Journal of Encapsulation and Adsorption Sciences
Vol.06 No.04(2016), Article ID:72751,14 pages
10.4236/jeas.2016.64011
Potentialities of the Poly(Aminoethyl Methacrylate) p(AMA) as Gelatin-Like Polymer in Complex Coacervation
Nicolas Esselin, Frédérique Portolan, Nouha Domloge, David Rees, Osama M. Musa, Jean-François Pilard
Ashland Specialties France, Sophia Antipolis, France

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: August 23, 2016; Accepted: December 11, 2016; Published: December 14, 2016
ABSTRACT
In order to overcome all encapsulation variations during a complex coacervation process, the replacement of gelatin cationic polymer has been performed using p(AMA). The synthesis of p(AMA) was realized through a random radical methodology. Under these conditions a polymer with 18,600 g/mol was found appropriate for optimal capsule yield and physico-chemical properties. Turbidity measurements performed during the coacervation reactions with different ratios of both CMC and p(AMA) allowed optimizing coacervation conditions. Coacervates characterizations particularly demonstrate the stability of the capsules exhibiting a break strength over 3 N/m2.
Keywords:
p(AMA), Complex Coacervation, Encapsulation, Polymerization

1. Introduction
Primary amine copolymers have been the target of tremendous activity demonstrating their involvement in numerous applications, including drug carriers [1] [2] [3] , functional colloidal particles for biomedical applications [4] [5] , preparations of sterically-stabilized polyaniline particles [6] and cationic latexes [7] , synthesis of polypeptide vesicles and micelles [8] , and also localized biomineralization of calcium carbonate and silica [9] [10] . This technology is used in food [11] , in medical, in home (laundry) [12] and in cosmetics [13] . Nevertheless, despite their use in various domains, such polymers have always been used through standard mixing process. To date, no development was reported using primary amine copolymers in a complex coacervation process.
Based on the separation of a colloidal system into two liquid phases, coacervation can occur either through a simple or a complex mode.
More particularly, complex coacervation occurs when two oppositely charged colloids interact with each other to form a dilute phase and a concentrated (coacervate) phase. In the complex coacervation process, there are phase separation processes based on the simultaneous desolvation of oppositely charged polyelectrolytes induced by media modifications. The protein and polysaccharide precipitation is obtained by changing pH and temperature or by the “salting-out” technique. The emergence of a dispersed phase of dense coacervates made of concentrated polyelectrolytes and a dilute equilibrium phase is dependent on pH, ionic strength and polyion concentrations. Although Tiebackx first reported on coacervation [14] , Bungenberg de Jong and Kruyt [15] coined the name complex coacervation in order to distinguish coacervation of a single polymer. They were the first to systematically investigate the complex coacervation phenomenon on the gum Arabic-gelatin system. This work was completed by Overbeek and Voorn [16] who developed the first theoretical model. Subsequent theoretical investigations were realized during the following 20 years by Veis et al. [17] [18] [19] [20] , Nakajima and Sato [21] , with Tainaka [22] [23] demonstrating the controversy of such a model. From an experimental and practical point of view, the gum arabic-gelatin system was extensively studied and applied in the famous patent on carbon-less copying paper [24] [25] . It is remarkable that the study of complex coacervates was rather scarce in view of their biological and practical relevance. In the last 10 years, interest in complex coacervation has considerably increased since it can be applied to various polyelectrolytes. The literature demonstrates that the phase separation process at the origin of the complex coacervation process mainly involves natural polymers.
More particularly, gelatin, a collagen hydrolysis product, is widely used. This natural copolymer is associated with different polysaccharides to neutralize its charge and thereby form a complex coacervate. Usually, gelatin is positively charged and the coacervation is induced by anionic colloids like pectin, alginate, Arabic gum or carboxymethylcellulose. However, the use of gelatin generates a safety problem due to the emergence of the prion diseases. Albumin, bovine serum albumin (BSA), and beta-lactoglobulin (BLG) were proposed as an alternative. They are commonly used as model proteins in the development of pharmaceutical, cosmetic, and food formulations but the bovine origin of the protein makes it still suspected. Moreover, all these biopolymers present a stability problem during the coacervation process and thus reproducibility in encapsulation cannot be optimized. In addition, since it is generally believed that electrostatic interactions dominate the association and the phase behaviour in the protein-polysaccharide complex, other important contributions from hydrogen bondings, covalent bondings and hydrophobic effects should not be overlooked.
