Paper Menu >>
Journal Menu >>
 Materials Sciences and Applications, 2010, 1, 223-246 doi:10.4236/msa.2010.14035 Published Online October 2010 (http://www.SciRP.org/journal/msa) Copyright © 2010 SciRes. MSA Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Levan Chkhartishvili1, Ivane Murusidze2 1Department of Physics, Georgian Technical University, Tbilisi, Georgia; 2Institute of Applied Physics, Ilia State University, Tbilisi, Georgia. Email: chkharti2003@yahoo.com Received August 4th, 2010; revised August 27th, 2010; accepted September 6th, 2010. ABSTRACT Molar binding energy of the boron nitride single-walled zigzag and armchair nanotubes is calculated within the quasi-classical approach. We find that, in the range of ultra small radii, the binding energy of nanotubes exhibit an oscillatory dependence on tube radius. Nanotubes (1,1), (3,0), and (4,0) are predicted to be more stable species among single-walled boron nitride nanotubes. The obtained binding energies of BN single-walled nanotubes corrected with zero-point vibration energies lies within the interval (12.01-29.39) eV. In particular, molar binding energy of the ul- tra-large-radius tube is determined as 22.95 eV. The spread of the molar zero-point vibration energy of BN nanotubes itself is (0.25-0.33) eV and its limit for ultra-large-radius tubes is estimated as 0.31 eV. The binding energy peak lo- cated at 2.691 Å corresponds to the equilibrium structural parameter of all realized stable BN nanotubular structures. Keywords: Binding Energy, Zigzag and Armchair Nanotubes, BN 1. Introduction Boron nitride with the chemical formula BN can be found in the form of one-dimensional diatomic molecules, two-dimensional nanotubes and fullerenes, three-dimen- sional crystals like the layered hexagonal h-BN and rhombohedral r-BN as well as turbostratic t-BN, cubic zinc-blende c-BN and wurtzite w-BN modifications as well as their nanostructures etc. Boron and nitrogen at- oms are surrounded tetrahedrally in both denser c-BN and w-BN crystals. Any constituent atom of an h-BN crystal, which is be- lieved to correspond to the boron nitride ground state, may be considered as a 3-coordinated atom because the strong chemical binding (covalent with some deal of ionic) occurs only within the layers, while weak van der Waals forces seem to be responsible mostly for interlayer binding. The h-BN crystal has a “graphitic” structure with a two-layer stacking sequence (r-BN is character- ized by a three-layer stacking). These layers consist of regular hexagons (i.e., 6-membered atomic rings) with vertexes alternatively occupied by B and N atoms. In the h-BN crystal, B atoms are placed directly above N atoms and vice versa. Therefore one might suppose that the ionicity contributes to interlayer bonding as well. How- ever, actually, the electrostatic component is insignificant due to the large interlayer distances. It is argued also by the existence of a layered r-BN crystal, in which each subsequent layer is turned around an angle of /3 , and also by the isolated plane defects and their bundles in- cluded in real h-BN crystals, in which any given atom can be placed above the same atom. In addition, it is pos- sible to obtain turbostratic t-BN and amorphous struc- tures in the form of mixes of various boron nitride crys- talline phases, and multi-walled nanotubular and multi- shelled fullerene-like BN structures. Strong chemical bonding between atoms in a given layer and weak inter- layer interaction in layered boron nitrides specify an op- portunity of physical and chemical intercalations by various atoms and molecules. Based upon the similarity of structures of the boron nitride layered phases with graphite, it was assumed [1] that along with carbon C nanotubes, stable BN nanotubes – fragments of hexagonal or mixed BN layers wrapped into cylinders – could also exist. Indeed, by means of arc discharge BN nanotubes had been obtained both from carbon nanotubes [2] and in carbon-free plasma [3]. The arc discharge methods were used to produce BxCyNz nanotubular structures identified by the high-resolution transmission-electron-microscopy  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 224 (TEM) together with K-edge electron-energy-loss-spec- trometry (EELS) determining the local atomic composi- tion, while, in a carbon-free plasma discharge area be- tween a BN-packed W-rod and a cooled Cu-electrode, multi-walled pure BN tubes were produced with inner diameters on the order of 1 to 3 nm and with lengths up to 200 nm; EELS of individual tubes yielded B/N ratio of approximately 1. At present various methods of synthesis of the BN nanotubular structures are developed. The arc discharge in a molecular nitrogen atmosphere between electrodes made of graphite and refractory bo- ron compounds, e.g., hafnium diboride HfB2, forms BN-C nanotubes with strong phase separation between BN and C layers along the radial direction [4,5]. If both elec- trodes are made of HfB2 rods, single- or double-walled chemically pure BN nanotubes are formed with a struc- ture close to stoichiometric B/N ~1 [6]. Most obtained tube ends are closed by flat layers perpendicular to the tube axis. A closure by a