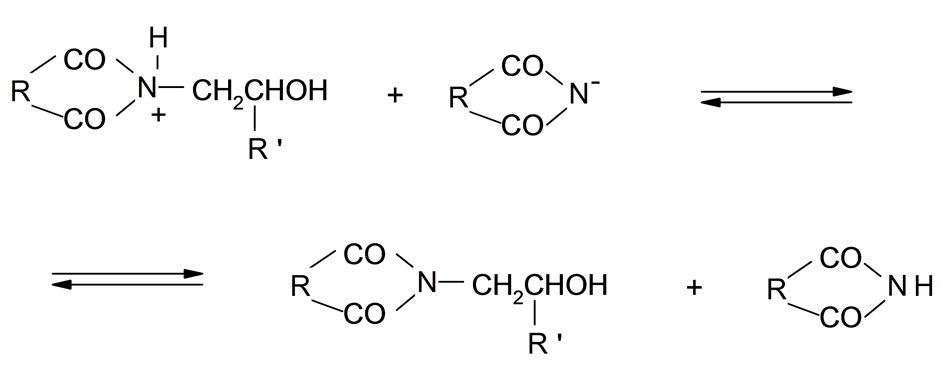
The mechanism of the reaction of imides with oxiranes can be deduced based on experimental kinetic law Equation (2). It was assumed that adducts (ion pairs or hydrogen bonded complexes of imides (AH) and catalyst (K) take part in elementary reactions. The crucial adduct is formed between imide group and nitrogen of TEA. The products are formed upon reaction of imide with adduct of triethylammonium cation and oxirane molecule (AHK) according to the scheme (the acronyms under the reaction were introduced to be later used in kinetic equation):
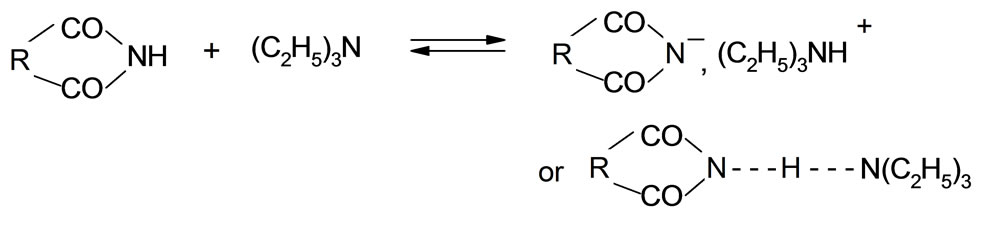
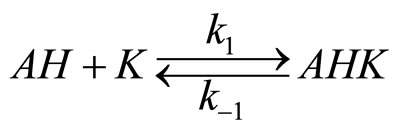 (7)
(7)
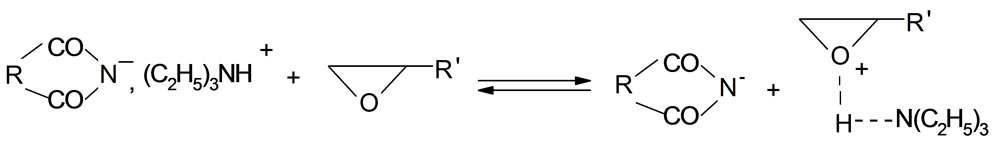
 (8)
(8)
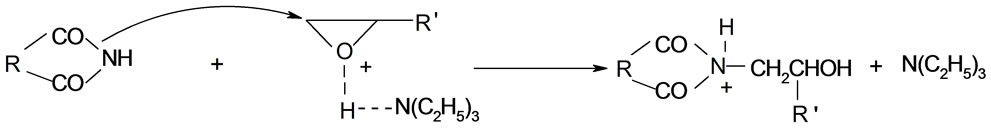
 (9)
(9)
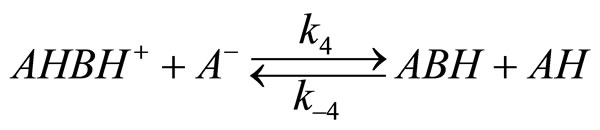 (10)
(10)
Thus the elementary steps are:
1) Formation of AHK adduct from imide and TEA (7), which plays the role of proton transfer agent from imide to oxirane;
2) Reaction of adduct with oxirane with formation of new adduct BHK+;
3) Reaction of BHK+ with next molecule of imide resulting in formation of the alcohol protonated on imide group and recovery of catalyst (9);
4) Neutralization of imide anion by proton transfer from protonated alcohol to imide anion (10).