In order to avoid any natural copolymers variability problems as well as reproducibility defects, our goal was to develop a synthetic gelatin alternative copolymer. Thus poly(aminoethyl methacrylate) was synthesized and its ability to form complex coacervates in the presence of polysaccharides was studied.
2. Materials and Methods
2.1. Materials
Methacryloyl chloride (>97%) (Aldrich, WI), ethanolamine hydrochloride (>98%) (Aldrich, WI), hydroquinone (99%) (Aldrich, WI), glutaraldehyde (99%) (Aldrich, WI) and D2O were all purchased from Aldrich and were used as received. Regenerated cellulose dialysis membrane (Spectra/Por 6, molecular weight cut-off 1000 Da) and 2-propanol (IPA) were purchased from Fisher and used as received. De-ionized water (pH 6) was used in all experiments. Sodium carboxymethylcellulose (CMC) was purchased from ASHLAND Inc.
The radical initiator used was 2,2’-Azobis(2-methylbutyronitrile) (VAZOTM 67) (Aldrich, WI). The solvent was Butanol (BuOH) (Aldrich, WI). N-Methyl-2-pyrrolidone (NMP) (Aldrich, WI) has been used as the internal standard for GC analysis. 20% w/w sodium hydroxide and 20% w/w hydrochloric acid solutions were prepared to adjust the pH of the solution during encapsulation. Mineral oil was used as Internal phase (IP) during encapsulation.
2.2. Synthesis of AMA Monomer
AMA monomer was synthesized using a previously reported protocol [26] . Briefly, 2-aminoethanol hydrochloride (65 g, 0.67 mol), methacryloyl chloride (100 mL, 0.96 mol), and hydroquinone (0.50 g) were added to a three-necked round-bottomed flask fitted with a condenser. The molten 2-aminoethanol hydrochloride salt was esterified in the melt at 90˚C - 95˚C under a nitrogen atmosphere for one hour and the reaction was maintained at 70˚C - 75˚C for a further two hours. The HCl gas evolved during this reaction was neutralized using an aqueous NaOH solution connected to the reaction vessel. The crude product was cooled to 40˚C, diluted with THF (50 mL) and precipitated into excess n-pentane (500 mL). The resulting creamy white precipitate was isolated by filtration, washed thoroughly with n-pentane (500 mL), and dried under vacuum. It was then recrystallized twice using a 7/3 (v/v) ethyl acetate/2-propanol mixture at 25˚C. The final product was dried under vacuum to produce a white solid of more than 99% purity as judged by 1H NMR (overall yield = 66%).
2.3. Synthesis of p(AMA) Homopolymer
75 g (0.454 mol) of AMA with 170 g of de-ionized water were introduced in the reactor at room temperature. The pH of the solution was adjusted to pH 4 by using acetic acid 99%. The solution was then heated to 83˚C and VAZOTM 67 radical initiator (1.25 g, 6.5 mmol) was added by 10 equal fractions (0.125 g, 0.65 mmol) over the first 9 hours. At the end of this period, the reaction mixture continued to be stirred at 83˚C for another hour. At this point, the residual level of AMA was reported by GC analysis to be 700 mg/kg. The final polymer remained in solution without any purification. The percentage of solid was 30% w/w. The melting point of the product was 110˚C.
2.4. Coacervates Formation
20 g of the p(AMA) were added to 330 g of de-ionized water and 2.5 g of CMC at 50˚C. The dissolution was helped by adding droplets of sodium hydroxide (0.3 g). Once the polymer was dissolved, a solution of acetic acid at 20% induces coacervation. An overhead laboratory stirrer equipped with a propeller and Russian turbine was installed in the vessel (Figure 1). 100 g of mineral oil was added as the internal phase of the microcapsules with the stirrer speed set at 75 - 90 rpm. The shear applied by the motor led to the formation of an oil-in-water emulsion, from which the oil droplets will eventually comprise the core of the microcapsules.
