Open Journal of Physical Chemistry
Vol. 2 No. 1 (2012) , Article ID: 17531 , 8 pages DOI:10.4236/ojpc.2012.21003
A Computational Evaluation of Bond Order and Charge Distributions in Isomeric Aminotroponiminiums and Their Benzo-Fused Derivatives
1Department of Organic Chemistry, Indian Institute of Science, Bangalore, India 2Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore, India
3Department of Chemistry, Indian Institute of Technology, Mumbai, India
4Department of Chemistry, Mahatma Gandhi University, Kottayam, India
Email: *snb@orgchem.iisc.ernet.in
Received July 6, 2011; revised August 24, 2011; accepted October 10, 2011
Keywords: Vinamidinium Element; Amino-Iminiotropylium Cations; Benzo-Fused Amino-Iminiotropylium Derivatives; Ab initio Hartree-Fock Procedures; Partial Bond Fixations; Net Charge Distributions; Stereochemical Factors
ABSTRACT
This paper reports the results on the nature of bond-order and net charge distributions predicted by ab initio HartreeFock procedures for 1-amino-2-iminio-, 1-amino-3-iminioand 1-amino-4-iminiotropylium cations that incorporate, in order, the 1,7-, 1,3- and 1,5-diazapentadienium (vinamidinium) elements. There appears to be very little contribution from tropylium-type charge distribution, the positive charges residing largely in the nitrogen atoms. The partial bond fixations and charge distributions show interesting variation in the three isomers. The 1,3-isomer in which the 1,3-diazapentadienium element is preserved in the favoured zigzag conformation appears to be relatively the best stabilized. The six isomeric benzo-fused derivatives arising from the three amino-iminiotropylium cations show similar differences in patterns of behaviour. Interestingly, the isomer in which a zigzag 1,3-diazapentadienium element is conjugated with a styrene moiety receives the deepest stabilization. While showing that the element largely contributes to the relative stabilization among the systems studied, contribution from certain stereochemical destabilizing factors may not be insignificant.
1. Introduction
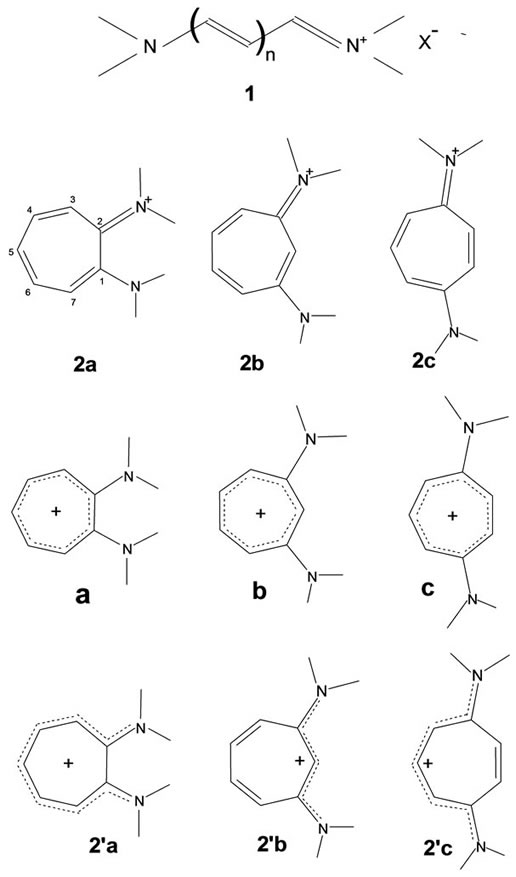
An evaluation of bond localizations and charge redistributions by theoretical methods when the amino (poly) vinyliminium (vinamidinium) structural element 1 (for the wide application of this element in chemistry see [1-11]) is made part of a cyclic system (e.g. aminotropon-2-, -3- & -4-iminiums 2a-c; n = 3, 1 & 2 in 1; the numbering indicated for 2a applies to all structures 2) was of interest because differences in energy content among these possibilities can arise for a number of reasons: While it is possible to conceive of a formal “diaminotropylium”-type charge distribution (a, b, c) for all three cases, only 2a, which has the N ipso-carbons directly linked, can participate as a whole in a vinamidinium-type delocalization (2’a). In the cases of its analogues 2b and 2c passage of p-information between the nitrogens for delocalization of that type would be, apparently, confined to their “odd” sides (2’b and 2’c) even while both have alternate vinylogous connectivity between the nitrogen ipso carbons.
• Available experimental evidence [12] has shown that vinamidinium chains 1, including those with 1,3-disubstitution, generally prefer anti-periplanar (i.e. zigzag) conformations. While such a conformation (2’b) can be maintained in the vinamidinium part of 2b, syn-periplanar conformations are forced on 2a and the vinamidinium part of 2c (2’c). Differences in the degree of destabilization may, therefore, be expected to depend on the delocalization that comes into effect on cyclization into forms 2a-c.
• Angle strain would be introduced when the internal angles open out beyond 120° to accommodate a planar 7-membered cyclic structure. Relief of strain can be expected to depend on the specific partial bond fixations that come about in 2a-c.
• Alternatively, bond localization could be such as would permit the 7-membered cycles to assume specific puckered conformations, minimizing angle strain while affecting the nature of the delocalization at the same time.
• The N-C-C-N array may not be uniplanar in the 1,2- system 2a in order that syn-periplanar interaction be relieved, causing it to be different from the 1,3- and 1,4-systems 2b and 2c both of which may prefer to be entirely planar.
