Stem Cell Discovery
Vol. 3 No. 3 (2013) , Article ID: 34690 , 13 pages DOI:10.4236/scd.2013.33022
Matrigel modulates a stem cell phenotype and promotes tumor formation in a mantle cell lymphoma cell line
![]()
1Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, USA 2School of Pharmacy, University of Nebraska Medical Center, Omaha, USA 3Department of Pathology, City of Hope Medical Center, Duarte, USA 4Department of Cell Biology, Genetics and Anatomy, University of Nebraska Medical Center, Omaha, USA; *Corresponding Author: jsharp@unmc.edu
Copyright © 2013 Abigail Hielscher et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 27 March 2013; revised 27 April 2013; accepted 27 May 2013
Keywords: Mantle Cell Lymphoma; Side Population; Tumor-Initiating Cells; Microenvironment
ABSTRACT
Tumors may be maintained by subpopulations of cells possessing stem cell-like properties. We evaluated the stem cell-like and tumor-forming properties of side population (SP) and CD133+/ CD44+ cells in Granta 519, a human mantle cell lymphoma cell line. The in-vitro Cobblestone Area Forming Cell (CAFC) assay, designed to detect stem and progenitor cells, revealed that SP cells contained the greatest proportion of stem cell-like cells. The addition of Matrigel to CAFC assays of SP and non-SP cells both increased their respective stem cell frequencies in comparison to those cultures without Matrigel, and additionally resulted in observed stem cell frequencies which were the same between SP and non-SP cells. Contrary, Matrigel decreased the stem cell frequencies of CD133+/CD44+ or CD133−/CD44− cells. In-vivo assays revealed tumor formation from Matrigel-mixed SP and nonSP cells, and in one instance, occurred with as few as one Matrigel-mixed SP cell. Vehicle-mixed injections of SP and non-SP tumor cells resulted in tumor formation from SP cells only. Tumor formation did not occur from Matrigel nor hyaluronan (cellular substrate for CD44-expressing cells)-mixed populations of CD133+/CD44+ or CD133−/CD44− cells. These data demonstrate that Matrigel modulates a stem cell phenotype and promotes tumor formation from SP and nonSP cells. The tumor micro-environmental niche and tumor cell to micro-environmental interactions may be important future targets for novel chemotherapeutic agents.
1. INTRODUCTION
Mantle cell lymphoma (MCL) is a type of non-Hodgkin’s Lymphoma, characterized by an aberrant proliferation of mature B lymphocytes in the mantle zones of lymphoid follicles. The disease has an aggressive clinical course with a median patient survival of only 3 - 4 years following diagnosis [1,2]. This dismal outcome is due to the advanced stage of disease upon diagnosis with several extranodal sites typically involved including the spleen [3] , bone marrow [4] and gastrointestinal tract [5]. Although a wide variety of chemotherapeutic agents are presently available to treat the disease, such therapeutic strategies have not significantly improved patient outcome [6-8] with patient relapse being a frequent occurrence [9,10] . Due to the aggressive nature of the disease and the high propensity for relapse, novel approaches for identifying the cell type(s) responsible for MCL initiation and progression are imperative for understanding and better combating the disease.
Increasing evidence has pointed to the existence of subsets of tumor cells proposed to be responsible for tumor initiation and propagation. Such tumor cells are referred to as tumor-initiating cells (TICs), rare subpopulations of tumor cells capable of tumor initiation when introduced in small numbers into immunocompromised mice, self-renewal upon serial transplantation into immunocompromised mice and differentiation into cells making up the bulk of the original tumor [11]. First described by Bonnet et al [12] in human leukemias, TICs have since been discovered in solid tumors including breast [13], prostate [14,15] , colorectal [16] , and liver carcinomas [17,18] amongst others. Regarding lymphomas, TICs have been identified in a mouse model of MCL [19] and have recently been prospectively identified in human MCL tumors [20]. In the latter study, it was reported that MCL cells lacking the mature B cell surface receptor CD19 were enriched for self-renewal and tumor-forming capabilities following xenotransplantation in immunodeficient animals [20]. Additional investigation is warranted to further define and characterize this population of putative MCL TICs so as to better identify and design chemotherapeutic agents aimed at eliminating these potentially rare and aggressive MCL subpopulations.
Validated methods, similar to those employed for tissue stem cells, are required for isolation and evaluation of putative MCL TICs. Such methods include the use of cell surface markers, aldehyde dehydrogenase activity, or the presence of a side population (SP). Of particular interest is the cell surface marker CD133, a hematopoietic stem cell (HSC) marker [21], and CD44, a cell surface adhesion receptor which facilitates cellular attachment to the extracellular matrix (ECM) [22,23] . Both CD133 and CD44, alone or in conjunction with other cell surface markers, have been assigned to cells having enhanced tumorigenic and stem cell-like properties [14-16,18,24] . The second cell population evaluated is the SP, a tailshaped cell population visualized as a result of ABCG2- mediated efflux of the Hoechst 33342 dye [25] . First described by Goodell et al. [26] as a novel method for isolating HSCs from murine bone marrow, the SP has been employed to isolate both tissue stem cells [27] and putative TIC populations [11,17,19,20] .
Tissue stem cells are known to reside in supportive niches or microenvironments, comprised of cellular and non-cellular components which together maintain stem cell homeostasis [28,29] . It is possible that the enhanced tumorigenicity and stem cell-like properties of TICs may be a result of their maintenance in supportive micro-environmental niches. Indeed, Vermeulen et al. [30] reported that the microenvironment was imperative for regulating Wnt activity, functionally assigned to colon TICs. Similarly, Calabrese et al. [31] discovered that brain TICs were localized to the brain vascular niche, which they demonstrated to be responsible for promoting self-renewal and accelerating tumor growth from brain TICs. Regarding lymphomas, it has been demonstrated that the lymph node microenvironment actively participates in the progression of follicular lymphoma [32].
In the present study, we sought to determine whether microenvironmental factors were necessary for accelerating self-renewal and tumor-formation capabilities of Granta MCL TICs isolated on the basis of either CD133/ CD44 co-expression or the SP functional pheno-type. While the stem cell niche is comprised of numerous cellular and non-cellular entities, the ECM is of particular interest as it is known to play an important role in regulating stem cell self-renewal and differentiation [33-37] . As such, we elected to utilize Matrigel, a commercially available conglomerate of ECM proteins derived from a murine sarcoma. Matrigel served as a biological substrate to promote MCL TIC xenograft tumor formation, previously demonstrated by others [38-40] . We demonstrate that Matrigel, rather than cell phenotype differences, affect the enrichment of stem cell-like cells invitro and expedite tumor development in-vivo for both SP and non-SP cells. Elucidating the role whereby the tumor microenvironment influences proposed MCL TIC behaviors is imperative for implementation of better and/ or novel therapeutics targeted toward both the tumor microenvironment and MCL to niche interactions.
2. MATERIALS AND METHOD
2.1. Cell Lines and Culture
Granta 519 (herein, Granta) cells were kindly obtained from Dr. Shantaram Joshi, UNMC and cultured in DMEM (Invitrogen; Grand Island, NY) containing 10% fetal bovine serum (FBS) and 1× penicillin/streptomycin (Invitrogen). Murine bone marrow stromal cells (MS5) [41] were kindly provided by Dr. John Jackson (Wake Forest Institute of Regenerative Medicine, Winston Salem, NC) and employed as described previously [42]. MS5 were cultured in RPMI (Invitrogen) containing 10% FBS, 1× penicillin/streptomycin and 1 μg/ml hydrocortisone (Sigma-Aldrich; Allentown, PA). MS5 cells were rendered non-adherent with 0.05% trypsin/EDTA (Invitrogen). Granta and MS5 cells were passaged every 2 - 3 days and were maintained at 37˚C in a humidified air atmosphere containing 5% CO2.