Coacervate droplets deposit onto the oil droplets and coalesce to form a continuous liquid coating. The temperature of the solution was cooled to 10˚C, 2.0 g of 50% glutaraldehyde solution (0.01 mol) were added, and the solution was left to stir for 6 hours to crosslink the capsule shells. Crosslinking renders the microcapsule shells stable, insoluble, and durable. The microcapsules were then filtered and washed using de-ionized water.
2.5. Characterization Techniques
2.5.1. 1H NMR Spectroscopy
All 1H NMR spectra were recorded in ether D2O, D2O/NaOD or D2O/DCl using a 400 MHz AV-400 Bruker spectrometer.
2.5.2. GC Analyses
1) Apparatus and conditions
A GC-7890A Gas Chromatograph (GC) equipped with a split/splitless injector, a 7693 auto-sampler, and a flame ionization detector (FID) from Agilent Technology Inc. were used in this study. The separation was carried out on a DB-624 capillary column (30 m × 0.53 mm i.d., 3.00 µm film thickness) from J and W Scientific. A 2.0 µL sample was injected in the split mode at a split ratio of 5:1 and with an injector temperature of 250˚C. GC oven temperature was initially set at 80˚C, then programmed to 130˚C at a rate of 5˚C/min and maintained for 40 minutes, finally raised to 250˚C at a rate of

Figure 1. Laboratory scale complex coacervation microencapsulation apparatus.
5˚C/min and maintained for 15 minutes. Ultra-high-purity helium was used as the carrier gas with a head pressure of 4.4 psi and constant flow rate of 33 mL/min measured into the detector at room temperature and uncorrected. The detector temperature was set at 250˚C.
2) Sample preparation
All of the samples were analyzed using N-methylpyrrolidone (NMP) as an internal standard.
A 1.0 g sample and 1.0 g of 0.5 g/L NMP were mixed in 10 mL butanol and centrifuged in a sealed centrifuge tube at 4200 rpm for 10 minutes at room temperature. The supernatant was then recovered for the analysis.
3) Calibration curve
1.0 g of various concentrations of monomers (2.0; 1.2; 0.4 g/L) and 1.0 g of 0.5 g/L NMP, respectively, were spiked in 10 mL butanol. The samples were prepared as described above and the calibration curve was constructed by plotting the peaks area ratio of monomers to NMP versus the ratio of their corresponding concentrations (1) [27] :
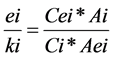 (1)
(1)
With Cei and Ci: mass concentrations of the internal standard (ei) and the solute (i) in solution
ki/ei: coefficient of the solute i relative to the internal standard ei.
Aei and Ai: peaks area of internal standard (Aei) and the solute (Ai).
Using the internal standard and the correlation factor, the residual level of each monomer is calculated according to Equation (2):
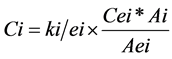 (2)
(2)
2.5.3. Zeta Potential
The electrical charge (Zeta potential) was determined against pH change at 25˚C ± 1˚C of individual biopolymer dispersions p(AMA) and CMC (0.01%, w/v) using a Zetasizer Nano (Malvern Instruments, Westborough, MA) capable of electrophoresis measurements. The calculations of electrophoretic mobility were automatically converted into zeta potential values dependent on the Smoluchowski model. The pH of the specific dispersion was acidified by the addition of HCl (0.25 M) and basified by adding NaOH (0.25 M). The final values were determined from two separately prepared samples, and the measurements were duplicated for each sample.
2.5.4. Aqueous GPC
The molecular weight and polydispersity of the p(AMA) homopolymer was determined by GPC at 30˚C; Shodex SB806 MHQ, SB-G columns connected in series to a Differential Refractive index detector. The polymer samples were prepared as 0.15% solution of 50/50 Methanol/Water, with 0.4 M LiNO3, 0.1 M Tris-2-chloroisopropyl phosphate, pH 9. The solution was mixed on a rotating wheel for several hours and then filtered (Millex-HV 0.45 µm) and injected into the GPC system for analysis. Molecular weight values were determined relative to PEO/PEG standards. The flow rate of the eluent was at 0.5 mL/min. The data was analyzed using Empower 2 software. The calibration was performed with a 4th order polynomial fit.