3.2. Kinetic Evidences for Proposed Mechanism
If one assume that:
1) The equilibrium between adduct (AHK) and TEA is reached very fast
2) The protonation constant of oxirane (K2) is low and equilibrium (8) establishes slower that that in reaction (7)
3) The rate determining step is reaction of AH with adduct BHK+ the rate of oxirane consumption equals the rate of disappearance of BHK+ adduct, i.e.:
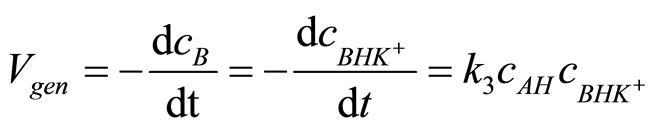 (11)
(11)
The concentration of cBHK can be found from stationary state condition [7]:
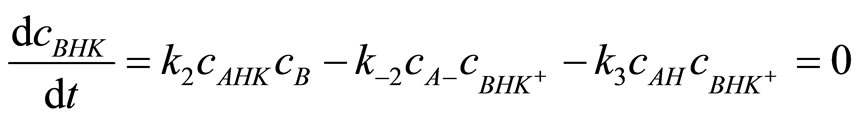 (12)
(12)
Because
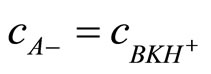 (13)
(13)
then:
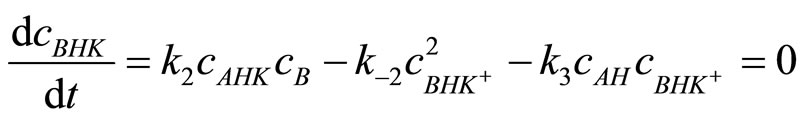 (14)
(14)
After rearrangement the Equation (14) is:
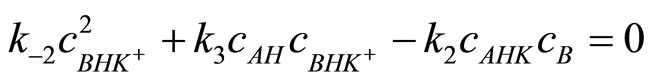 (15)
(15)
The solution for cBHK gives:
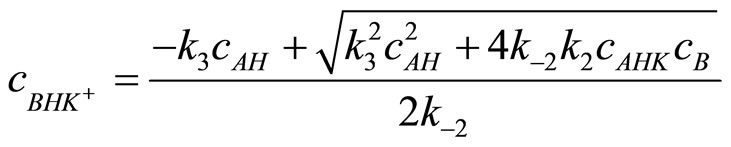 (16)
(16)
Taking into account the Assumption 3 the relationship:
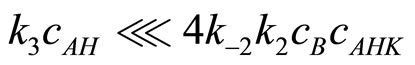
is obvious, which furthermore allows to simplity Equation (16) to:
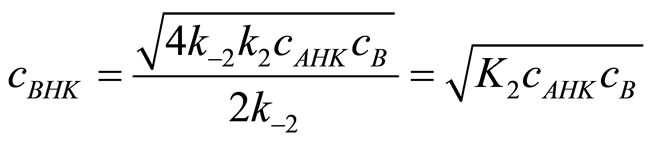 (17)
(17)
Putting (17) into (11) the rate of reaction becomes:
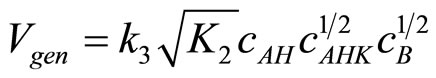 (18)
(18)
As before, the concentration of adduct AHK can be obtained from stationary state condition:
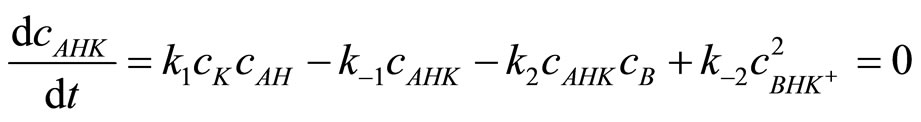 (19)
(19)
Introducing Equation (17) for 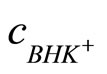 the relationship:
the relationship:
 (20)
(20)
is obtained, from which:
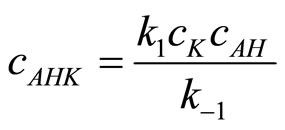 (21)
(21)
or:
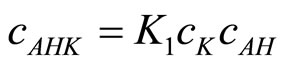 (22)
(22)
Thus:
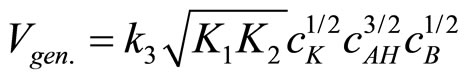 (23)
(23)
when
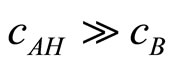 (24)
(24)
the latter equation can be simplified into:
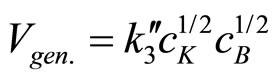 (25)
(25)
where:
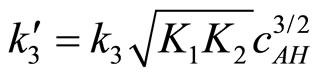 (26)
(26)
The obtained kinetic equation is in agreement with the experimental one, which suggests that proposed mechanism operates in the studied systems.