triangular facet resulting from three 120-disclinations was proposed to account for this specific shape. For the most part, the multi-walled BN nanotubes are formed with electrodes made of zirconium diboride ZrB2 [7,8]. In this case most of the nanotubes have diameters from 3 to 40 nm and lengths on the order of 100 nm. Single-layer tubes with diameters of 2 to 5 nm are also formed rarely. The morphology of the tube tips suggests the presence of pentagons and heptagons which are energetically less favorable compared with squares. A laser melting of the solid-state BN (of any crystal structure, not only layered but also amorphous) at high nitrogen pressure, (5-15) GPa, forms nanotubes free from inclusions, containing from 3 up to 8 walls, and having a characteristic outer diameter of (3-15) nm [9]. BN nanotubes have been also obtained by pyrolysis of the molecular precursor with the use of Co catalysts [10]. Bundles of single-walled (or containing few layers) BN nanotubes with almost stoichiometric structure can be formed in substitution reactions – by thermal treat- ment of a mixture of boron trioxide or trichloride, and bundles of single-walled C-nanotubes at high tempera- tures, (1250-1350)℃, in a nitrogen flow [11,12]. BN nanotubes also grow in solid-state process that in- volves neither deposition from the vapor phase nor che- mical reactions [13]. The nanotubes were produced by first ball-milling of the layered h-BN powder to generate highly disordered or amorphous nanostructures and fo- llowed by the product annealing at temperatures up to 1300℃. The annealing leads to the nucleation and growth of hexagonal BN nanotubes both of cylindrical and bam- boo-like morphologies. Multi-walled BN nanotubes have been obtained by carbothermal reduction of the ultra-dispersive amorphous boron oxide B2O3 at simultaneous nitriding at high tem- peratures, (1100-1450)℃ [14,15]. For large tubes, it is found that the ratio of length to radius is preserved. Besides of arc-melting, pyrolysis, and chemical reac- tions, boron nitride nanotubular structures were created by means of ballistic nuclear displacements caused in a h-BN layered crystal structure by electron irradiation in TEM [16,17]. High growth temperatures (above 1100℃), a low pro- duction yield, and impurities have prevented progress in applications of BN nanotubes in the past decade. Rather recently, it has been shown that these tubes can be grown on substrates at lower temperatures (of about 600℃) [18]. High-order tubular structures were constructed, which can be used without further purification. For synthesizing BN nanotubular material, some meth- ods were inspired from carbon. In particular, these in- clude techniques such as laser ablation and non-ablative laser heating [19]. Transformation of the compressed powders of the fine-grained h-BN into nanotubular form can be induced [20] by the concentrated light energy in nitrogen flow. Fiber-like clusters synthesized by evapo- ration of the layered BN in a nitrogen atmosphere and obtained in powders formed on substrate or chamber surfaces contained nanotubes with diameters and lengths equal to (0.05-200) and (100-3000) m, respectively. Applying TEM, there were obtained their associations in tree- and coral-like aggregates. BN nanotubes can be grown from a nanococoon seed as well [21]. Recently the development of a new method for pro- ducing long, small-diameter, single- and few-walled, BN nanotubes in macroscopic quantities has been reported [22]. The pressurized vapor/condenser (PVC) method produces highly crystalline, very long, small-diameter nanotubes without catalysts. Their palm-sized, cotton- like masses of raw material were grown by this technique and spun directly into centimeters-long yarn. Nanotube lengths were observed to be ~100 times that of those grown by the most closely related method. Soon after synthesizing the first BN nanotubes, it was proposed a number of their possible applications in tech- nique [3]. For example, a system of the collinear BN nanotubes forms a boron nitride fiber. At the same time, the theory [23] developed for structural and electronic properties of nanotubular heterojunctions, in which one of the layers is nanotubular boron nitride (namely, for C/BN and BC2N/BN systems), leads to a conclusion that on basis of it a different electronic devices can be de- signed. In particular, nanostructures with C-layers both in the center and at the periphery separated by a few BN- layers may allow the creation of sandwich nanotubular  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 225 devices [4]. Within the special semiempirical approach [24] C/BN superlattices and isolated junctions have been investigated as specific examples by the wide variety of electronic devices that can be realized using such nano- tubes. The bottom of the conduction bands in pure BN nanotubes is controlled by a nearly-free-electron-state localized inside the tube suggesting interesting electronic properties under doping. Other opportunities of application of the BN nanotubes are connected with the features of their phonon spectrum [25]. Such dielectric tubes without inversion center can be used as a phonon