2.5.5. Fourier Transform Infrared Spectroscopy (FTIR)
Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) spectra were acquired on a Spectrum 100 spectrometer by ATR over a total range of (7800 - 370 cm−1) as the sum of 25 accumulations with 0.5 cm−1 resolution using a LiTaO3 (lithium tantalate) detector. The velocity of the moving mirror was 0.6329 cm/s. The ambient temperature and the relative humidity for the spectrometer were respectively set between 18˚C and 25˚C and less than 30% humidity. A background was performed and all the aliquots were analyzed without further polymer precipitation.
2.5.6. Turbidity Measurement
The solutions of p(AMA)-CMC were gently poured into the 1.0 cm cuvette and the turbidity was then measured with a spectrophotometer (Unocal, UV 2000, Japan) at 600 nm.
2.5.7. Microscopic Observation
A Nikon Eclipse E200 was used to observe the microcapsules with an objective ×10 - ×20 magnification.
3. Results and Discussion
3.1. Monomer
AMA is commercially available as a “technical” grade, but its purity is only around 90%. Thus we synthesized this monomer on a 75 g scale in high purity (99%) using a literature protocol [28] .
AMA monomer is relatively unstable in its free amine form (pKa ≈ 8.8), since it undergoes internal rearrangement to generate the thermodynamically more stable isomer, 2-hydroxyethyl methacrylamide. In contrast, AMA monomer remains stable in its hydrochloride salt form [29] .
3.2. Polymer
Synthesis of p(AMA)
Optimized reaction conditions for the homopolymerization of AMA via reversible addition-fragmentation chain transfer (RAFT) have been recently reported [30] . In this condition, the optimized solvent composition was determined to be an 80:20 IPA/H2O mixture at 50˚C, since this formulation gave short reaction times, high conversions, and relatively low final polydispersity [30] . The authors had molecular weight until 15,000 g/mol with a small value of polydispersity (1.7). In our study, we have privileged the random polymerization which facilitated the production of higher molecular weight above 50,000 g/mol polymers with higher polydispersity.
These characteristics were needed to observe the formation of complex coacervates. With a lower molecular weight, the interactions are just ionic bond and not complex formation. On the contrary, if the molecular weight is too high, the interactions lead to the formation of precipitates. The molecular weight control by a random polymerization seems a good opportunity [30] .
Therefore, we focused on a statistical polymerization and used VAZOTM 67 as initiator at pH 4.0. No other initiator is known to be soluble in water besides AIBN, classically used in this process. An AMA monomer conversion of 98.5% was achieved within 10 hours (Mw/Mn 4.7). A number-average molecular weight (Mn) of 18,600 was obtained by aqueous GPC.
Monomer conversions were calculated by 1H NMR spectroscopy by comparing the integrated intensity of the vinyl signals at δ 5.4 - 6.3 to that of the methacrylic backbone signals at δ 0.5 - 1.0 and by GC with an internal standard.
3.3. Degradation of p(AMA)
The degradation of AMA monomer and the p(AMA) synthetized by RAFT is known to occur in alkaline media as its primary amine groups become deprotonated. The effect of temperature, pH, and polymer concentration on the rate of p(AMA) degradation in dilute aqueous solution has been examined [31] . The degradation of p(AMA) synthetized by random polymerisation is also observed in basic condition. The p(AMA) degradation was observed by 1H NMR spectroscopy in basic condition, or with the temperature (Figure 2).
1H NMR studies indicated that p(AMA) degradation is complex. The analyses

Figure 2. 1H NMR spectra recorded for a 5.0 w/w% p(AMA) solution in D2O/NaOD (pH 8) aged at 50˚C for 168 hours.
confirm the presence of anionic carboxylate groups, which suggests that such elimination is due to ester saponification. The degradation of p(AMA) is expected to proceed by the following reactions as explained in the literature [28] (Intramolecular Amidation and the formation of hydroxyethyl methacrylate, Intramolecular amidation (II) and the intramolecular crosslinking, or an Intermolecular amidation and the intermolecular crosslinking).
The p(AMA) ester groups may undergo saponification by hydroxide anion, producing anionic carboxylate groups and 2-aminoethanol. Clearly this reaction is more likely to occur at a more alkaline pH.