2.2. Flow Cytometry and Sorting
Granta cells were stained 1:11 with PE-CD133 (Miltenyi; Gergisch Gladbach, Germany) and 1:89 with APC-CD44 (e-Bioscience; San Diego, CA) for 20 minutes at 4˚C. For SP analyses, Granta cells were re-suspended in 1 ml of Hoechst IMDM (Invitrogen; IMDM media supplemented with 2% FBS, 1μM Hepes, and 1x penicillin/streptomycin) for incubation at 4˚C overnight, adjusted to 1.0 × 106 cells/ml and stained with 6μg/ml Hoechst 33342 (Sigma-Aldrich) at 37˚C for 30 minutes. For confirmation of the SP, Granta cells were stained with 40 μM of verapamil (Sigma-Aldrich) for 15 minutes at 37˚C prior to incubation with Hoechst 33342. Analyses were conducted in the cell analysis facilities at UNMC and Creighton University, Omaha, NE. Data was analyzed using FlowJoTM. Granta TIC and non-TIC cells were isolated using Fluorescence Activated Cell Sorting (FACS).
2.3. Cobblestone Area Forming Cells Assays
Granta populations were sorted in limiting dilutions into 96 well plates containing Murine Stromal 5 (MS5) cells. CD133+/CD44+ and CD133−/CD44− populations were adjusted to six dilutions ranging from 407 to 1 cell/ well. SP and non-SP populations were adjusted to 10 dilutions ranging from 33,300 to 1 cell/well. All dilutions were made at a factor of 3 with each dilution being conducted in a total of 20 or 30 wells. In some instances, Granta populations were overlaid with a final concentration of 275 μg/ml of Matrigel (BD Biosciences; San Jose, CA). The co-cultures were maintained for 5 weeks. Phase contrast microscopy was used to assess for the presence of phase dark cobblestones (groups of 3 or more cells growing in a colony beneath the stromal layer). Using a semi-logarithmic scale, the frequency of stem cell-like cells in each population was plotted and measured as the inverse of the number of seeded cells corresponding to 37% negative wells [43]. Images were captured at 10× magnification using a Nikon P5000 digital camera.
2.4. Tumorsphere Assays
Granta SP and non-SP fractions were sorted at dilutions of 1, 10, 100 and 1000 cells/well into 96 well plates containing 150 µl of DMEM/HamsF12 media supplemented with either 20 ng/ml of bFGF (Invitrogen) and 20 ng/ml of EGF (Invitrogen) or 20 ng/ml bFGF and 10 ng/ml of EGF. Cultures were maintained for a total of 6 weeks in a 95% humidified air atmosphere at 37˚C and 5% CO2. The presence of tumorspheres was evaluated at weekly intervals using phase contrast microscopy at 20× magnification. Images were photographed using a Nikon P5000 digital camera.
2.5. Enzyme-Linked Immunosorbent Assay
Supernatant from Granta cells, MS5 cells, Granta/ MS5 co-cultures and Granta TIC and non-TIC/MS5 cocultures were collected and quantified for interleukin 8 (IL-8) using enzyme-linked immunosorbent assay (ELISA). The Quantikine Immunoassay Kit (R&D Systems, Minneapolis, MN) was utilized and all procedures were followed according to the manufacturer’s protocol. The lower limit of sensitivity for the assay is 3.5 pg/ml with a dynamic range between 3.5 pg/ml and 2000 pg/ml. Intra-assay coefficient of variation ranges from 5.4% - 6.5% and inter-assay coefficient of variation from 6.1% to 9.7%. For analyses of IL-8 secretion from Granta and MS5 cells, 2.0 × 106 cells/ml were used. These cultures were maintained for 3 days prior to collection of supernatants. The cells were then re-suspended in fresh media and maintained in culture for another 3 days prior to collection of additional supernatant. For Granta/MS5 cocultures, 1.0 × 106 MS5 cells were plated and maintained as previously described. Once MS5 cells reached 75% confluency, 1.0 × 106 Granta cells were plated and grown in standard growth media as previously described. The supernatants from Granta/MS5 co-cultures were collected at 1, 3, 7 and 14 day intervals for the assessment of IL-8 levels over time. All supernatant was passed through a 70 µM strainer for removal of residual cell debris. For each analysis, the supernatants from triplicate cultures were evaluated.
2.6. In-Vivo Analyses
All procedures on animals were followed in compliance with the UNMC Animal Care and Use Committee. Three-five animals were maintained in a stainless steel cage and were given food and water ad libitum. Granta TIC and non-TIC populations were subcutaneously injected into the hind flanks of 3 - 5 week old female NOD-scid IL2Rgnull (NSG) mice (Jackson Laboratories; Bar Harbor, ME). Dilutions of 10, 100 and 1000 SP and non-SP cells and 10, 100 and 500 CD133+/CD44+ and CD133−/CD44− cells were prepared as described by Quintana et al (2008). Additionally, 10, 100 and 1000 CD133+/CD44+ and CD133−/CD44− cells were resuspended at a 1:8 dilution in hyaluronan (Hyaluronex; Lexington, KY). Single SP or non-SP cells were sorted using FACS into 96 well plates and were re-suspended at a 1:4 dilution in Matrigel. When tumors reached 1 - 1.5 cm, animals were necropsied. Tumor volume was assessed using the formula for an ellipsoid: 4/3 πr1r2r3. As a control, Matrigel and vehicle were injected alone into either the right or left shoulders of 2 NSG mice.
2.7. Histolopathology and Flow Cytometry
Murine tissues including the lung, right femur, and kidney and portions of the spleen, small intestine, liver and kidney were fixed in 10% neutral buffered formalin for at least 24 hours and subjected to histopathological analysis using hematoxylin and eosin (H&E) staining (Eppley Cancer Center Histology Core Facility, UNMC). The remaining murine tissues were evaluated for human CD20 expression using flow cytometry. The lung was cut into 5 mm pieces and digested in 1 mg/ml collagenase V (Sigma-Aldrich) and 500 µg/ml elastase (Sigma-Aldrich) for 30 minutes at 37˚C. Bone marrow from the right femur was extracted using a 22 gauge syringe. Remaining tissues were subjected to mechanical disruption using a blunt end syringe. Tissues were filtered in 70 μM cell strainers. Between 3.0 × 105 and 1.0 × 106 cells were stained 1:11 with PE-CD20 (BD Biosciences). Flow cytometry assessed CD20 species cross-reactivity in the tissues from a control NSG mouse. H&E staining and SP analyses were performed as previously described.
2.8. Statistical Analyses
All data are presented as the mean ± SEM. Linear regression (Sigma Plot v 9.0; San Jose, CA) produced the line of best fit for CAFC assays. Graphs were made in Sigma Plot v 9.0 or GraphPad Prism v4.02 (GraphPad Software Inc. La Jolla, CA). Student’s t test was utilized for all remaining analyses. A P value ≤ 0.05 was considered significant.