2. Structures with Aminotroponiminium Elements
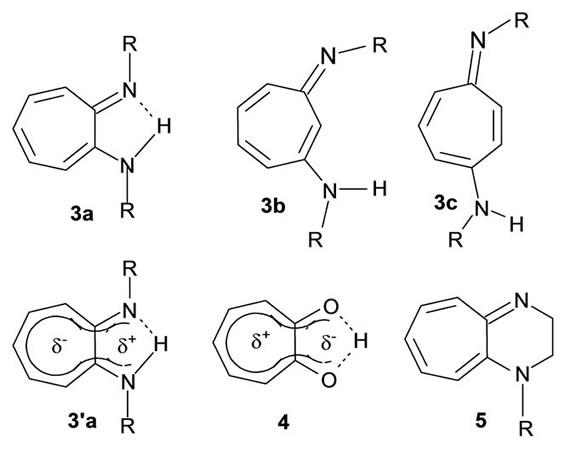
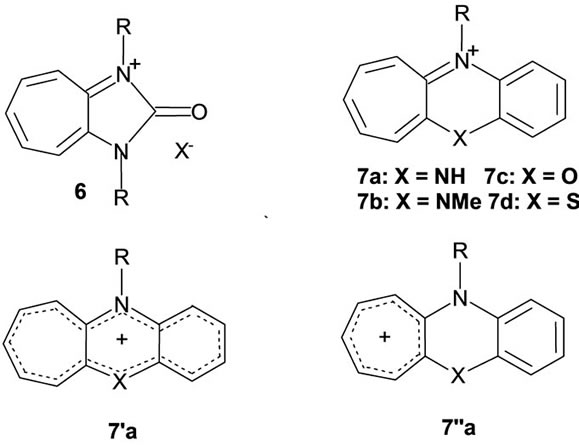
Attempts to synthesize the aminotroponimines structured like 2a-c were made at a time when interest in comparing them, in order, with a-, band g-tropolones was running high [13-17]. Ionic representation 2a/2’a would definitely have been that of the N,N’-tetramethylated primary product when one of the methods developed for the synthesis of the 1,2-free base N,N’-dimethyl derivative 3a (R=Me) and salts thereof was suitably adapted [13]. The products were found to be highly unstable compared with 3a which shows unusual stability and appreciable “aromatic” character: it undergoes facile nuclear electrophilic substitution and shows IR absorption bands for the presence of strong intramolecular N-H---N bridging. However, it exhibits its ring proton resonances upfield of those of a-tropolone (4) and its dipole moment is in the opposite direction. An initial interpretation of these pieces of evidence was that they were consistent with the establishment of a 10p-electron circuit (3a’) involving hybridization of non-bonding electrons of the NHR substituent. (cf. the case of N-tert. butyl-3-(tert. butylamino)-2-nitroprop-1-en-1-amine for which X-ray evidence [18] can be said to accord with “through-resonance” of this type.) It should be noted, however, that a canonical structure (akin to 4) where negative charge resides mostly on oxygen and the positive charge is distributed in a “tropylium” manner is not a large contributor to resonance in tropone. X-ray diffraction measurements and analysis of dipole moments and NMR spectra have disclosed that tropones and tropolones display appreciable partial bond fixation and must be regarded essentially as non-aromatic although some aromatic character can be accorded because of a susceptibility to aromatictype substitution. The 1H and 13C spectra of the N,N’- isopropyl derivative of corresponding to 3a (R=iPr), have been stated to show better consistency with the presence of rapid tautomeric equilibrium in the solution state. However, X-ray crystallographic data have disclosed that the two C-N bonds differ significantly in length (N=C: 1.314 Å; N-C: 1.342 Å), showing that in the solid state the structure is not symmetric on the whole even while the seven-membered ring and the two nitrogen atoms are nearly uniplanar.
Structures 3b and 3c can only be just presumed to have been those of the primary products in their attempted synthesis [16,17]. The instability of 3b and 3c is in accord with the absence of possibility in them of intramolecular N-H---N bridging. While a hydrochloride of the 1,3-system 3b is hygroscopic and unstable, both the 1,3- and 1,4-systems (3b and 3c) are said to give stable, crystlline picrates associated with waters of crystallization. The authors of reference [16] managed to record the 1H NMR spectrum of the 1,3-system 3b and found that the ring protons appear at fields higher than the “aromatic region”, the shifts being even less than those of the protons of b-tropolone. While this may be seen as according well with an absence of possibility of establishing a H-bond mediated peripheral circuit the authors do not say explicitly whether the 1H NMR spectrum is or is not consistent with a symmetric structure [16,17]. The ethylene bridged free base 5, where N-H---N bridging is not possible, has been synthesized [19]. The cycloheptimidazolonium salt 6, where the two nitrogen atoms that bear partial positive charges are bonded to a carbonyl, is an interesting precursor in a synthesis of N,N’-disubstituted 1-amino-2-imino-cycloheptatrienes [20]. This system and the cycloheptaquinoxalinium salts 7 [15], are among those that formally retain the 1-aminotropon-2-iminium cationic element 2a per se. The electronic structure of cation 7a has spawned an interesting debate. It was first put forward as an example of a stable anti-aromatic with a peripheral 16p-electron circuit (form 7’a) [21]. However, an estimation of resonance and circuit resonance energies and ring currents (employing graph theoretical methods [22-25]) showed that it would be more realistic to regard it and its other hetero-analogues 7b-d as 6ptropylium-6p-benzo systems 7”a. p-Electron distributions calculated by MNDO and 13C NMR chemical shifts [26-28] have been found to accord with this structure.