These studies encouraged the control of the pH during the reaction of polymerization and the process of encapsulation.
3.4. Formation of Coacervates
Complex coacervates form when two oppositely charged polyelectrolytes interact with each other to form a complex coacervate. Coacervates differ from a precipitate because they are at a liquid phase due to their high water content and relatively weak electrostatic interaction. Hydrogen bondings also contribute to the formation of coacervates.
In the case of coacervation between gelatine and gum arabic, the interactions between the protonated amine moieties in gelatine and the anionic carboxylic moieties of gum arabic can be observed using FTIR. In our case, we have produced complex coacervates between CMC and p(AMA) (Figure 3).
The FTIR spectrum of p(AMA) (Figure 3) shows absorption bands related to C=O stretching at 1734 cm−1 (ester groups) and C-O bending at 1636 cm−1 (ester). The absorption band at 1387 cm−1 was attributable to the C-N stretching and the 3300cm−1 band is from N-H bending (primary amine) vibrations.
The IR spectrum of CMC shows a broad absorption band at 3410 cm−1 due to the stretching frequency of the -OH group. The band at 2920 cm−1 is due to C-H stretching vibration. The presence of a strong absorption band at 1603 cm−1 confirms the presence of COO−. The bands around 1424 and 1328 cm−1 are assigned to -CH2 scissoring and -OH vibration, respectively. The band at 1057 cm−1 is due to CHO-CH2 stretching.
The p(AMA)-CMC coacervates displayed a band at 1652 cm−1 and 1565 cm−1 attributed to -COO− and -NH3 groups stretching vibrations. The coacervation between the two biopolymers changed the FTIR spectrum in the carbonyl-amide region. The NH3 groups (band at 1594 cm−1) and asymmetric and -COO− stretching vibrations at 1644 and 1536 cm−1 respectively disappeared, indicating the electrostatic interaction between the carboxyl groups of CMC (COO−) and amine groups of p(AMA) (NH3).
Additionally, the spectrum of p(AMA)-CMC coacervates showed a broad band at around 3000 - 3600 cm−1, indicating enhanced hydrogen bonding compared to those of p(AMA) and CMC. This implied that hydrogen bonding was also involved in the interaction between p(AMA) and CMC.
The primary amine seems to be a good candidate to have interaction with the

Figure 3. FTIR of (a) coacervates of p(AMA)-CMC with a ratio of 1:0.4, (b) p(AMA), and (c) CMC.
carboxylic function of the CMC, and allows for a large degree of hydrogen bonding.
A study of the turbidity (Figure 4) depending on the pH at different ratios of CMC with p(AMA) was made to determine the best pH and best ratio to achieve the optimal coacervate yield. We can conclude at which ratio and which pH the capsules are formed:
We observed that at a ratio lower than 0.4 of p(AMA)-CMC, the yield of coacervation is not optimum and the pH of coacervation is around 6.5. Moreover, at this pH, cross-linking with the glutaraldéhyde is impossible. With a ratio of p(AMA)-CMC above 0.4, the interaction was too much important and the risk to form a precipitate was close. Above the ratio of 0.4, the pH of complex formation was around 4.5. We concluded that the optimum ratio was 0.4 and the optimum pH of complex formation was 4.5.
Zeta potential was measured (Figure 5) for different ratios of polymer. It appeared that the complex coacervates that formed were positively charged within the pH range where coacervation occurred. We observed that the pH of electrical equivalence PEE (Zeta Potential = 0) decreased as the ratio of CMC increased. In fact, the addition of more anionic polymer directly influenced the PEE of the complex.
At PEE pH, the interactions between the two polymers were too strong and formed a solid precipitate instead of coacervates. Thereafter, encapsulation was investigated at the optimum pH observed by turbidity, and not at the equivalence point indicated by Zeta-potential measurements. In the case of p(AMA)-CMC at a ratio of 1:0.4 (w:w) and at pH 4.5, the coacervates formed and were positively charged.

Figure 4. Evaluation of the turbidity depending on the ratio of polymers.

Figure 5. Evaluation of the zeta potential depending on the ratio of polymers.