3. RESULTS
3.1. SP and CD133+/CD44+ Cells Are Present in Granta
Initially, we sought to identify whether Granta cells possessed populations of cells, identified through use of Hoescht exclusion, (e.g. the SP) or expression of the hematopoietic stem cell marker CD133 [21] in conjunction with CD44, widely used markers for identification of TICs ADDIN EN.CITE ADDIN EN.CITE.DATA [14-16,18,24] . The SP phenotype was observed in Granta and was represented at an average of 6.4% ± 1.8% (range: 0% - 35.2%) (Figure 1(Aa)). Validation of the SP was confirmed through the incorporation of verapamil, an ABCG2 transport blocker (Figure 1(Ab)). Granta cells additionally possessed a small population of CD133+/CD44+ cells which represented 0.06% ± 0.03% (range: 0% - 0.39%) of the gated population. Examples of the unstained Granta control (Figure 1(Ac)) and Granta CD133+/CD44+ cells (Figure 1(Ad)) are shown. Overall, these analyses suggest that Granta possess subpopulations of cells bearing stem cell-like properties and points to the SP as being a more robust marker of putative TICs.
3.2. SP Cells Are Enriched for Cobblestone Area-Forming Cells
The cobblestone area forming cell (CAFC) assay is considered one of the best in-vitro techniques for the identification of stem cells [44,45] . The presence of stem and progenitor cells is visualized when a stem or progenitor cell migrates below a stromal cell layer and selfrenews to form a colony of cells, which resemble a cobblestone in appearance ADDIN EN.CITE ADDIN EN.CITE.DATA [44,45] . Utilizing this technique, limiting dilutions of Granta SP and non-SP cells and CD133+/CD44+ and CD133−/CD44− cells were plated onto a semi-confluent monolayer of MS5 cells, chosen for its known role in supporting long term maintenance of HSCs in-vitro [45,46] and evaluated at weekly intervals for the presence of cobblestones. At the end of the 5 week analysis, the CAFC frequencies of Granta CD133+/ CD44+ and CD133−/CD44− cells were not significantly different: 1/415 and 1/755 cells (P = 0.09) (Table 1), respectively. Granta SP cells, however, were enriched for stem cell-like cells, having a CAFC frequency of 1/970 cells while non-SP cells had a CAFC frequency of 1/4350 cells (P = 0.02) (Table 1). It is important to note that cell dilutions corresponding to 33,300, 11,100 and 3700 cells/well were not included in SP and non-SP CAFC assays as they yielded non-responsive wells from non-SP cells, resulting in a shifted CAFC curve which could not be evaluated. It is possible that these high cell numbers may have overwhelmed the conditions of the assay. Figures 1(Ba) and 1(Bb) illustrate the presence and absence of cobblestones from Granta SP and non-SP cells, respectively.
Interestingly, 1000 SP cells were observed to form spheres in the tumorsphere assay, a technique utilized to detect self-renewal of both stem cell populations and TICs [47-50] . These spheres were detected as early as 2 weeks in culture (Figure 1(C)). Lower limits of SP cells (1, 10 and 100) and all tested dilutions of non-SP cells (1, 10, 100 and 1000) were not observed to form tumorpheres. An example illustrating the absence of spheres and identifiable cells is shown for a culture of 1000 non-SP cells (Figure 1(C)). Together, the CAFC and tumorsphere results suggest that Granta SP cells are enriched for self-renewing TICs at a dilution which corresponds to a frequency of ~1000 cells.
3.3. Matrigel Enhances the CAFC Frequencies of SP and Non-SP Cells
Since ECM components have been shown to maintain certain stem cell populations in an undifferentiated state [35,51,52] , we hypothesized that the addition of Matrigel would enhance the number of self-renewing TICs and thus the CAFC frequencies of putative SP and CD133+/ CD44+ TICs. Limiting dilutions of Granta SP and non-SP cells (33,300 cells/well to 1 cell/well) and CD133+/ CD44+ and CD133−/CD44− cells (407 cells/ well to 1 cell/well) were plated onto MS5 cells and the cultures were overlaid with a final concentration of 275 μg/ml of Matrigel. Interestingly, the addition of Matrigel to Granta SP and non-SP/MS5 co-cultures not only significantly increased the CAFC frequencies of both populations, but resulted in intersecting CAFC frequencies of 1/175 for both SP and non-SP cells (P > 0.05) (Table 1) (Figure 1(D)). Surprisingly, the addition of Matrigel to CD133+/CD44+ and CD133−/CD44−/MS5 co-cultures significantly decreased the CAFC frequencies to 1/4850

Figure 1. Granta possess populations of cells with stem cell-like properties. (Aa) The SP phenotype was identified in Granta cells and occurred at a frequency of 6.4% ± 1.8% (range: 0% - 35.2%) of the population. (Ab) Verapamil confirmed the presence of SP cells. (Ac) Quadrants for the analysis of CD133/CD44-expressing cells were defined in unstained Granta cells. (Ad) CD133+/CD44+ cells, indicated by the oval in Q2, were present in Granta cells and occurred at a frequency of 0.06% ± 0.03% (range: 0% - 0.39%). (B) Images illustrating the presence of phase-dark cobblestones (indicated by the black arrows) from a 2 week culture of SP/MS5 cells and absence of cobblestones from non-SP/MS5 cells maintained in culture for 5 weeks. Images were captured at ×100 magnification. (C) The presence of a tumorsphere was observed from 1000 SP cells maintained in culture for 2 weeks. Tumorsphere formation was not observed from 1000 non-SP cells maintained in culture for 6 weeks. Images were captured at ×200 magnification. (D) The CAFC frequencies of Granta SP and non-SP cells overlaid with 275 μg/ml Matrigel is demarcated by the line representing 37% non-responding wells and was determined to be 1/175 cells for both cell populations. 95% confidence intervals are indicated by the error bars. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
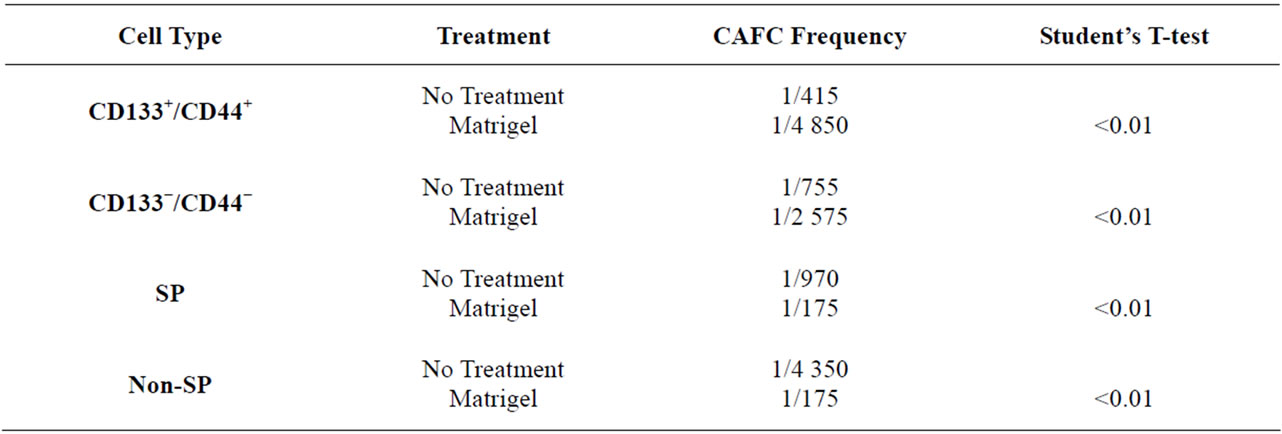
Table 1. Granta TIC and non-TIC CAFC frequencies.
and 1/2575, respectively, in comparison to co-cultures without Matrigel (P < 0.01) (Table 1). Together, these data not only demonstrate that Matrigel has opposing effects on the CAFC frequencies of SP and non-SP cells and CD133+/CD44+ and CD133−/CD44− cells, but additionally demonstrates that micro-environmental components, rather than phenotype differences, may be a more important factor in promoting stem cell-like behaviors of Granta SP and non-SP cells.