3. Computational
3.1. Aminotroponiminiums
Ab initio MO procedures were applied to systems 2 [with amino (-NH2) and iminium ( ) groups] in the attempt to obtain information on the possibilities raised above. Algorithms of the well-known GAUSSIAN procedures of various levels HF/3-21G, HF/6-31G*and MP2/6-31G* (including one that takes into account electron correlation, all available in our laboratories) were used for implementing the calculations. The MO procedures yield somewhat different relative energies for each case (Table 1) for the possible reason that they take into account electron correlation in different ways. MO procedures are generally not regarded as suitable for direct comparison of stabilities on the basis of (small) differences in energy content. We hope to show, at least by implication, that the on-the-average lesser stabilization of the 1,2-system 2a has significance.
) groups] in the attempt to obtain information on the possibilities raised above. Algorithms of the well-known GAUSSIAN procedures of various levels HF/3-21G, HF/6-31G*and MP2/6-31G* (including one that takes into account electron correlation, all available in our laboratories) were used for implementing the calculations. The MO procedures yield somewhat different relative energies for each case (Table 1) for the possible reason that they take into account electron correlation in different ways. MO procedures are generally not regarded as suitable for direct comparison of stabilities on the basis of (small) differences in energy content. We hope to show, at least by implication, that the on-the-average lesser stabilization of the 1,2-system 2a has significance.
3.2. Benzo Fusion
A strategy adopted in the investigation of mechanisms of reactions of tropolones has been predicated on a possibility that double bond character can be localized at certain positions within the 7-cycle by benzo-fusion [29].
Benzo-fusion to systems 2a-c yields six isomeric benzo-analogues, formally represented by particular sets of canonical structures 8a-f shown in Figure 1 below, that may be expected to differ in the modes of electron delocalization.
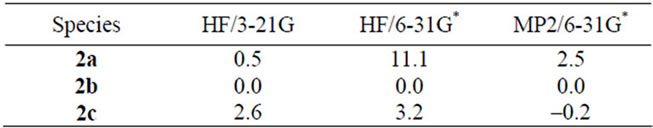
Table 1. Ab initio relative energies (kcal/mol) of aminotroponiminiums 2a-2c.
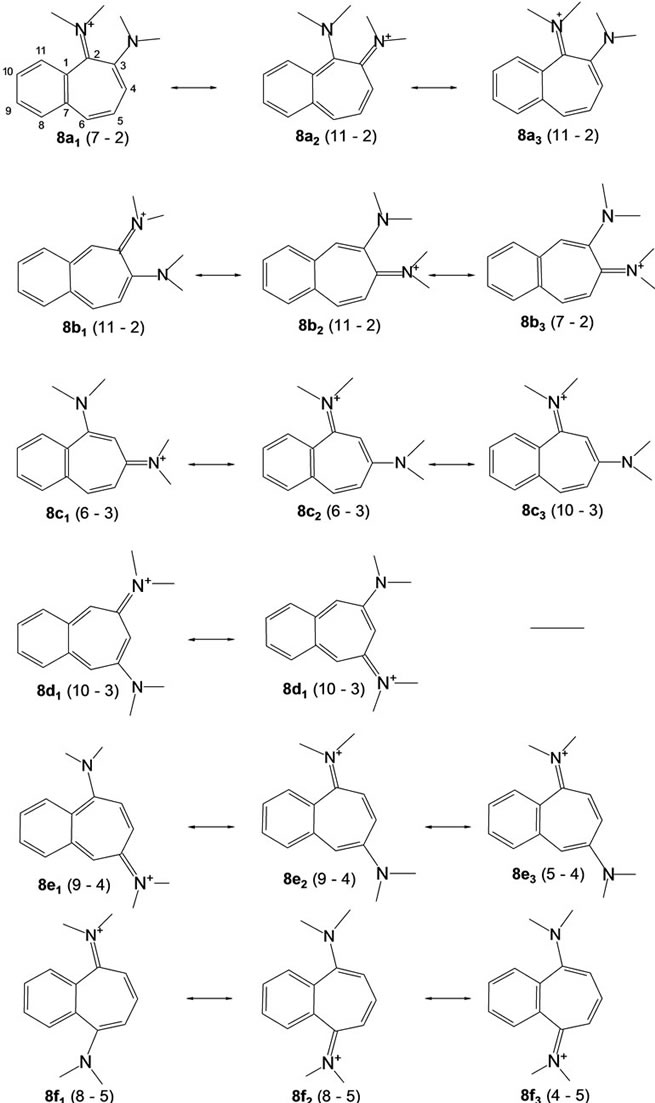
Figure 1. Structures resulting from benzo-fusion of 1, 2-, 1, 3- and 1,4-amino-iminiocycloheptatrienes. Odd numbers in parentheses denote possible “vinamidic” pathways. The numbering indicated for 8a applies also to 8b-f.
It was of interest to examine in what way the MO procedures would reflect the electron redistributions and structural features. The relative energies of the isomeric systems 8 are assembled in Table 2.
We discuss possible factors that may underlie why the non-symmetric 1,3-diamino system 8c, derived from the diaminotropylium 2b, the most stabilized among the monocyclic systems, is predicted also to be the most stabilized one. The structures were geometry optimized in the two sets of cases. Net atomic charges and HF calculated bond lengths (Å; in italics), shown in the structures in Figures 2 and 3 below, were chosen to serve as the parameters relevant to making qualitative comparisons among each set of the systems.