At this step the small differences of reactivity of PI and SI (cf. Part I) with oxiranes can be explained on the basis of proposed mechanism. Higher reactivity of PI is electronic in origin; phtalimide anion is mesomerically stabilized, thus the lifetime of AHK is larger in case of phtalimide, which enables the reactions (8) and (9).
Different values of entropy of transition states obtained from thermodynamic calculations (cf part I, Table 7) suggest slightly different structure of transition state of rate determining step. The value of –150 J/mol s can be attributed to transition State (II) which results straightforward from reaction (9), while the values < –200 J/mol s suggest the cyclic transition State (III) exists, in which imide anion participates: participates:

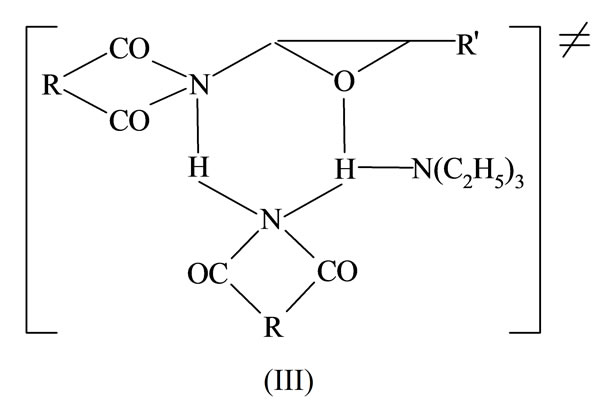
It has been observed, that the studied reaction are the fastest in DMF in comparison to other solvents (DMSO or dioxane; see Part I., Table 8). Presumably the reason is an involvement of solvent molecule in hydrogen cation transfer from imide to oxirane, i.e. the same as the catalytic role of TEA in reactions (7) and (8).
3.3. Non-Kinetics Evidences
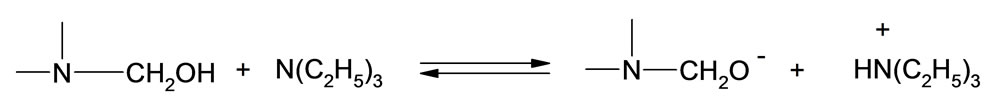
The crucial points in proposed mechanism are the formation of 1:1 imide-TEA adduct and fast equilibria. Conductivity of aqueous solution of TEA is related to the equilibrium according the to scheme of reaction:
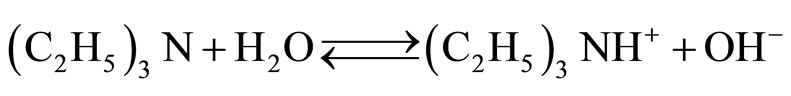 (27)
(27)
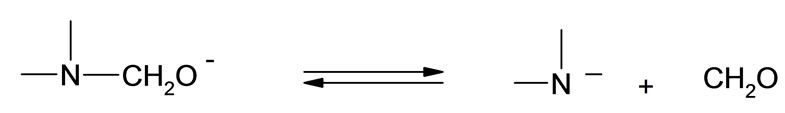
Introducing the imide or HMSI to the solution of TEA results in considerable increase of conductivity (Table 1), which is not additive, i.e. it is higher than the sum of conductivities of single components. This suggests that reaction (28) occurs:
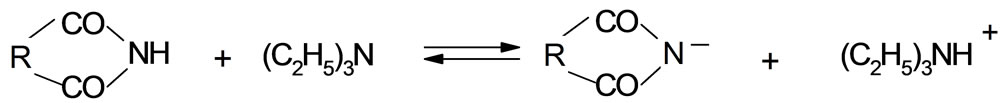 (28)
(28)
The measurements performed with variable composition indicated that in imide—TEA mixtures the maximum conductivity is reached immediately at equimolar solution of imide and TEA for the SI-TEA system (Figure 1). This was expected result for the stoichiometry of adduct.