laser in GHz − THz range or hyper- sound quantum generator. Because of the presence of special nanotubular oscillatory modes, there is a strong enhancement of electron-phonon interaction in compari- son with a bulk material. It is not excluded that close- packed one-dimensional BN nanotubes will serve as high-temperature superconductors. The GHz oscillatory behavior of double-walled BN nanotubes was also pre- dicted [26,27]. This system can also be employed for making good shock absorbers because application of low pressure leads to its significant compression. The band gap progression with BN nanotube diameter (which is of crucial importance for device applications) was presented and analyzed in detail in [28]. In zigzag BN nanotubes, radial deformations that give rise to transverse pressures decrease the gap from 5 to 2 eV, allowing for optical applications in the visible range [29]. Importantly, both the zigzag and armchair tubes are found [30] (see also [31]) to exhibit large second-order nonlinear optical behavior with the second-harmonic generation and linear electro-optical coefficients being up to 15-times larger than that of bulk BN in both denser zinc-blende and wurtzite structures. This indicates that BN nanotubes are promising materials for nonlinear op- tical and optoelectronic applications. The electronic structure of BN nanotubes can be tuned within a wide range through covalent functionalization [32] (see also [33]). The ultraviolet (UV) and visible ab- sorption spectra indicate that their electronic structure undergoes drastic changes under functionalization. First principle calculations revealed that the covalently func- tionalized BN nanotubes can be either n- or p-doped de- pending on the electronegativity of molecules attached. Their energy gap can be adjusted from UV to visible optical range by varying concentration of functionalizing species. One-dimensional crystals of potassium halides, inclu- ding KI, KCl, and KBr, were inserted into BN nanotubes [34]. High-resolution TEM and energy-dispersive X-ray spectrometry were used to characterize their microstruc- tures and compositions. The fillings are usually single crystals with lengths up to several m. The wetting properties (static contact angles of the liquids and surface tension) of individual BN nanotubes were studied [35] experimentally using a nanotube-based force to measure the interactions between nanotubes and liquids in situ. First principles simulations on the interaction of mo- lecular hydrogen H2 with the native and substitutional defects in small-diameter (8,0) BN nanotubes were per- formed in [36]. The adsorption of H2 in structures found to be endothermic with respect to dissociation, with the small-diameter nanotube possessing the smaller barrier. Although chemisorption along the tube axis is energeti- cally preferred, the barrier for dissociation is lower for chemisorption across the tube axis. This implies that chemisorbed hydrogen can be kinetically trapped in a higher energy state. Dopants that maximize the localiza- tion of the higher-occupied-molecular-orbital (HOMO) and lower-unoccupied-molecular-orbital (LUMO) states maximize hydrogen binding energies. C-dopants do not enhance H2 binding, whereas Si-dopants substituting for N provide H2 binding energies of 0.8 eV, at the upper end of the range required for hydrogen storage. The for- mation energy of most defects is reduced with increasing curvature except for the C-substitutionals. Vacancies do not reduce the barriers for H2 dissociation for strongly curved nanotubes. The surface stress induced by the nanotube curvature boosts the hydrogen storage capabili- ties of vacancies with the nitrogen vacancy chemisorbing 4H and allowing a H2 molecule to enter the interior of the tube. The hydrogen binding properties of BN systems strongly depend on existing defects and dopants. Pre- treating of these systems so as to partially remove nitro- gen should enhance H2 adsorption properties. The hy- drogen absorption capacity of Ti-covered single-walled BN nanotube was investigated using first principles plane-wave (PW) method [37]. The weak interaction of H2 molecules with the outer surface of bare nanotube can be significantly enhanced upon functionalization by Ti atoms: each Ti atom adsorbed on tube can bind up to four H2 molecules with average binding energy suitable for room temperature storage. The morphology of BN nanotubes with a collapsed structure has been discovered by a metal-catalyzed treatment [38]. The collapse causes the dramatic enlarge- ment of a specific surface area of BN nanotubes and re- markably enhances the hydrogen storage capacity of BN nanotubes. It was reported [39] that proteins are immobilized on boron nitride nanotubes. There is a natural affinity of a protein to BN nanotube: it can be immobilized on tube directly, without using of an additional coupling reagent. Besides, boron nitride nanotubes may be dissolved in organic solvents by wrapping them with a polymer [40].  