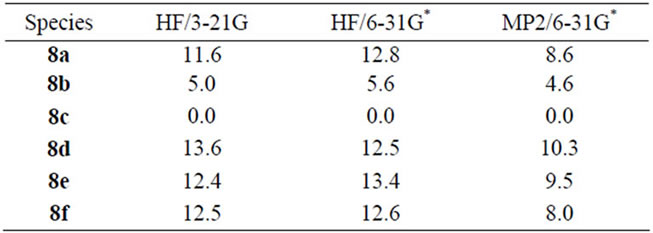
Table 2. Ab initio relative energies (kcal/mol) of Aminobenzotroponiminiums 8a-8f.
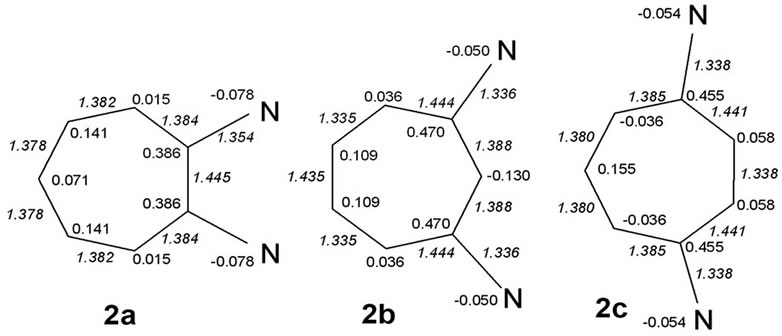
Figure 2. HF bond lengths and net charge distributions in aminotroponiminiums.
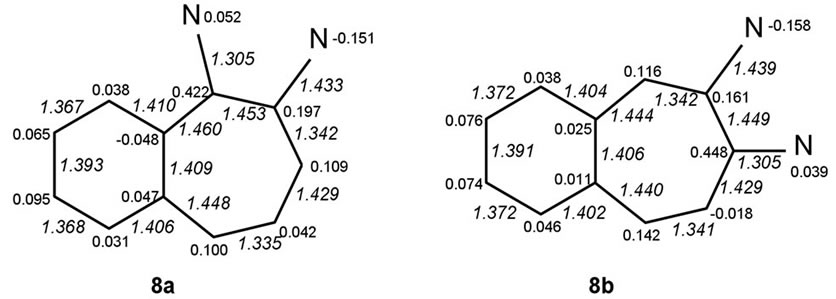
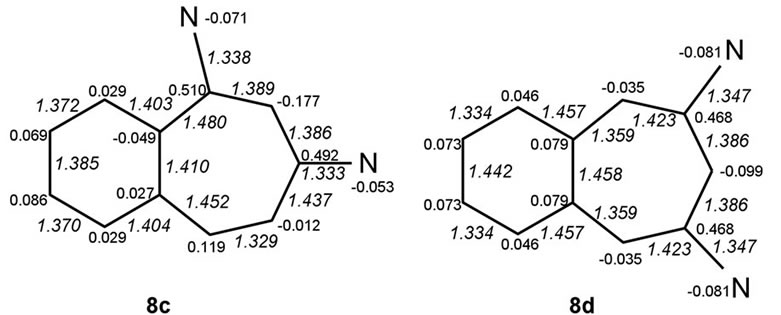
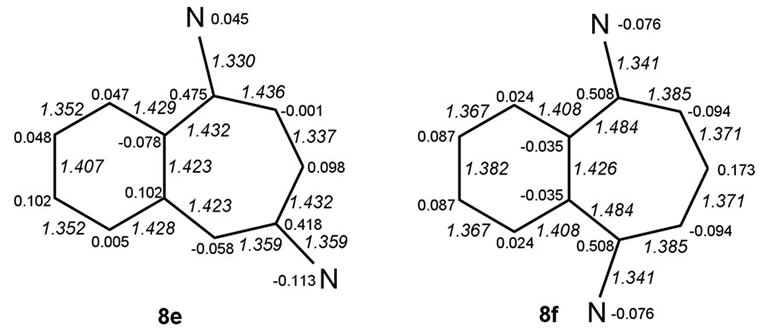
Figure 3. HF bond lengths and net charge distributions in benzo-aminotroponiminiums.
4. Results and Discussion
4.1. The Aminotroponiminiums 3
There seems to be good indication that stabilization correlates inversely with the length of the vinamidinium chain (2’a > 2’c > 2’b). Differences in the energies of formation of systems 2 apparently depend on maintenance of the zigzag conformation, as is the case in 2b. Energy changes on the account that one and two synperiplanar conformations are forced on the vinamidinium chains in 2c and 2a may not be strictly additive, however.
Factors of further interest are:
• Protonation of nitrogen draws electron density away from the nitrogen-ipso carbons to appreciable extents in all these aminotroponiminiums. High positive charges at the nitrogen-ipso carbons and build-up of electron density at the a carbons are seen.
• Consequent “vinyl cation” delocalization leads to charge alternation on the vinamidinium parts, the “odd” sides of aminotroponiminiums 2b and 2c.
• High bond orders are found at adjacent bonds in 2b and 2c.
• Vinamidinium character is well-preserved in 2b and 2c.
• Partial bond fixation is clearly seen on the “even” sides of 2b and 2c in the form of appreciable shortening of the C(4)-C(5) & C(6)-C(7) bonds in 2b (1.335 Å) and the C(2)-C(3) bond in 2c (1.338 Å).