Yet another kind of association takes place in nonaqueous solvents. Cryoscopic measurements revealed that all mixtures of imides with TEA follow the nonequality ce < ct, thus in these conditions the single species (ionic pair or hydrogen bonded adduct) is formed at the 5.2 - 7.7 mol% level in equimolar solutions (Table 2). The association is the highest at the PI-TEA system due to mesomeric stabilization of phtalimide anion. This causes shifting of the equilibrium (7) to the right. It should also be noted that association occurs also in the system
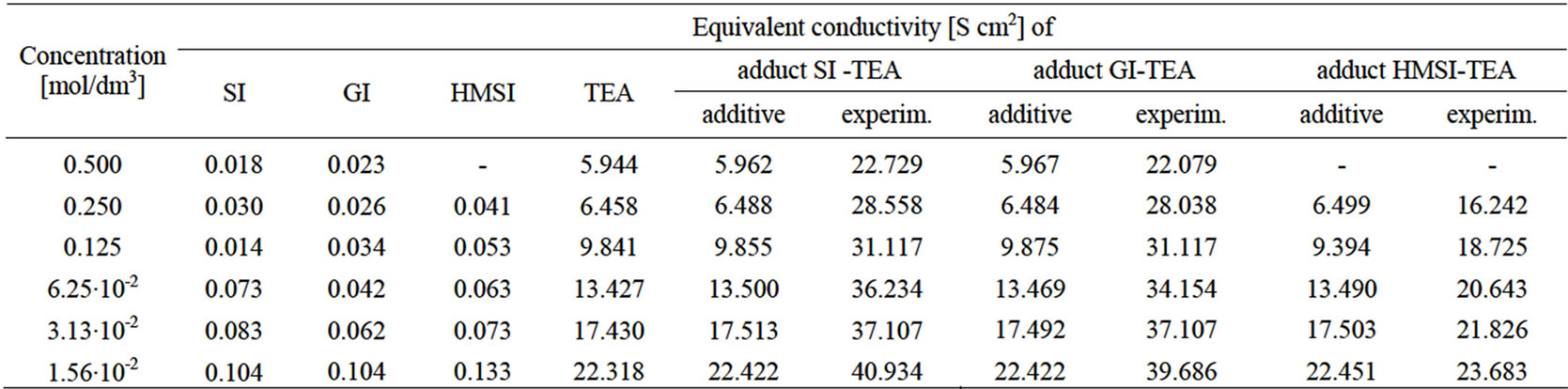
Table 1. Changes of conductivity of aqueous solutions of imides, TEA, and equimolar mixtures imide—TEA.
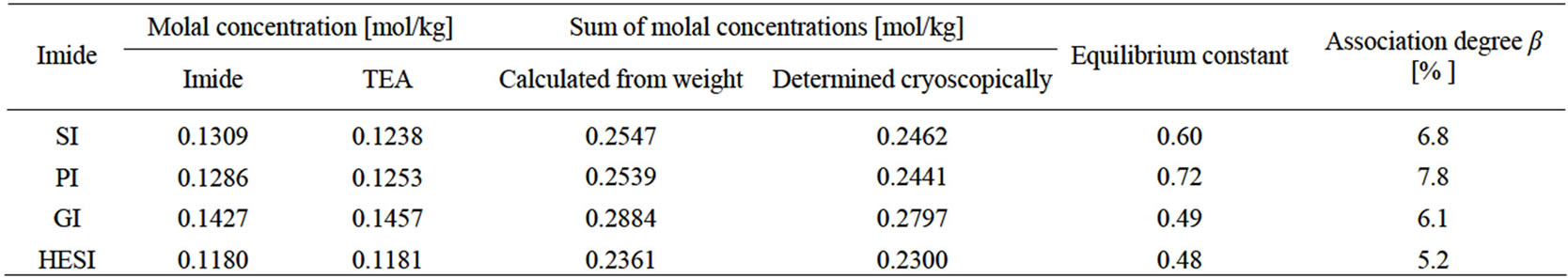
Table 2. Analysis of imide—TEA adduct in dioxane by cryoscopic method.