3.5. Final Obtention of Coacervates
The optimal yield of coacervates was obtained at pH 4.5 with the p(AMA)-CMC (ratio 1:0.4, w:w) at 50˚C.
Crosslinking of the capsules was achieved through covalent bonding of the amines functionality of p(AMA) using glutaraldehyde, a di-functional aldehyde.
3.6. Formation of Microcapsules
The capsules were obtained after the formation of coacervates at pH 4.5 with the p(AMA)-CMC (ratio 1:0.4, w:w) at 50˚C.
After coacervats formation, the internal phase was added and the temperature was decreased to allow the formation of the capsule shell. The last step of encapsulation is the cross linking. This chemical reaction allowed the formation of covalent bonding by reaction of a primary amine of the p(AMA) on an aldehyde of the glutaraldéhyde.
3.7. Evaluation of Microcapsules
After all the evaluations in encapsulation, the capsules were observed by microscopy to show the size and the permeability of the wall (Figure 6).
The break strength of the microcapsules was evaluated after each encapsulation. In the studies of p(AMA)-CMC microcapsules, the break strength was around 3 N/m2. The required break strength is determined by the application and desired release properties. Concerning the food application, the break strength has to be higher than 6 N/m2; for the paintings application, the break strength has to be lower than 3 N/m2. The break strength could be increased by adding higher levels of cross linker, or by increasing the quantity of available amine groups in the polymer. This can be achieved by increasing the ratio of p(AMA) to CMC. Other studies were realized using p(AMA) with another anionic polymer, the break strength was higher than 4.
The sizes of the microcapsules are related to the speed of the stirrer, and the angle of the motor.
With these parameters (stirrer of rotation at 75 - 95 rpm, pH of coacervation at 4.5, ratio of polymer 1:0.4… we can linked to have microcapsules from 20 µm to 2000 µm depending of on the final use.
4. Conclusion
We have successfully prepared a cationic polymer that can be used to replace natural polymers, e.g., gelatin. The use of a synthetic polymer could be a good alternative to optimize the stability and the reproducibility of the capsules. The process of p(AMA) synthesis was performed to provide a better stability of the polymer. The break strength of the capsules was increased to allow the application of these capsules in few domains

Figure 6. Image of the microcapsules.
(medicine, food, cosmetic, painting). All the parameters to better control the encapsulation were evaluated to optimize the yield of coacervates and the capsules (including FTIR, turbidity, pH, coacervate yield, and Zeta-potential). The presence of amine functions on the polymer shows a significant difference to increase the properties of the capsules.
Cite this paper
Esselin, N., Portolan, F., Domloge, N., Rees, D., Musa, O.M. and Pilard, J.-F. (2016) Potentialities of the Poly(Aminoethyl Methacrylate) p(AMA) as Gelatin-Like Polymer in Complex Coacervation. Journal of Encapsulation and Adsorption Sciences, 6, 147-160. http://dx.doi.org/10.4236/jeas.2016.64011
References
- 1. Boussif, O., Lezoualc’h, F., Zanta, M.A., Mergny, M.D., Scherman, D., Demeneix, B. and Behr, J.P. (1995) A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in Vivo: Polyethylenimine. Proceedings of the National Academy of Sciences USA, 92, 7297-7301.
https:/doi.org/10.1073/pnas.92.16.7297 - 2. Kukowska-Latallo, J.F., Bielinska, A.U., Johnson, J., Spindler, R., Tomalia, D.A. and Baker, J.R. (1996) Efficient Transfer of Genetic Material into Mammalian Cells Using Starburst Polyamidoamine Dendrimers. Proceedings of the National Academy of Sciences USA, 93, 4897-4902.
https:/doi.org/10.1073/pnas.93.10.4897 - 3. Sakaki, S., Tsuchida, M., Iwasaki, Y. and Ishihara, K. (2004) Water-Soluble Phospholipid Polymer as a New Biocompatible Synthetic DNA Carrier. Bulletin of the Chemical Society of Japan, 77, 2283-2288.
https:/doi.org/10.1246/bcsj.77.2283 - 4. Ganachaud, F., Sauzedde, F., Elaissari, A. and Pichot, C. (1997) Emulsifier-Free Emulsion Copolymerization of Styrene with Two Different Amino-Containing Cationic Monomers. I. Kinetic Studies. Journal of Applied Polymer Science, 65, 2315-2330.