• The pairs connecting the vinylic part with the vinamidic part [C(3)-C(4) & C(7)-C(1) in 2b (1.444 Å) and C(1)-C(2) and C(3)-C(4) in 2c (1.441 Å)] remain long (>1.40 Å), emphasizing suppression of delocalization on the “even” sides.
These characteristics are not strictly shared by the vinamidinium system that encompasses the whole of 2a. While the bond connecting the nitrogen-ipso carbons [C(1) & C(2)] is long (1.445 Å) and there is a progresssive decrease in bond lengths along the half cycle, there is no charge alternation but only an alternate placement of low and high residual positive charges, in the form of equal partial positive charges on the symmetrically placed carbons. A similar polarization is found on the “even” sides of 2b and 2c: high positive charges at the nitrogen-ipso carbons cause a build-up of electron density at the a carbons but successive carbons bear decreasing amounts of positive charges; there is no charge alternation.
“Steric interactions” between the two vicinal nitrogens seem clearly present: atom co-ordinates show out-ofplane movements of the two nitrogen atoms (Figure 4). Puckering of the ring preserves C2v symmetry. The C-N bonds are lengthened compared with the values in 2b or 2c (1.395 Å in 2a vs 1.363 Å in 2b and 1.365 Å in 2c). The increase in the negative charge at the nitrogens
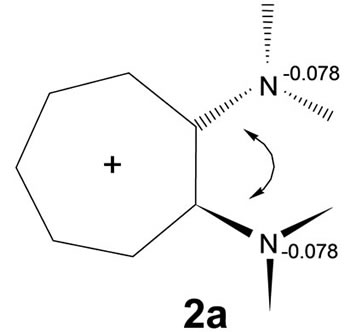
Figure 4. Steric interaction between vicinal amino groups in 1,2-aminotroponiminium.
indicates a more amine-like and pyramidal than iminiolike and planar character.
While these factors can account for some of the differences in the energies of formation among systems 2 it appears that the roughly inverse correlation of stabilization with the length of the vinamidinium chain (2b @ 2c > 2a) must derive from what was anticipated in the “Introduction” section. While the zigzag conformation is maintained in 2b, one and two syn-periplanar conformations are forced on the vinamidinium chains when they take the cyclic forms, respectively 2c and 2a, the proviso being energy changes on this account may not be strictly additive, as already noted.
4.2. The Benzo-Aminotroponiminiums
For carrying through qualitative comparisons conveniently features of systems 8 salient from the MO calculations have been assembled in Table 3 overleaf.
• System 8c, the nonsymmetric one of the two 1,3- systems, emerges as the most stabilized. All systems 8 share the feature of having the C(8)-C(9) and C(10)- C(11) bonds short and C(7)-C(1) bonds long making it appear that partial bond fixation in the benzo parts accords with a large contribution from o-quinonoid canonical structures, including the cases 8c and 8f for which such structures cannot be set down without charge separation.
• That the fixation is really not o-quinonoid in any of the cases except 8d is clear from the increase in the lengths of both C(1)-C(2) and C(6)-C(7) bonds.
• While N(A) in 8a and N(B) in 8b bear partial positive charges, the partial charges at N(B) in 8a and N(A) in 8b are negative.
• The negative charges are much larger than at the negatively charged nitrogens of the other systems. The C-N bond lengths associated with the negative nitrogens are much longer than the ones associated with the positive nitrogens C(A)-N(A)—1.305 & C(B)-N(B)—1.433 Å in 8a and C(A)-N(A)—1.439 & C(B)-N(B)—1.305 Å in 8b, betokening amino and
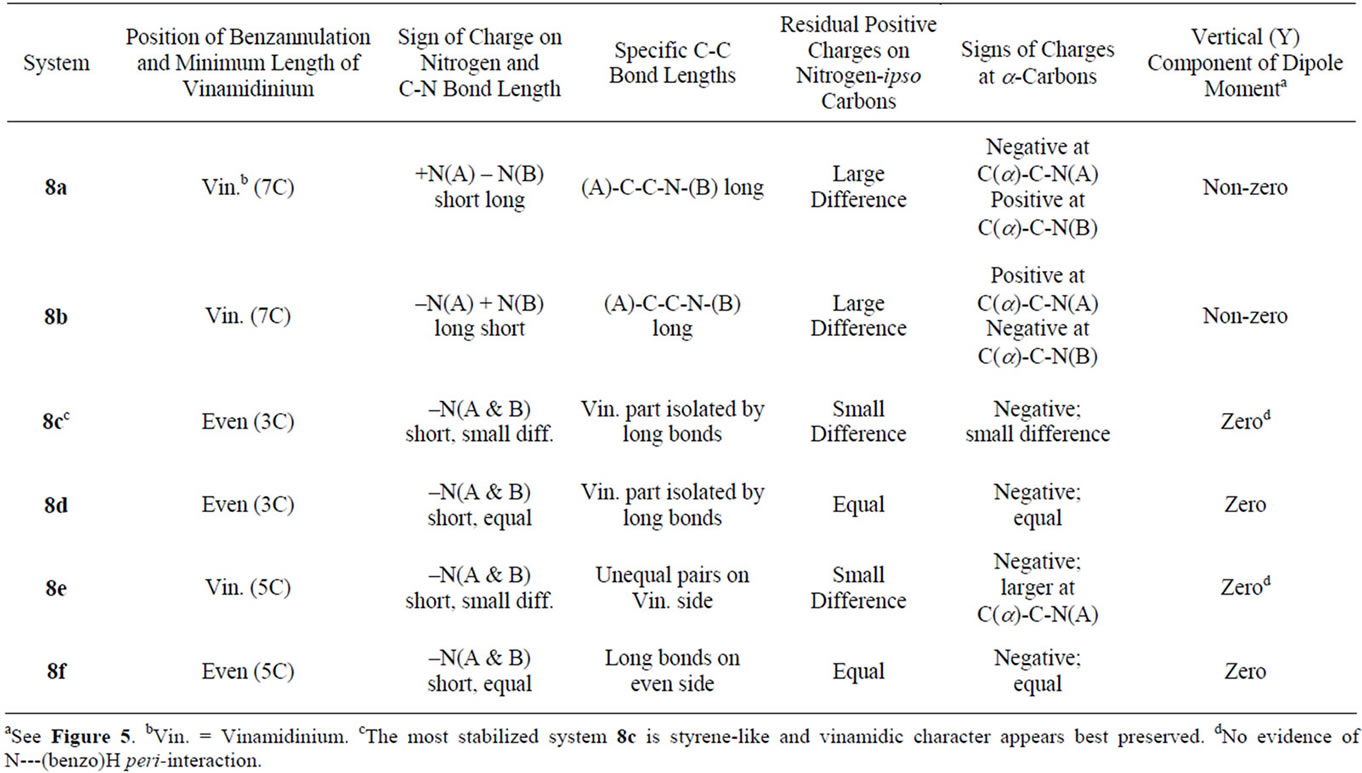
Table 3. Features of systems 8 qualitatively compared.