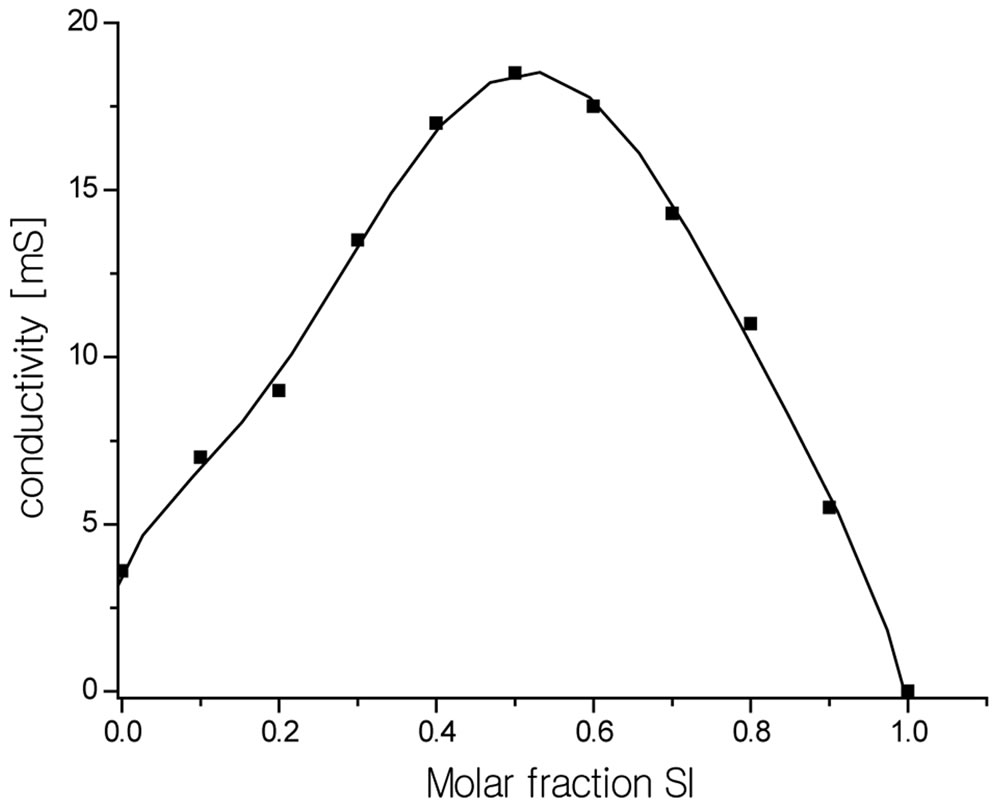
Figure 1. Changes of conductivity of solutions for the SI—TEA system of variable composition.
HESI-TEA. This suggests that proton donor to oxirane can be not only the imide but also its product of reaction, the alcohol, while the TEA catalyst mediates the transfer of proton from imide or alcohol to oxirane.
It has been noted that upon addition of oxirane to HMSI partial release of formaldehyde from N-hydroxymethyl group occurred accompanied by its migration to the end of hydroxyalkyl chain. Based upon cryoscopic data and previous studies it was possible to determine the mechanism of this rearrangement. Previously we have focused on kinetic models for initial steps of oxirane reactions with N-(hydroxymethyl) derivatives of some azacyclic compounds (barbituric, isocyanuric, and uric acids) leading to the formation of polyetherols [8-11]. The percentage of N-hydroxymethyl and groups in HMSI and O-hydroxymethyl groups in the product of its reaction with oxirane was determined as weakly bound formaldehyde. According to the data the HMSI contained 1 mole of formaldehyde per mole of HMSI (%CH2Ocalc = 23.3, %CH2Ofound = 23.2). It was also found that upon addition of 1 mole of EO to HMSI ca 0.3 mole of unstable bonded formaldehyde as O-(hydroxymethyl) groups remains, but after addition of 2 moles EO practically whole formaldehyde is blocked by oxirane. These results suggest, that either O-(hydroxymethyl) groups are formed as the result of partial dissociation of formaldehyde from N-(hydroxymethyl) groups of HMSI and then added to N-(hydroxyalkyl) group according to the scheme:
 (29)
(29)
 (30)
(30)
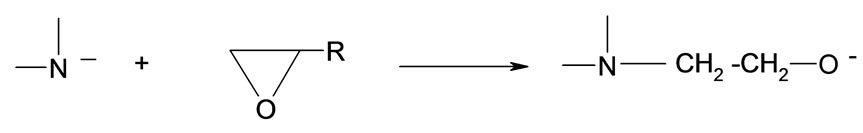 (31)
(31)
 (32)
(32)
 (33)
(33)
or first the oxirane is attached to N-(hydroxymethyl) group with formation of (IV), from which the formaldehyde is released and rearranged into the end of chain according to the scheme:
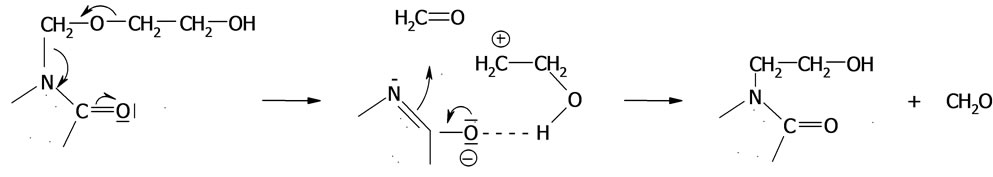 (34)
(34)
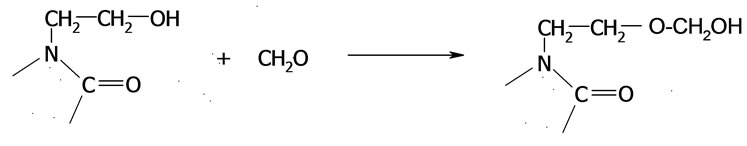 (35)
(35)
Cryoscopic determination of molar mass of HMSI in dioxane and DMSO indicated, that in both solvents the molar mass of HMSI was equal to calculated one (128.9 and 128.5 g/mol, respectively; calc. 129.1). This suggests that no dissociation of formaldehyde from N-(hydroxymethyl) groups occurs and the formaldehyde rearrangement takes place after blocking with oxirane (reaction 34). This corroborate well with our previous studies on the reaction of hydroxymethylated derivatives of uric acid with oxiranes [11]. The system HMSI-TEA was also studied by cryoscopic method (Table 2). As in other cases the relation ce < ct, was kept valid, what confirms that dissociation of formaldehyde from alcoholate did not occur upon TEA.