3.4. Matrigel Expedites Tumor Formation of Granta SP and Non-SP Cells
We sought to determine whether Matrigel enhanced the tumorigenic properties of Granta SP and CD133+/ CD44+ cells, a property reported by others ADDIN EN.CITE ADDIN EN.CITE.DATA [38-40] . Palpable tumors from Matrigel injections of 10, 100 and 1000 SP and non-SP cells were evident in 100% of tested cases and arose within an average time of 34 ± 0.9 days following cell injection. Examples of tumors derived from the injections of 10 Matrigel-mixed SP and 10 Matrigel-mixed non-SP cells are shown (Figures 2(Aa) and 2(Ab)). Remarkably, a tumor arose from the injection of a single Matrigel-mixed SP cell at 70 days post injection (Figures 2(Ac) and 2(B)). Tumors did not; however, arise from single cell injections of Matrigel-mixed nonSP cells at 160 days post-injection. Overall, significant differences in tumor volumes between Matrigel-mixed SP and non-SP cells were not observed (Figure 2(C)). In the absence of Matrigel, tumors arose in two animals injected with 1000 and 100 vehicle-mixed SP cells; however, the tumor volumes were 2.4× and 1.4× less than their counterpart Matrigel-mixed tumors, respectively. Tumors did not develop from vehicle-injected non-SP cells nor did they form from control Matrigel or vehicle injections, demonstrating that these inoculums were not tumorigenic alone. Figure 2(D) depicts the frequency of tumor formation in Matrigel and vehicle-mixed SP and non-SP cells. CD133+/CD44+ and CD133−/CD44− cells were also evaluated for their tumor-forming capabilities. Both cell populations were injected into NSG mice in the presence and absence of Matrigel or hyaluronan, chosen as CD44 preferentially interacts with hyaluronan [22]. Table 2 provides a summary of findings from Matrigel and vehicle-mixed injections of SP and non-SP cells. Surprisingly, tumors did not arise by 160 days post-injection from Matrigel and hyaluronan-mixed CD133+/ CD44+ and CD133−/CD44− cells (data not shown). Similarly, vehicle-mixed injections of CD133+/CD44+ and CD133−/CD44− cells did not give rise to tumors at 160 days post-injection. Taken together, these results suggest that phenotypic differences in tumor formation from SP and non-SP cells may be irrelevant in the presence of Matrigel but may be necessary for tumor establishment and growth in the absence of Matrigel. CD133+/CD44+ and CD133−/CD44− cells are not capable of tumor formation under the tested conditions.
In order to validate that tumors obtained from SP and non-SP cells were from Granta cells, flow cytometry was employed to quantify cell surface CD20. Flow cytometry detected enriched levels of CD20 positive cells from SP and non-SP-derived tumors (Figures 2(Ea) and (Eb)). SP and non-SP-derived tumors had an average tumor CD20 expression of 82% ± 6%. Together, these data confirm that tumor specimens were derived from human MCL cells.
3.5. SP Fractions Were Identified in SP and Non-SP Derived Tumors
Flow cytometry was utilized to assess the presence of the SP in Matrigel-mixed SP and non-SP-derived tumors. SP fractions were found in tumors arising from injections of both SP (Figure 2(Ec)) and non-SP cells (Figure 2(Ed)). The mean SP expression was the same for tumors derived from Matrigel-mixed SP and non-SP cells: 1.1% ± 0.5% and 1.1% ± 0.3%, respectively (P = 0.4) and was 1.4% ± 0.7% in vehicle-mixed SP cells (P > 0.05: Matrigel-mixed non-SP and SP-derived tumors). Together, these data strongly suggest that the SP phenotype is malleable and may be modulated by microenvironmental factors.
3.6. SP and Non-SP Derived Tumors Cells Are Capable of Spread to Distant Organs
In order to evaluate the metastatic capabilities of SP and non-SP cells, several murine organs including the bone marrow, spleen, lung, small intestine, liver and kidney were harvested and assessed for the presence of tumor cells using H&E staining and flow cytometry. H&E staining did not reveal the presence of distinct tumor nodules in any of the tissues examined (data not shown). Given the limitations of H&E/immunostaining to small regions of tissue sections, we elected to utilize flow cytometry on intact, prepared tissues to investigate whether human CD20 cells, indicative of metastasizing Granta cells, were present in the aforementioned organs. Human CD20-positive cells were detected by flow cytometry in all evaluated tissues, with the greatest level of detection observed in the small intestine (Figure 3), a common metastatic site for non-Hodgkin’s lymphomas
[53]. Taken together, these results indicate that while identifiable nodular tumors were not detected in potential metastatic sites, human CD20 expression was observed and quantified in murine tissues indicating that Granta cells from SP and non-SP injections most likely migrated to these distant sites.
3.7. IL-8 Levels Are Significantly Elevated in Granta/MS5 Co-Cultures