iminio C-N partial bond fixation in the respective cases.
• The ionic positive charge appears redistributed over the entirety of systems 8. However, the largest residual positive charges are concentrated at the nitrogen-ipso carbons. While these charges are equal in the symmetric cases (8d & 8f) and not very different in their non-symmetric analogues 8c & 8e, systems 8a & 8b are exceptional in that the charge differences are large, those at carbons ipso to the negative (amino-type) nitrogens, C(3) in 8a and C(4) in 8b, being much lower. The gathering of positive charges at both the nitrogen and carbon of the iminio-type C-N groups could be a destabilizing factor in these systems.
• Carbons a to the nitrogen-ipso carbons bear small negative charges in all cases. A part of this feature could be normal to the vinamidinium-type charge redistribution but there are three exceptions: carbons α to those bearing low positive charges in systems 8a and 8b and C(4) on the “even” side of 8e bear positive charges.
• Neighbouring carbons on the “even” side of 8f [C(1) & C(7)] bear negative charges.
• Low bond order (length > 1.45 Å) of the N(A)-CC-N(B) bond, found in the precursor 2a, is maintained in both of the benzo systems 8a and 8b derived from it. The lengthening arises, presumably, from an accumulation of high residual positive charges at vicinal carbons, C(2) & C(3) in 8a and C(3) & C(4) in 8b. It is manifest in a different form in systems 8c-8f, leading to virtual isolation of the “odd” vinamidinium part from the “even” part—bonds C(1)-C(2) & C(4)- C(5)in 8c, C(2)-C(3) &C(5)-C(6) in 8d, C(2)-C(3) & C(4)-C(5) in 8e and C(1)-C(2) & C(6)-C(7) in 8f are long.
• The lengthening of the C(1)-C(7) bridging bond in systems 8, coupled with partial bond fixation within the benzo ring, may or may not always accord with the bond order distribution natural to vinamidinium in those cases where benzo-substitution is on the vinamidinium side, as in 3e.
• This factor is important if systems 8c and 8e or 8e and 8f are compared. 8c is stabilized relative to 8d despite the cyclizing entity being styrene-like in 8c and o-quinonoid-like in 8d.
• Absence of an iminio-type C-N group, with its inplane hydrogens is presumably necessary for any outof-plane movement of the constituent atoms or puckering of the 7-membered cycle. Systems other than 8a and 8b show zero orthogonal component of dipole moment (Table 4) even though the possibility of peri-interaction exists [30,31], one in each of 8c and 8e and two in 8f.
• A component of dipole moment orthogonal to the general planes is present in both 8a and 8b and absent in the others (Figure 5). Atom co-ordinate data have indicated that the component arises both from a puckering of
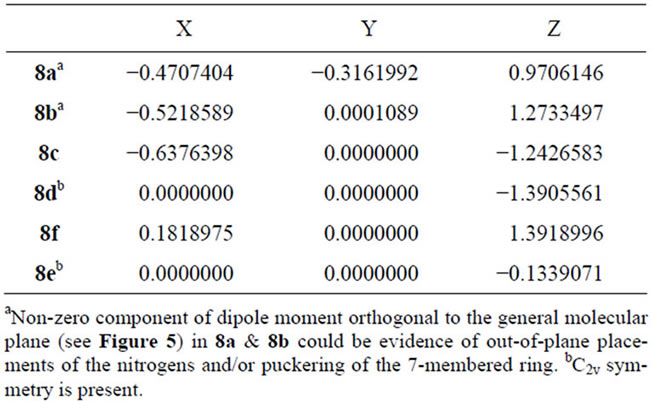
Table 4. Dipole moment components (debyes).