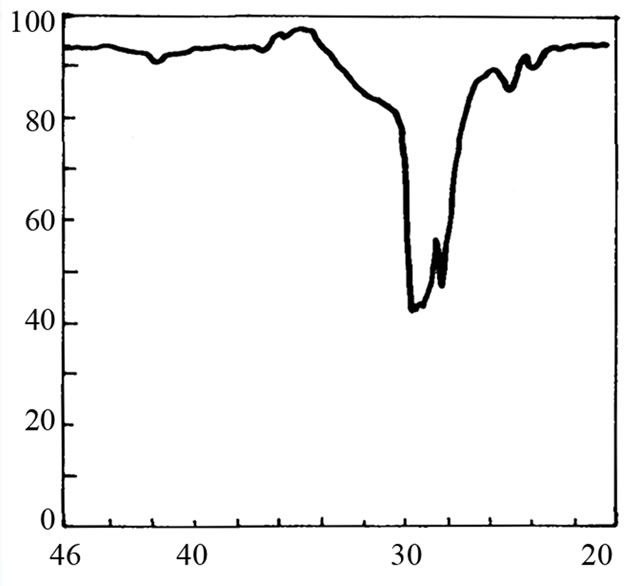
It is not possible to define the character of the bond in adduct on the basis of cryoscopic measurements. Therefore the IR and 1H-NMR spectra were recorded. The IR spectrum of SI shows the valence band of NH at about 3200 cm–1 (Figure 2(b)). When 3-fold excess of TEA was added, the band disappeared (Figure 2(d)) accom-panied by appearance of weak band from ammonium cation (C2H5)3NH+ in the region 2300 - 2500 cm–1. Undo-ubtedely, the proton transfer to TEA from imide took place. This band was more pronounced in the mixture of HESI and TEA (Figures 2(c) and (e)). The spectral features were: the disappearance of hydroxyl valence band at 3500 cm–1, and simultaneous appearance of absorption at 2400 cm–1. Thus, the product of reaction can also be the proton donor, which agrees well with cryoscopic data.