https:/doi.org/10.1002/(SICI)1097-4628(19970919)65:12<2315::AID-APP6>3.0.CO;2-C - 5. Basinska, T. and Slomkowski, S. (1995) Attachment of Horseradish Peroxidase (HRP) on to Thepoly(styrene/acrolein) Latexes and on to Their Derivatives with Amino Groups on the Surface; Activity of Immobilized Enzyme. Colloid and Polymer Science, 273, 431-438.
https:/doi.org/10.1007/BF00656887 - 6. Armes, S.P. and Aldissi, M.J. (1989) Novel Colloidal Dispersons of Polyaniline. Journal of the Chemical Society, Chemical Communications, 88-89.
https:/doi.org/10.1039/c39890000088
Armes, S.P., Aldissi, M., Agnew, S. and Gottesfeld, S. (1990) Aqueous Colloidal Dispersions of Polyaniline Formed by Using Poly(vinylpyridine)-Based Steric Stabilizers. Langmuir, 6, 1745-1749.
https:/doi.org/10.1021/la00102a007 - 7. Ramos, J., Martin-Molina, A., Sanz-Izquierdo, M.P., Rus, A., Borque, L., Hidalgo-Alvarez, R., Galisteo-Golzalez, F. and Forcada, J. (2003) Amino-Functionalized Latex Particles Obtained by a Multistep Method: Development of a New Immunoreagent. Journal of Polymer Science Part A: Polymer Chemistry, 41, 2404-2411.
https:/doi.org/10.1002/pola.10782
Sharma, G. and Ballauff, M. (2004) Spherical Polyelectrolyte Brushes as Nanoreactors for the Generation of Gold Particles. Macromolecular Rapid Communications, 25, 547-552.
https:/doi.org/10.1002/marc.200300107 - 8. Deming, T.J. (1997) Facile Synthesis of Block Copolypeptides of Defined Architecture. Nature, 390, 386-389.
https:/doi.org/10.1038/37084
Wong, M.S., Cha, J.N., Choi, K.S., Deming, T.J. and Stucky, G.D. (2002) Assembly of Nanoparticles into Hollow Spheres Using Block Copolypeptides. Nano Letters, 2, 583-587.
https:/doi.org/10.1021/nl020244c
Holowka, E.P., Pochan, D.J. and Deming, T.J. (2005) Charged Polypeptide Vesicles with Controllable Diameter. Journal of the American Chemical Society, 127, 12423-12428.
https:/doi.org/10.1021/ja053557t - 9. Euliss, L.E., Trnka, T.M., Deming, T.J. and Stucky, G.D. (2004) Design of a Doubly-Hydrophilic Block Copolypeptide That Directs the Formation of Calcium Carbonate Microspheres. Chemical Communications, 10, 1736-1737.
https:/doi.org/10.1039/b403211j - 10. Jin, R.H. and Yuan, J.J. (2005) Synthesis of Poly(ethyleneimine)s-silica Hybrid Particles with Complex Shapes and Hierarchical Structures. Chemical Communications, 21, 1399-1401.
https:/doi.org/10.1039/B417351A - 11. Oxley, J.D. (2012) Stability and Prediction of Shelf-Life for Microencapsulated Ingredients. Agro Food Industry Hi Tech, 23, 60-63.
- 12. William, R.M. (1990) The Procter & Gamble Company, Perfume Microcapsules for Use in Granular Detergent Compositions. CA 2004270 A1
- 13. Cheng, S.Y., Yuen, C.W.M., Kan, C.W. and Cheuk, K.K.L. (2008) Development of Cosmetic Textiles Using Microcapsulation Technology. Research Journal of Textile and Apparel, 12, 41-51.
https:/doi.org/10.1108/RJTA-12-04-2008-B005 - 14. Tiebackx, F.W.Z. (1911) Gleichzeitige Ausflockung zweier Kolloide. Colloid and Polymer Science, 8, 198-201.
https:/doi.org/10.1007/bf01503532 - 15. Bungenberg de Jong, H.G. and Kruyt, H.R. (1929) Coacervation (Partial Miscibility in Colloid Systems). Proceedings of the Koninklijke Nederlandse Akademie Van Wetenschappen, 32, 849-856.