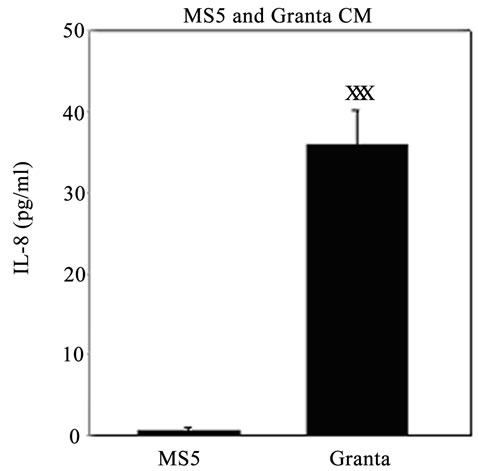
We examined IL-8 levels, chosen due to its implications in tumor invasion and metastasis [54], in the conditioned media (CM) of Granta and MS5 cells after 3 and 6 days in culture. IL-8 was also evaluated in Granta MS5 co-cultures at several days along the culture period and was further investigated in co-cultures of Granta TIC and non-TIC populations with MS5 cells. IL-8 was found to be secreted at negligible levels from MS5 cells after 3 (Figure 4(A)) and 6 days (data not shown) in culture and highly secreted from Granta cells after 3 (Figure 4(A))
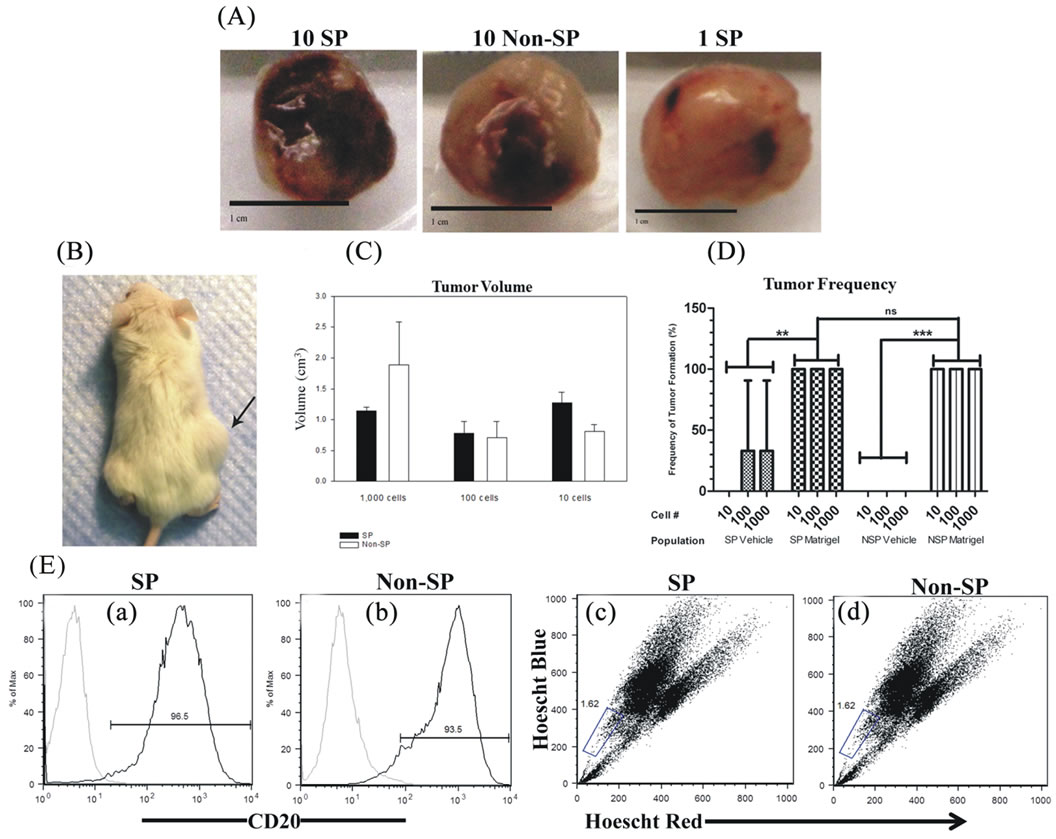
Figure 2. Tumor specimens and characteristics of Matrigel-mixed SP and non-SP cells. (A) Tumor specimens obtained from Matrigel-mixed 10 SP cells, 10 non-SP cells and 1 SP cell. Scale bars represent 1 cm. (B) NSG mouse harboring a tumor from the injection of a single Matrigel-mixed SP cell. (C) Tumor volumes were not statistically significantly different between SP and non-SP-derived tumors at corresponding dilutions. (D) The frequency of tumor formation was the same for all tested dilutions of Matrigel-mixed SP and non-SP cells and occurred 100% of the time. Tumor formation of vehicle-mixed SP cells occurred 33% of the time and did not result from vehicle-mixed non-SP cells. (E) Flow cytometry demonstrated the presence of cell surface CD20 expression ((a) and (b)) and SPs ((c) and (d)) in tumors arising from 10 SP and 10 non-SP cells.
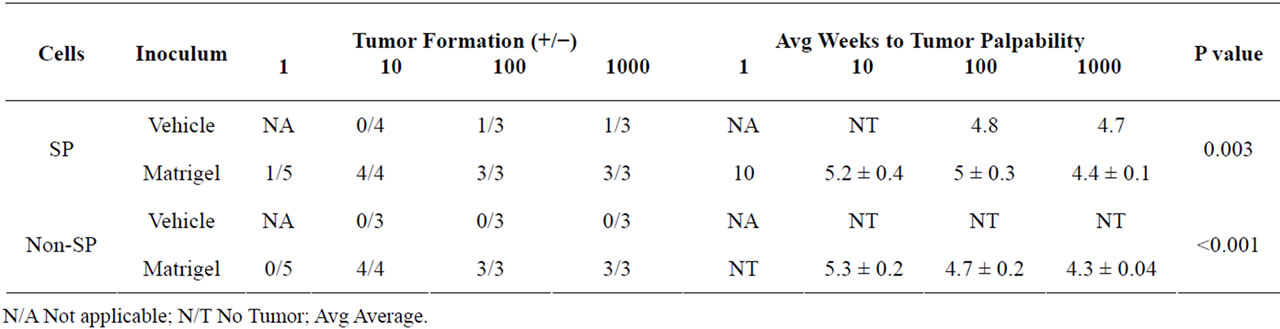
Table 2. Tumor Formation from Granta SP and non-SP cells.
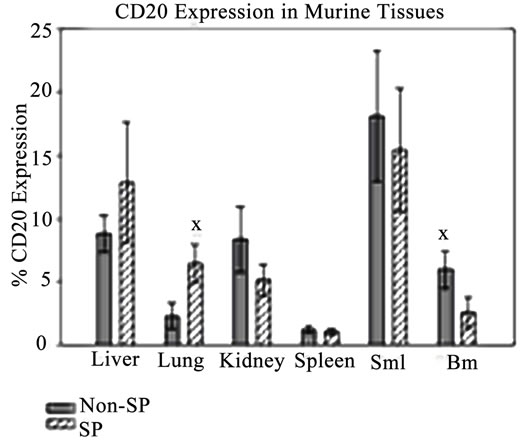
Figure 3. Granta cells spread to distant murine tissues. CD20-positive cells were detected in all murine tissues from SP and non-SP-injected NSG mice. All values are averages and presented as a percentage of the total CD20-positive cells minus control. SMI: Small Intestine. BM: Bone Marrow. XP ≤ 0.05.
and 6 days (data not shown) in culture. Interestingly, it was noted that the greatest level of IL-8 secretion oc curred in a time dependent manner in Granta/MS5 cocultures (Figure 4(B)). Surprisingly, it was found that IL-8 secretion was significantly higher in SP/MS5 cocultures than in non-SP/MS5 co-cultures where IL-8 was not detectable (Figure 4(C)). Regarding CD133+/ CD44+ and CD133−/CD44− cells co-cultured with MS5, IL-8 was not significantly different (Figure 4(C)). Together, these studies suggest that MS5 cells alone do not secrete appreciable levels of IL-8, but significantly contribute to IL-8 levels when cultured in direct contact with Granta cells. Moreover, these results further suggest that IL-8 is predominantly secreted from co-cultures of SP/MS5, but not appreciably from co-cultures of MS5 with non-SP, CD133+/CD44+ and CD133−/CD44− cells.
4. DISCUSSION
Here, we report that the Granta MCL cell line possesses subpopulations of cells expressing stem cell functions and markers. For the purpose of more closely mimicking the in-vivo environment, we evaluated the stem cell-like and tumor-forming capabilities of Granta SP and CD133+/CD44+ cells in the presence of microenvironmental components found in Matrigel. While numerous reports have documented the existence of rare sub-populations of TICs in a variety of carcinomas, the majority of these studies have failed to consider the role of the microenvironment in modulating the stem cell-like and tumor-forming properties of such cells. Thus, the question remains as to whether properties associated with TICs are mediated through mechanisms solely related to intrinsic or extrinsic factors, or some combination of both.
Our results demonstrate that when assessed for the presence of stem and progenitor cells, evaluated using the CAFC assay, Granta SP cells were highly enriched for stem cell-like cells. The ability of Granta cell populations to form cobblestones in the CAFC assay is in contrast to observations from Kurtova et al. (2009) who reported that Granta cells were incapable of cobblestone formation. It’s possible that this discrepancy may have arisen due to the type of fibroblasts used in these assays. For instance, it has been documented that certain murine and human fibroblast cell lines are incapable of supporting the long-term maintenance of human HSCs in-vitro [46, 55,56] . Since the MS5 cell line has been shown to reliably support the quantification and long term maintenance of HSCs in-vitro [45,46] , we employed this cell line for analyses of stem and progenitor cell frequencies and believe it to be a valid cell line for this purpose. In this manner, our data not only demonstrates that the SP isolation technique better enriches for cells possessing stem cell-like properties in Granta, but warrants caution in the choice of fibroblast cell lines for the purpose of identifying and/or supporting putative stem cell populations.