IR spectra rather preclude the formation of hydrogen bonded adduct; in such a case the broadening of the N-H
 (a)
(a)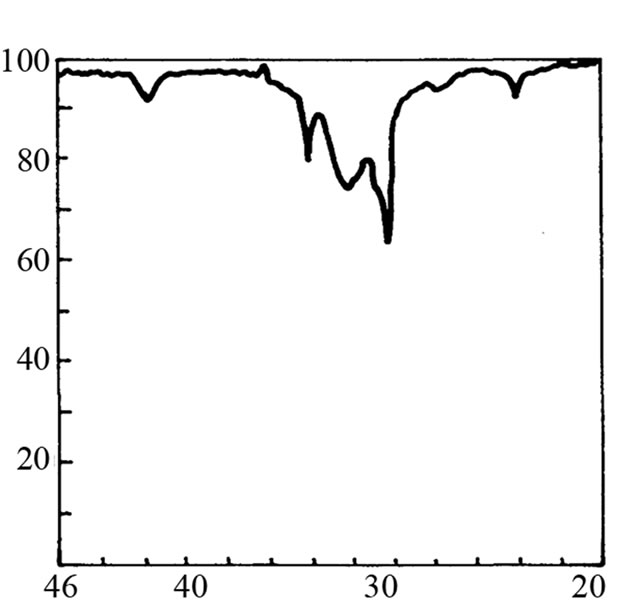 (b)
(b)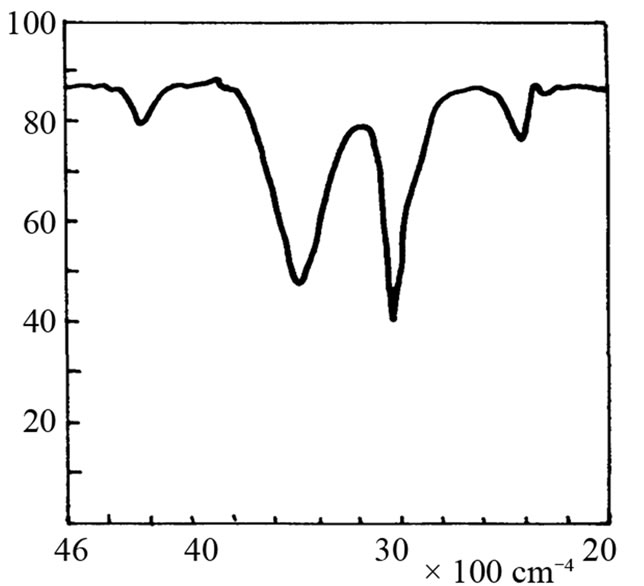 (c)
(c)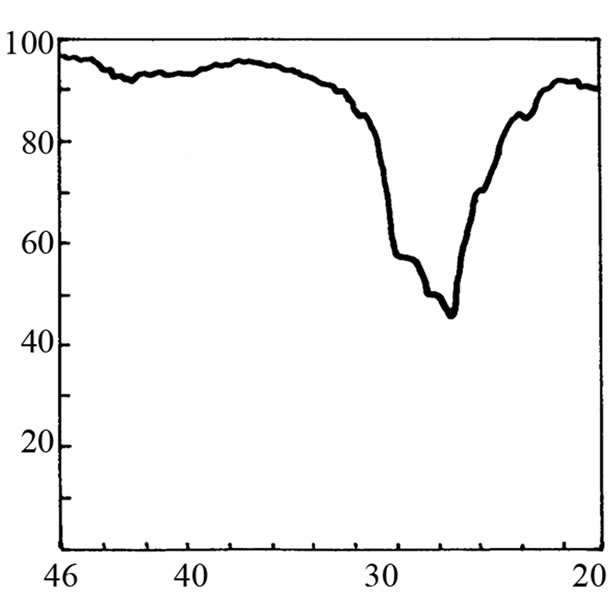 (d)
(d)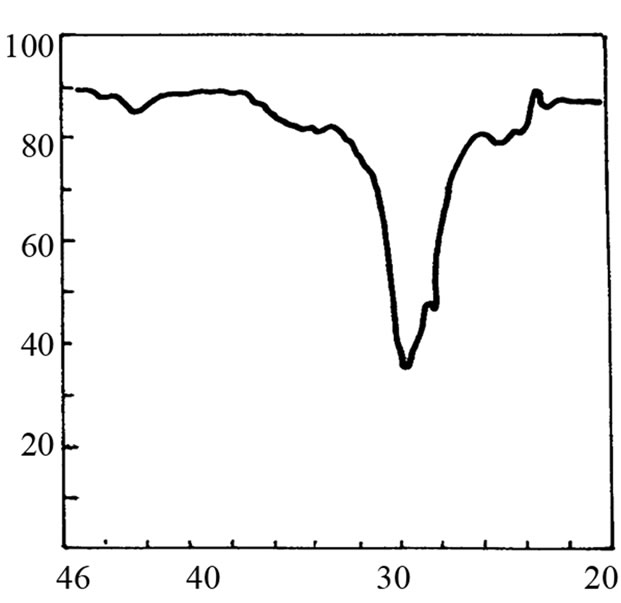 (e)
(e)
Figure 2. The relevant region of IR spectrum of TEA (a), SI (b), HESI (c), and mixtures of TEA with SI (d) and HESI (e).
band should be observed, while only the disappearance was the case. 1H NMR spectra provide further evidences on the presence of ion pair. Addition of TEA to imide solution in DMSO-d6 resulted in disappearance of imide group proton resonance >NH and simultaneous grow of resonance from (C2H5)3NH+ cation shifted upfield. The resonances are separated by 4.2 ppm in SI/SI-TEA system and by 6.1 ppm PI/PI-TEA. This spectroscopic picture also indicates the formation of ion pair instead of hydrogen bonding. Individual chemical shifts for the system SI/SI-TEA and PI/PI -TEA are as below:
SI (2.8, 4H, CH2; 11.4, 1H, NH); SI/TEA ( 1.05, 9HCH3; 2.55, 6H, CH2 from TEA, 2.8, 4H, CH2 from SI; 7.2, 1H, NH+).