- 16. Overbeek, J.T.G. and Voorn, M.J. (1957) Phase Separation in Polyelectrolyte Solutions, Theory of Complex Coacervation. Journal of Cellular and Comparative Physiology, 49, 7-26.
https:/doi.org/10.1002/jcp.1030490404 - 17. Veis, A. and Aranyi, C. (1960) Phase Separation in Polyelectrolyte Systems. I. Complex Coacervates of Gelatine. Journal of Chemical Physics, 64, 1203-1210.
https:/doi.org/10.1021/j100838a022 - 18. Veis, A. (1961) Phase Separation in Polyelectrolyte Solutions. II. Interaction Effects. Journal of Physical Chemistry, 65, 1798-1803.
https:/doi.org/10.1021/j100827a026 - 19. Veis, A. (1963) Phase Separation in Polyelectrolyte Systems. III. Effect of Aggregation and Molecular Weight Heterogeneity. Journal of Physical Chemistry, 67, 1960-1964.
https:/doi.org/10.1021/j100804a004 - 20. Veis, A., Bodor, E. and Mussell, S. (1967) Molecular Weight Fractionation and the Self-Suppression of Complex Coacervation. Biopolymers, 5, 37-59.
https:/doi.org/10.1002/bip.1967.360050106 - 21. Nakajima, A. and Sato, H. (1972) Phase Relationships of an Equivalent Mixture of Sulfated Polyninyl Alcohol and Aminoacetalyzed Polyvinyl Alcohol in Microsalt Aqueous Solution. Biopolymers, 10, 1345-1355.
https:/doi.org/10.1002/bip.1972.360110704 - 22. Tainaka, K. (1979) Study of Complex Coacervation in Low Concentration by Virial Expansion Method. I. Salt Free Systems. Journal of the Physical Society of Japan, 46, 1899-1906.
https:/doi.org/10.1143/JPSJ.46.1899 - 23. Tainaka, K. (1980) Effect of Counterions on Complex Coacervation. Biopolymers, 19, 1289-1298.
https:/doi.org/10.1002/bip.1980.360190705 - 24. Green, B.K. and Schleicher, L. (1956) Manifold Record Material. US 2 730 456.
- 25. Green, B.K. and Schleicher, L. (1957) Oil-Containing Microscopic Capsules and Method of Making. US 2 800 457.
- 26. He, L., Read, E.S., Armes, S.P., Adams, D.J. and Lönngren, J. (2007) Synthesis of Controlled-Structure Primary Amine-Based Methacrylic Polymers by Living Radical Polymerization. Macromolecules, 40, 4429-4438.
https:/doi.org/10.1021/ma070670q - 27. Kromidas, S. and Kuss, H. (2009) Quantification in LC and GC: A Practical Guide to Good Chromatographic Data. John Wiley & Sons, Weinheim.
- 28. Narain, R. and Armes, S.P. (2003) Synthesis and Aqueous Solution Properties of Novel Sugar Methacrylate-Based Homopolymers and Block Copolymers. Biomacromolecules, 4, 1746-1758.
https:/doi.org/10.1021/bm034166e - 29. He, L., Read, E.S., Adams, D.J. and Armes, S.P. (2007) Macromolecules, 40, 4429-4438.
https:/doi.org/10.1021/ma070670q - 30. Frugier, D. and Audebert, R. (1994) Interaction between Oppositely Charged Low Ionic Density Polyelectolytes: Complex Formation or Simple Mixture? In: Dubin, P.L., Bock, J., Davis, R., Schultz, D.N. and Thies, C., Eds., Macromolecular Complexes in Chemistry and Biology, Springer, Berlin, 135-149.
https:/doi.org/10.1007/978-3-642-78469-9_9 - 31. Thompson, K.L., Read, E.S. and Armes, S.P. (2008) Chemical Degradation of Poly(2-Aminoethyl Methacrylate). Polymer Degradation and Stability, 93, 1460-1466.
https:/doi.org/10.1016/j.polymdegradstab.2008.05.013