Further investigating the in-vitro stem cell-like properties of putative Granta TIC cells, we observed that Matrigel had discordant effects on SP and CD133+/ CD44+ cells, enhancing and depressing the CAFC frequencies, respectively. Interestingly, Matrigel endowed both SP and non-SP cells with a concordantly enriched
 (a)
(a)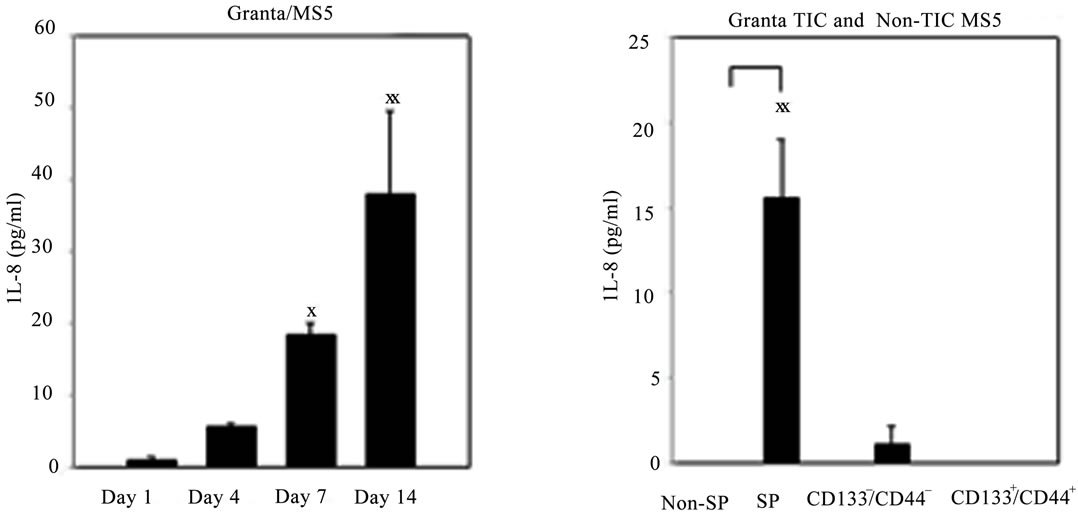 (b)
(b)
Figure 4. IL-8 secretion was significantly elevated in Granta and co-cultures of Granta and Granta SP with MS5. Secreted IL-8 was significantly elevated in (A) Granta CM (B) Granta/MS5 CM, with increasing IL-8 observed during the culture period. All comparisons were made against baseline (day 1) IL-8 secretion. (C) IL-8 secretion was significantly elevated in SP/MS5 co-cultures and was negligibly detected in non-SP/MS5 co-cultures. Detectable IL-8 was observed from CD133−/CD44− /MS5 co-cultures, but was negligibly secreted from CD133+/CD44+ /MS5 co-cultures . XP ≤ 0.05; XXP ≤ 0.01; XXXP ≤ 0.001.
proportion of cells exhibiting a stem cell phenotype. A similar observation was made by Cao et al. [57] who reported that non-SP cells in nasopharyngeal carcinomas could be induced to express the SP phenotype when cultured in the conditioned media of macrophage-like cells, a cell type comprising the microenvironment. Our results suggest that the microenvironment plays an important role in modulating a stem cell-like phenotype of Granta SP and non-SP cells. Matrigel did not, however, enrich for stem cell-like cells in the CD133+/CD44+ population. It’s possible that Matrigel may not have provided the requisite ECM constituents for facilitating a stem cell phenotype. For instance, the main ligand for CD44 is HA [22], a polysaccharide ECM component reported to be enriched in the tumor microenvironment [58]. Since Matrigel does not contain HA, it’s possible that the Matrigel niche does not provide the cues necessary to support the expression of a stem cell-like phenotype from CD133+/CD44+ cells. However, it should be noted that CD133+/CD44+ may not enrich for a population of MCL TICs as evaluated in the CAFC assay.
Recent reports have documented the tumor-forming capabilities of Matrigel-injected TICs ADDIN EN.CITE ADDIN EN.CITE.DATA [38-40] . Utilizing similar techniques, we demonstrated that both Matrigelmixed SP and non-SP cells were tumorigenic in NSG mice at dilutions of 10 or more cells. Indeed, the microenvironment-mediated control of tumorigenesis is a widely acknowledged phenomenon for unselected tumor cells [59,60] and was reported for thyroid [61] and mesenchymal [62] TICs. In the latter studies, Matrigelmixed SP and non-SP cells were reported to form tumors in immunocompromised mice [61,62] . While these authors did not attribute their observations to a microenvironment-mediated effect, it is likely, given our data, that the ECM factors found in Matrigel is a more important determinant for influencing tumor formation than SP and non-SP cell phenotype differences. In the absence of Matrigel, intrinsic differences may be important as tumor formation was documented in two instances from vehicle-injected SP cells. Together, these observations would suggest that both intrinsic and extrinsic properties may govern tumor-forming capabilities of Granta SP and non-SP cells. A remarkable observation was the finding that a tumor arose from 1 Matrigel-injected SP cell. While tumors arose in 1/5 cases from the injections of single Matrigel-mixed SP cells, our results are consistent with that of Quintana et al [60], who observed a ~20% tumor formation frequency from the injection of single Matrigel-mixed melanoma cells. Regarding the absence of tumors from single cell injections of Matrigel-mixed non-SP cells, we speculate that intrinsic mechanisms may provide a tumorigenic advantage to SP cells. This potentially suggests that interactions and communications between tumor cells may be an important means by which non-SP cells form tumors. In future studies, it will be necessary to address the limiting number of non-SP cells which can reliably form tumors in NSG mice.
The presence of a SP denotes a population of cells capable of self-renewal ADDIN EN.CITE ADDIN EN.CITE.DATA [27] . We discovered that SP cells were found in tumors derived from Matrigel-mixed SP and non-SP injections. The presence of the SP in Matrigel-mixed non-SP tumors was unexpected and may relate to the transformation ability of Matrigel as previously demonstrated in thyroid cancer cells [61]. This assessment is reasonable when taking into consideration the in-vitro results whereby Matrigel significantly enhanced the CAFC frequency of non-SP cells. Together, these data strongly suggest that the SP phenotype is malleable and may be modulated by micro-environmental factors. It will be necessary to determine whether this phenomenon is due to the microenvironment-mediated release of cytokines/chemokines or the acquisition of epigenetic changes facilitating cellular transformation of the target cells.
Metastasis is the greatest determinant of cancer morbidity. Although solid tumor nodules were not detected, diffuse infiltrates of MCL cells were present in all murine tissues examined. While it is unknown as to what caused the discrepancy between the lack of nodular tumor formation in spite of the cellular presence of Granta MCL cells, we hypothesize that cellular to microenvironment interactions likely play a role. In other words, Granta SP and non-SP cells were incapable of forming the appropriate cellular to cellular or non-cellular associations due to host microenvironment-mediated incompatibilities, a postulate set forth by Kelly et al. [63]. In this manner, tumor development could not take place despite the presence of MCL cells. It will be important to address whether an orthotopic introduction of a carefully selected human-derived stromal cell line, with Granta SP and non-SP cells may overcome potential species incompatibility constraints.
Neither CD133+/CD44+ nor CD133−/CD44− cells comixed with Matrigel or hyaluronan formed tumors. It is possible that either the cell populations themselves and/ or the microenvironmental niches were not favorable for growth of these cell populations into tumors. Since hyaluronan principally mediates cellular interactions with CD44 [22], it is puzzling as to why tumors did not develop from CD133+/CD44+ cells. Given the absence of sequestered growth factors in the hyaluronan matrix, it is possible that hyaluronan in combination with growth factors is necessary for tumor formation from CD133+/ CD44+. In future studies, it will be important to evaluate how a hyaluronan-rich matrix containing growth factors influence tumor development from Granta CD133+/ CD44+ cells.