PI (7.8, 4H, Ph; 11.6, 1H, NH); /PI-TEA (1.05, 9H, CH3; 2.55, 6H, CH2 from TEA, 7.8, 4H, Ph; 5.5, 1H, NH+).
4. Summary and Conclusions
1) Based upon experimental kinetic the mechanism of the reaction of cyclic imides and oxiranes was proposed and verified by instrumental and analytical methods.
2) The initial step of reaction is the formation of 1:1 imide: TEA adduct, which intermediates the proton transfer from imide to oxirane. The adducts could not be isolated, although their presence in solutions was demonstrated experimentally.
3) The crucial bond in adduct has ionic character; in non-aqueous solvents it is present as ion pair, while in water the adduct dissociate and free ions are present.
4) The suggested mechanisms can be applied in expalantion of first stages of obtaining of polyetherols in reaction of oxiranes with cyclic imides containg a couple of imide groups such as isocyanuric acid, parabanic acid and barbituric acid etc.
REFERENCES
- J. Lubczak, “Hydroxyalkylation of Cyclic Imides with Oxiranes. Part I. Kinetics of Reaction in Presence of Triethylamine as Catalyst,” Open Journal of Physical Chemistry, Vol. 2, No. 2, 2002, p. 2.
- L. Shechter and J. Wynstra, “Glycidyl Ether Reactions with Alcohols, Phenols, Carboxylic Acids, and Acid Anhydrides,” Industrial and Engineering Chemistry, Vol. 48, No. 1, 1956, pp. 86-93. doi:10.1021/ie50553a028
- S. Blum, P. Walsh and R. Bergman, “Epoxide-Opening and Group-Transfer Reactions Mediated by Monomeric Zirconium Imido Complexes,” Journal of American Chemical Society, Vol. 125, No. 47, 2003, pp. 14276-14277. doi:10.1021/ja037267t
- L.-Z. Dai and M. Shi, “A Gold(I)-Catalyzed Intramolecular Reaction of Propargylic/Homopropargylic Alcohols with Oxirane,” Chemistry—A European Journal, Vol. 14, No. 23, 2008, pp. 7011-7018. doi:10.1002/chem.200701954
- N. Iranpoor, H. Firouzabadi, R. Azadi and F. Ebrahimzadeh, “Regioselective Synthesis of vic-Halo Alcohols and Symmetrical or Unsymmetrical vic-Dihalides from Epoxides Using Triphenylphosphine N-Halo Imides,” Canadian Journal of Chemistry, Vol. 84, No. 1, 2006, pp. 69-75. doi:10.1139/v05-261
- J. F. Larrow and E. N. Jacobsen, “Asymmetric Processes Catalyzed by Chiral (Salen)Metal Complexes,” Topics in Organometallic Chemistry, No. 6, 2004, pp. 123-152. doi:10.1007/b11772
- K. Schwetlick, “Kinetische Methoden zur Un-tersuchung von Reactionsmechanismen”, VEB, Deutscher Verlag der Wissenschaften, Berlin 1971.
- I. Cisek-Cicirko and J. Lubczak, “Reactions of Hydroxymethyl Derivatives of Uric Acid with Oxiranes. II. an Analysis of Reaction Course and Product Structure,” Journal of Applied Polymer Science, Vol. 83, No. 9, 2002, pp. 1955-1962. doi:10.1002/app.10108
- A. Ślączka and J. Lubczak, “Hydroxyalkylation of Barbituric Acid. II. Synthesis of Polyetherols with Pyrimidine Ring,” Journal of Applied Polymer Science, Vol. 106, No. 6, 2007, pp. 4067-1074. doi:10.1002/app.26742
- M. Kucharski, J. Lubczak and E. Rokaszewski, “Addition of Oxiranes to Hydroxymethyl Derivatives of Isocyanuric Acid,” Chemia Stosowana, Vol. 27, No. 1-2, 1983, pp. 65-77.
- J. Lubczak, “Reactions of Hydroxymethyl Derivatives of Uric Acid with Oxiranes: Recognition of Mechanism Based on Kinetic Studies,” International Journal of Chemical Kinetics, Vol. 38, No. 5, 2006, pp. 345-350. doi:10.1002/kin.20167