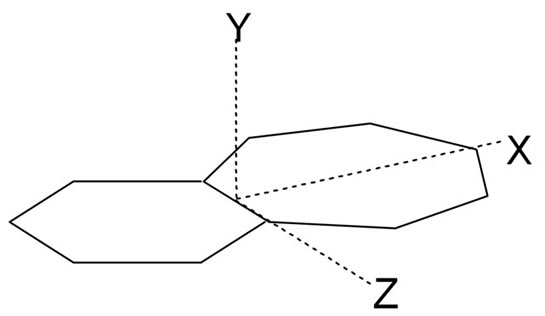
Figure 5. Co-ordinate system for the benzo-aminotropo niminiums.
the 7-membered ring and from out-of-plane placement of the nitrogens. The greater magnitude of the component in 8a could arise from a larger out-of-plane movement of N(A) from a need to minimize not only peri-interaction of the syn iminio hydrogen on planar N(A) with the C(11) hydrogen but also the interaction of the anti iminio hydrogen with the amine-like N(B). However, the need to maintain uniplanarity of HN(A)-H with C(1)-C(2)-C(3) in the system seems met partly by puckering of the ring whereby N(A) is moved above the general plane and partly by lengthening of the C(11)-C(1) bond.
• A point of similarity exists between systems 3f and 3d even though o-quinonoid bond order distribution, available to 3d, is not very likely for 3f. The symmetrically placed bonds between the vinamidinium and the rest of the system are lengthened; the latter are the longest bonds encountered in all of systems 8. This symmetric distortion of the 7-membered cycle may prevent the accumulation of angle strain.
5. Conclusion
The three formalisms employed for the computations HF/3-21G, HF/6-31G* and MP2/6-31G* are in agreement in predicting system 2b as relatively the best stabilized among the three isomeric aminotroponiminiums compared and system 8c as the most stabilized among their six positional isomeric benzo analogues. It appears safe to state that the 3-carbon length of the vinamidinium moiety and the freedom it has to assume the preferred zigzag conformation are paramount among the stabilizing factors. The destabilization of 2a and 8a relative to the others of its kind likely arises from twists around bonds of low bond order. The lengthening of the C(1)- C(7) bond in all of the systems 8, coupled with the implied partial bond fixation within the benzo ring, may or may not always accord with the bond order distribution natural to the vinamidinium chain in cases where benzosubstitution is on the vinamidinium side. This appears to be a factor when system 8c is compared with system 8e or 8e with 8f. The stabilization of 8c relative to 8d is of particular interest because the cyclizing entity is vinyl benzene-like (styrene-like) in the former whereas it is o-quinonoid in the latter. Of equal interest is a point of similarity between systems 8f and 8d even though bond order distribution of the o-quinonoid type, available to the latter, is not very likely for the former. The implied symmetric distortion of the 7-membered cycle seems to prevent either system from accommodating angle strain in a manner that prevents them from reaching the level of energy minimization possible for 8c on this account.
REFERENCES
- D. Lloyd and H. McNab, “Vinamidines and Vinamidinium Salts—Examples of Stabilized Push-Pull Alkenes,” Angewandte Chemie International Edition, Vol. 15, No. 8, 1976, pp. 459-468. doi:10.1002/anie.197604591
- S. S. Malhotra and M. C. Whiting, “Researches on Polyenes. VII. Preparation and Electronic Absorption Spectra of Homologous Series of Simple Cyanines, Merocyanines, and Oxonols,” Journal of the Chemical Society, Vol. 756, 1960, pp. 3812-3822. doi:10.1039/jr9600003812
- C. Jutz, “C-C Bond Forming Reactions of Chloromethinium Ions,” In: H.-G. Viehe and H. Bohm, Eds., Iminium Salts in Organic Chemistry, Wiley, New York, Vol. 9, 1976, pp. 225-343.
- J. Zemlicka and Z. Arnold, “Synthetic Reactions of Dimethylformamide. XI. Polyformylation of Ketones of Other Types and the Problem of the Reaction Course,” Collection of Czechoslovak Chemical Communications, Vol. 26, 1961, pp. 2852-2864.
- Z. Arnold, “Synthetic Reactions of Dimethyl Formamide. XII. Formylation of Some Carboxylic Acids and Their Derivatives,” Collection of Czechoslovak Chemical Communications, Vol. 26, 1961, pp. 3051-3057.
- I. M. Mallick and S. N. Balasubrahmanyam, “σ-Correlations and Steric Effects,” Organic Magnetic Resonance, Vol. 19, No. 3, 1982, pp. 160-163. doi:10.1002/mrc.1270190311
- S. N. Balasubrahmanyam, B. Jeyashri and I. N. N. Namboothiri, “Selectivities in the Formation of Pyridines and Pyrimidines by Ammonia-Induced Cyclocondensations of Vinamidiniums,” Tetrahedron, Vol. 50, No. 27, 1994, pp. 8127-8142. doi:10.1016/S0040-4020(01)85295-9
- R. Gompper and C. Schneider, “New (Dialkylamino)Allenes, -Thiophenes, and -Pyrroles from Vinamidinium Salts,” Synthesis, No. 3, 1979, pp. 213-215. doi:10.1055/s-1979-28624