In conclusion, our results demonstrate that the ECM constituents found in Matrigel are pivotal for stem celllike and tumor-forming properties of Granta SP and nonSP cells. Additionally, these data point to potential niche specifications regarding the manifestation of tumor formation and stem cell-like properties associated with given subpopulations of tumor cells. Importantly, these data warrant caution in assigning tumor-forming capabilities exclusively to rare TICs.
5. ACKNOWLEDGEMENTS
We gratefully thank Dr. Shantaram Joshi UNMC, Omaha, NE for provision of Granta 519 cells and Dr. John D. Jackson Wake Forest Institute of Regenerative Medicine, Winston Salem, NC for provision of the MS5 cell line. We also thank Dr. Anathbandu Chaudhuri UNMC, Omaha, NE for assistance with animal injections, Valerie Shostrum for assistance with statistical analyses, and Sue Brusnahan UNMC, Omaha, NE and Dr. Sharon Gerecht Johns Hopkins University, Baltimore, MD for critical reading of the manuscript. This work was supported in part by: UNMC Eppley Cancer Center Support Grant P30CA036727; NIA Grant AG024912; Nebraska Tobacco Settlement Biomedical Research Development Fund.
REFERENCES
- Campo, E., Raffeld, M. and Jaffe, E.S. (1999) Mantle-cell lymphoma. Seminars in Hematology, 36, 115-127.
- Zucca, E., et al. (1995) Patterns of survival in mantle cell lymphoma. Annals of Oncology, 6, 257-262.
- Pittaluga, S., et al. (1996) “Small” B-cell non-Hodgkin’s lymphomas with splenomegaly at presentation are either mantle cell lymphoma or marginal zone cell lymphoma. A study based on histology, cytology, immunohistochemistry, and cytogenetic analysis. American Journal of Surgical Pathology, 20, 211-223. doi:10.1097/00000478-199602000-00010
- Cohen, P.L., Kurtin, P.J., Donovan, K.A. and Hanson, C.A. (1998) Bone marrow and peripheral blood involvement in mantle cell lymphoma. British Journal of Haematology, 101, 302-310. doi:10.1046/j.1365-2141.1998.00684.x
- Marts, B.S., Longo, W.E., Maluf, H. and Vernava 3rd, A.M. (1994) Intermediate lymphocytic lymphoma of the small intestine. Mantle cell lymphoma. Journal of Clinical Gastroenterology, 18, 161-162. doi:10.1097/00004836-199403000-00018
- Howard, O.M., et al. (2002) Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: Molecular complete responses are not predictive of progression-free survival. Journal of Clinical Oncology, 20, 1288-1294. doi:10.1200/JCO.20.5.1288
- Nickenig, C., et al. (2006) Combined cyclophosphamide, vincristine, doxorubicin, and prednisone (CHOP) improves response rates but not survival and has lower hematologic toxicity compared with combined mitoxantrone, chlorambucil, and prednisone (MCP) in follicular and mantle cell lymphomas: Results of a prospective randomized trial of the German Low-Grade Lymphoma Study Group. Cancer, 107, 1014-1022. doi:10.1002/cncr.22093
- Weisenburger, D.D., et al. (2000) Mantle cell lymphoma. A clinicopathologic study of 68 cases from the Nebraska Lymphoma Study Group. American Journal of Hematology, 64, 190-196. doi:10.1002/1096-8652(200007)64:3<190::AID-AJH9>3.0.CO;2-B
- Dreyling, M., et al. (2005) Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: Results of a prospective randomized trial of the European MCL Network. Blood, 105, 2677-2684. doi:10.1182/blood-2004-10-3883
- Freedman, A.S., et al. (1998) High-dose chemoradiotherapy and anti-B-cell monoclonal antibody-purged autologous bone marrow transplantation in mantle-cell lymphoma: No evidence for long-term remission. Journal of Clinical Oncology, 16, 13-18.
- Ward, R.J. and Dirks, P.B. (2007) Cancer stem cells: At the headwaters of tumor development. Annual Review of Pathology, 2, 175-189. doi:10.1146/annurev.pathol.2.010506.091847
- Bonnet, D. and Dick, J.E. (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine, 3, 730-737. doi:10.1038/nm0797-730
- Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J. and Clarke, M.F. (2003) Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of USA, 100, 3983-3988. doi:10.1073/pnas.0530291100
- Patrawala, L., et al. (2006) Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene, 25, 1696-1708. doi:10.1038/sj.onc.1209327
- Vander Griend, D.J., Karthaus, W.L., Dalrymple, S., Meeker, A., DeMarzo, A.M. and Isaacs, J.T. (2008) The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Research, 68, 9703-9711. doi:10.1158/0008-5472.CAN-08-3084
- Du, L., et al. (2008) CD44 is of functional importance for colorectal cancer stem cells. Clinical Cancer Research, 14, 6751-6760. doi:10.1158/1078-0432.CCR-08-1034
- Chiba, T., et al. (2006) Side population purified from hepatocellular carcinoma cells harbors cancer stem celllike properties. Hepatology, 44, 240-251. doi:10.1002/hep.21227
- Yin, S., et al. (2007) CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. International Journal of Cancer, 120, 1444-1450.
- Vega, F., et al. (2010) Side population of a murine mantle cell lymphoma model contains tumour-initiating cells responsible for lymphoma maintenance and dissemination. Journal of Cellular and Molecular Medicine, 14, 1532-1545. doi:10.1111/j.1582-4934.2009.00865.x
- Chen, Z., et al. (2010) Prospective isolation of clonogenic mantle cell lymphoma-initiating cells. Stem Cell Research, 5, 212-225.
- Yin, A.H., et al. (1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood, 90, 5002-5012.
- Aruffo, A., Stamenkovic, I., Melnick, M., Underhill, C.B. and Seed, B. (1990) CD44 is the principal cell surface receptor for hyaluronate. Cell, 61, 1303-1313. doi:10.1016/0092-8674(90)90694-A
- Denhardt, D.T., Giachelli, C.M. and Rittling, S.R. (2001) Role of osteopontin in cellular signaling and toxicant injury. Annual Review of Pharmacology and Toxicology, 41, 723-749. doi:10.1146/annurev.pharmtox.41.1.723
- Haraguchi, N., et al. (2008) CD133+CD44+ population efficiently enriches colon cancer initiating cells. Annals of Surgical Oncology, 15, 2927-2933. doi:10.1245/s10434-008-0074-0
- Scharenberg, C.W., Harkey, M.A. and Torok-Storb, B. (2002) The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood, 99, 507-512. doi:10.1182/blood.V99.2.507
- Goodell, M.A., Brose, K., Paradis, G., Conner, A.S. and Mulligan, R.C. (1996) Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. Journal of Experimental Medicine, 183, 1797-1806. doi:10.1084/jem.183.4.1797
- Mao, Q. and Unadkat, J.D. (2005) Role of the breast cancer resistance protein (ABCG2) in drug transport. American Association of Pharmaceutical Scientists Journal, 7, E118-E133.
- Moore, K.A. and Lemischka, I.R. (2006) Stem cells and their niches. Science, 311, 1880-1885. doi:10.1126/science.1110542
- Morrison, S.J. and Spradling, A.C. (2008) Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell, 132, 598-611. doi:10.1016/j.cell.2008.01.038
- Vermeulen, L., et al. (2010) Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature Cell Biology, 12, 468-476. doi:10.1038/ncb2048
- Calabrese, C., et al. (2007) A perivascular niche for brain tumor stem cells. Cancer Cell, 11, 69-82. doi:10.1016/j.ccr.2006.11.020
- Calvo, K.R., et al. (2008) IL-4 protein expression and basal activation of Erk in vivo in follicular lymphoma. Blood, 112, 3818-3826. doi:10.1182/blood-2008-02-138933
- Chen, X.D. (2010) Extracellular matrix provides an optimal niche for the maintenance and propagation of mesenchymal stem cells. Birth Defects Research Part C: Embryo Today, 90, 45-54. doi:10.1002/bdrc.20171
- Chen, X.D., Dusevich, V., Feng, J.Q., Manolagas, S.C. and Jilka, R.L. (2007) Extracellular matrix made by bone marrow cells facilitates expansion of marrow-derived mesenchymal progenitor cells and prevents their differentiation into osteoblasts. Journal of Bone Mineral Research, 22, 1943-1956. doi:10.1359/jbmr.070725
- Kanatsu-Shinohara, M., et al. (2005) Long-term culture of mouse male germline stem cells under serum-or feeder-free conditions. Biology of Reproduction, 72, 985-991. doi:10.1095/biolreprod.104.036400
- Salasznyk, R.M., Williams, W.A., Boskey, A., Batorsky, A. and Plopper, G.E. (2004) Adhesion to vitronectin and collagen I promotes osteogenic differentiation of human mesenchymal stem cells. Journal of Biomedicine and Biotechnology, 2004, 24-34.
- Salasznyk, R.M., Williams, W.A., Boskey, A., Batorsky, A. and Plopper, G.E. (2004) Adhesion to vitronectin and collagen I promotes osteogenic differentiation of human mesenchymal stem cells. Journal of Biomedicine and Biotechnology, 1, 24-34. doi:10.1155/S1110724304306017
- Stabenfeldt, S.E., Munglani, G., Garcia, A.J. and LaPlaca, M.C. (2010) Biomimetic microenvironment modulates neural stem cell survival, migration, and differentiation. Tissue Engineering Part A, 16, 3747-3758.
- Charafe-Jauffret, E., et al. (2009) Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Research, 69, 1302-1313. doi:10.1158/0008-5472.CAN-08-2741
- Hansford, L.M., et al. (2007) Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Research, 67, 11234-11243. doi:10.1158/0008-5472.CAN-07-0718
- Vassilopoulos, A., Wang, R.H., Petrovas, C., Ambrozak, D., Koup, R. and Deng, C.X. (2008) Identification and characterization of cancer initiating cells from BRCA1 related mammary tumors using markers for normal mammary stem cells. International Journal of Biological Sciences, 4, 133-142. doi:10.7150/ijbs.4.133
- Itoh, K., et al. (1989) Reproducible establishment of hemopoietic supportive stromal cell lines from murine bone marrow. Experimental Hematology, 17, 145-153.
- Weekes, C.D., Kuszynski, C.A. and Sharp, J.G. (2001) VLA-4 mediated adhesion to bone marrow stromal cells confers chemoresistance to adherent lymphoma cells. Leukemia and Lymphoma, 40, 631-645. doi:10.3109/10428190109097661
- Taswell, C. (1981) Limiting dilution assays for the determination of immunocompetent cell frequencies. I. Data analysis. Journal of Immunology, 126, 1614-1619.
- Ploemacher, R.E., van der Sluijs, J.P., Voerman, J.S. and Brons, N.H. (1989) An in vitro limiting-dilution assay of long-term repopulating hematopoietic stem cells in the mouse. Blood, 74, 2755-2763.
- Robinson, S.N., Seina, S.M., Gohr, J.C., Kuszynski, C.A. and Sharp, J.G. (2005) Evidence for a qualitative hierarchy within the Hoechst-33342 “side population” (SP) of murine bone marrow cells. Bone Marrow Transplant, 35, 807-818. doi:10.1038/sj.bmt.1704881
- Issaad, C., Croisille, L., Katz, A., Vainchenker, W. and Coulombel, L. (1993) A murine stromal cell line allows the proliferation of very primitive human CD34++/CD38- progenitor cells in long-term cultures and semisolid assays. Blood, 81, 2916-2924.
- Eramo, A., et al. (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death and Differentiation, 15, 504-514. doi:10.1038/sj.cdd.4402283
- Hermann, P.C., et al. (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell, 1, 313-323. doi:10.1016/j.stem.2007.06.002
- Ponti, D., et al. (2005) Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Research, 65, 5506-5511. doi:10.1158/0008-5472.CAN-05-0626
- Ricci-Vitiani, L., et al. (2007) Identification and expansion of human colon-cancer-initiating cells. Nature, 445, 111-115. doi:10.1038/nature05384
- Adams, J.C. and Watt, F.M. (1989) Fibronectin inhibits the terminal differentiation of human keratinocytes. Nature, 340, 307-309. doi:10.1038/340307a0
- Xu, C., et al. (2001) Feeder-free growth of undifferentiated human embryonic stem cells. Nature Biotechnology, 19, 971-974. doi:10.1038/nbt1001-971
- Levine, M.S., Rubesin, S.E., Pantongrag-Brown, L., Buck, J.L. and Herlinger, H. (1997) Non-Hodgkin’s lymphoma of the gastrointestinal tract: Radiographic findings. American Journal of Roentgenology, 168, 165-172. doi:10.2214/ajr.168.1.8976941
- Waugh, D.J. and Wilson, C. (2008) The interleukin-8 pathway in cancer. Clinical Cancer Research, 14, 6735-6741. doi:10.1158/1078-0432.CCR-07-4843
- Kodama, H., et al. (1992) In vitro proliferation of primitive hemopoietic stem cells supported by stromal cells: Evidence for the presence of a mechanism(s) other than that involving c-kit receptor and its ligand. Journal of Experimental Medicine, 176, 351-361. doi:10.1084/jem.176.2.351
- Torok-Storb, B., Iwata, M., Graf, L., Gianotti, J., Horton, H. and Byrne, M.C. (1999) Dissecting the marrow microenvironment. Annals of the New York Academy of Sciences, 872, 164-170. doi:10.1111/j.1749-6632.1999.tb08461.x
- Cao, J.X., et al. (2010) Pluripotency-associated genes in human nasopharyngeal carcinoma CNE-2 cells are reactivated by a unique epigenetic sub-microenvironment. BMC Cancer, 10, 68. doi:10.1186/1471-2407-10-68
- Itano, N., Zhuo, L. and Kimata, K. (2008) Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Science, 99, 1720-1725. doi:10.1111/j.1349-7006.2008.00885.x
- Mbeunkui, F. and Johann Jr., D.J. (2009) Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemotherapy and Pharmacology, 63, 571-582. doi:10.1007/s00280-008-0881-9
- Quintana, E., Shackleton, M., Sabel, M.S., Fullen, D.R., Johnson, T.M. and Morrison, S.J. (2008) Efficient tumour formation by single human melanoma cells. Nature, 456, 593-598. doi:10.1038/nature07567
- Mitsutake, N., et al. (2007) Characterization of side population in thyroid cancer cell lines: Cancer stem-like cells are enriched partly but not exclusively. Endocrinology, 148, 1797-1803. doi:10.1210/en.2006-1553
- Wu, C., et al. (2007) Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Research, 67, 8216-8222. doi:10.1158/0008-5472.CAN-07-0999
- Kelly, P.N., Dakic, A., Adams, J.M., Nutt, S.L. and Strasser, A. (2007) Tumor growth need not be driven by rare cancer stem cells. Science, 317, 337. doi:10.1126/science.1142596