- J. T. Gupton, D. K. Krolikowski, R. H. Yu, S. W. Riesinger and J. A. Sikorski, “Application of 2-Substituted Vinamidinium Salts to the Synthesis of 2,4-Disubstituted Pyrroles,” Journal of Organic Chemistry, Vol. 55, No. 15, 1990, pp. 4735-4740. doi:10.1021/jo00302a047
- K. Hafner, K. F. Bangert and K. Orgafos, “1,3-Bis(dimethylamino)pentalene,” Angewandte Chemie International Edition, Vol. 6, No. 5, 1967, pp. 451-452. doi:10.1002/anie.196704511
- J. T. Gupton, J. Coury, M. Moebius and S. Fitzwater, “The Reaction of Organometallic Reagents with Unsymmetrically Substituted Vinamidinium and Chloropropeniminium Salts,” Synthetic Communications, Vol. 16, No. 12, 1986, pp. 1575-1586. doi:10.1080/00397918608056411
- M.-L. Filleux-Blanchard and D. le Botlan, “Conformational Isomerism about the Carbon-Carbon Bond in Propenylylidene Ammonium Salts,” Organic Magnetic Resonance, Vol. 9, 1977, pp. 618-620. doi:10.1002/mrc.1270091103
- W. R. Brasen, H. E. Holmquist and R. E. Benson, “Novel Analogs of Tropolone,” Journal of the American Chemical Society, Vol. 82, No. 4, 1960, pp. 995-996. doi:10.1021/ja01489a055
- W. R. Brasen, H. E. Holmquist and R. E. Benson, “N,N’-Disubstituted 1-Amino-7-imino-1,3,5-cyclo Heptatrienes, a Non-Classical Aromatic System,” Journal of the American Chemical Society, Vol. 83, 1961, pp. 3125-3135. doi:10.1021/ja01475a031
- N. Soma, J. Nakazawa, T. Watanabe, Y. Sato and G. Sunagawa, “Seven-Membered Ring Compounds. XV. Preparations of Troponimine Derivatives,” Chemical & Pharmaceutical Bulletin, Vol. 13, 1965, pp. 457-464. doi:10.1248/cpb.13.457
- M. Cavazza and F. Pietra, “Synthesis of 3-Aminocycloheptatrienylidenamines,” Journal of the Chemical Society, Chemical Communications, Vol. 11, 1976, 412-413. doi:10.1039/c39760000412
- M. Cavazza and F. Pietra, “A Synthetic Approach to 4-Amino-1-imino-2,4,6-cycloheptatrienes,” Synthesis, No. 3, 1977, 204-205. doi:10.1055/s-1977-24326
- D. L. Ostercamp, L. M. Preston and K. D. Onan, “The Synthesis and Crystal Structure of N-Tert-butyl-3-(tertbutylimino)-2-nitropropen-1-amine,” Zeitschrift für Naturforschung B, Vol. 48, 1993, pp. 1138-1142.
- T. Nozoe, S. Ishikawa and K. Shindo, “Synthesis and Reactions of 2,3-Dihydro-cyclohepta[b][1,4] Thiazines and 2,3-Dihydro-1H-cyclohepta [b] Pyrazines,” Heterocycles, Vol. 28, 1989, pp. 733-744.
- H. Nakao, “Seven-Membered Ring Compounds. XVIII. Reactions of Quaternary Ammonium Salts of Cycloheptimidazole Derivatives,” Chemical & Pharmaceutical Bulletin, Vol. 13, No. 7, 1965, pp. 810-818. doi:10.1248/cpb.13.810
- T. Nozoe, “Cyclohepta[b][1,4]benzoxazine and Related Compounds—Some Novel Aspects in Troponoid Chemistry,” Pure and Applied Chemistry, Vol. 54, 1982, pp. 975-986. doi:10.1351/pac198254050975
- J. Aihara, “A New Definition of Dewar-Type Resonance Energies,” Journal of the American Chemical Society, Vol. 98, No. 7, 1976, pp. 2750-2758. doi:10.1021/ja00426a013
- J. Aihara, “Resonance Energies of Nonbenzenoid Hydrocarbons,” Bulletin of the Chemical Society of Japan, Vol. 51, No. 12, 1978, pp. 3540-3543. doi:10.1246/bcsj.51.3540
- J. Aihara, “Graph-Theoretical Formulation of London Diamagnetism,” Journal of the American Chemical Society, Vol. 101, No. 20, 1979, pp. 5913-5917. doi:10.1021/ja00514a009
- J. Aihara and T. Horikawa, “Graph-Theoretical Formula for Ring Currents Induced in a Polycyclic Conjugated System,” Bulletin of the Chemical Society of Japan, Vol. 56, No. 6, 1983, pp. 1853-1854. doi:10.1246/bcsj.56.1853
- T. Kurihara, K. Shindo, S. Ishikawa and T. Nozoe, “Benzo[b]cyclohepta[e][1,4]thiazines and Their Diazine Analogues. III. π-Electronic Structures of 5H,11H+- Cyclohepta[b]quinoxalinium Ions and Their Oand S-Analogues,” Bulletin of the Chemical Society of Japan, Vol. 64, No. 9, 1991, pp. 2668-2676. doi:10.1246/bcsj.64.2668
- T. Nozoe, T. Kitahara, K. Takase and M. Sasaki, “Syntheses of Benzo[b]tropazines,” Proceedings of the Japan Academy, Vol. 32, No. 5, 1956, pp. 349-352.
- J. Aihara, “A New Definition of Dewar-Type Resonance Energies,” Journal of the American Chemical Society, Vol. 98, No. 10, 1976, pp. 2750-2758. doi:10.1021/ja00426a013
- F. Pietra, “Revival of Troponoid Chemistry,” Accounts of Chemical Research, Vol. 12, 1979, pp. 132-138. doi:10.1021/ar50136a004
- I. N. N. Naboothiri and S. N. Balasubrahmanyam, “Transformations in Bromoand Alkoxybenzotropones,” Journal of the Indian Institute of Science, Vol. 74, 1994, pp. 473-486.
- I. N. N. Namboothiri and S. N. Balasubrahmanyam, “On the Temperature-Dependence of Regioselectivity in the Amination of Bromobenzotropones,” Indian Journal of Chemistry—Section B, Vol. 32, 1994, pp. 1029-1034.
NOTES
*Corresponding author.