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 226 It was proposed [41] that BN polymers, having the structures similar to organic polymers, can serve as a cheap alternative to inorganic semiconductors in design- ing modern electronic devices. Some related potential innovations, including band gap tuning, were also dem- onstrated. The observed giant Stark effect significantly reduces the band gap of BN nanotubes and thus greatly enhances their utility for nanoscale electronic, electromechanical, and optoelectronic applications [42]. In particular, this effect may be important for tuning the band gap of BN nanotubes for applications as a nanoscale field-effect- transistor (FET). Boron nitride nanotubes have also manifested stable currents in field emission geometry and may be more stable than carbon nanotubes at high temperatures [43,44]. As it was mentioned, boron nitride nanotubes exhibit many similarities with the carbon ones (such as high Young modulus etc.) and might have superior unique mechanical, thermal, and electronic properties [45]. In addition, BN nanotubes are characterized by chemical inertness and poor wetting [46]. The factor that distinguishes BN from C is partial het- eropolarity of the chemical bonding. For this reason, one more sphere of possible applications of BN nanotubes can be new pyroelectric and piezoelectric materials promising for applications in nanometer-scale sensors and actuators. The 3-fold symmetry of a BN sheet, the III-V analog to graphite, prohibits an electric polarization in its ground state. However, this symmetry is broken when the sheet is wrapped to form a BN tube. It was shown [47] that this leads to an electric polarization along the nanotube axis which is controlled by the quan- tum-mechanical boundary conditions of its electronic states around the tube circumference. Thus, the macro- scopic dipole moment has an intrinsically nonlocal quan- tum-mechanical origin from the wrapped dimension. Combining first principles, tight-binding methods and analytical theory, the piezoelectricity of heteropolar (in particular, BN) nanotubes was found [48] to depend on their chirality and radius. This effect can be understood starting from the piezoelectric response of an isolated sheet along with a structure specific mapping from the sheet onto the tube surface. It was demonstrated that a linear coupling between the uniaxial and shear deforma- tions occurs for chiral nanotubes, and the piezoelectricity of nanotubes is fundamentally different from its coun- terpart in a bulk material. First principles calculations of the spontaneous polarization and piezoelectric properties of BN nanotubes have shown [49] that they are excellent piezoelectric systems with response values larger than those of piezoelectric polymers. The intrinsic chiral sy- mmetry of the nanotubes induces an exact cancellation of the total spontaneous polarization in ideal, isolated nanotubes of arbitrary indexes. But the breaking of this symmetry by the intertube interaction or elastic deforma- tions induces spontaneous polarization comparable to that of wurtzite bulk semiconductors [50]. Multielement nanotubes comprising multiple SiC-core, an amorphous SiO2-intermediate layer, and outer shells made of BN and C layers separated in the radial direction with diameters of a few tens of nm and lengths up to 50 m were synthesized by means of reactive laser ablation [51]. They resemble a coaxial nanocable with a semi- conductor-insulator-semiconductor (SIS) geometry and suggest applications in nanoscale electronic devices that take advantage of this self-organization mechanism for multielement nanotube formation. A theoretical description of electron irradiation of sin- gle-walled BN nanotubes was presented in [52]. As a first step, the anisotropy of the atomic emission energy threshold was obtained within extended molecular-dyna- mical (MD) simulations based on the density-functional- theory (DFT) tight-binding method. As a second step, total cross section for different emission sites as a func- tion of the incident electron energy was numerically de- rived. Two regimes were then described: at low irradia- tion energies (below 300 keV), atoms are ejected mostly from the upper and lower parts of the tube while at high energies (above 300 keV) atoms are ejected mostly from the side walls. Typical values of the total cross section of knock-on processes are obtained to vary from a fraction of barn (at side wall for 150 keV electrons) up to around 20 barns (for 1 MeV electrons). In BN nanotubes, the emission energy threshold maps were reported to show B sputtering to be more favorable for low irradiation ener- gies, while N sputtering is more favorable at high ener- gies. These calculations of the total knock-on cross sec- tion for nanotubes can be used as a guideline for TEM experimentalists using high energy focused beams to shape nanotubes, and also, more generally, if electron irradiation is used to change nanotube properties such as their optical behavior or conductivity. Such wide field of possible technical and technological applications of boron nitride nanotubes makes useful theoretical research determining their main physical characteristics. In particular, for purposeful design of some materials and devices based on nanotubular BN, like the fibrous composites, tubular heterojunctions, other nanoelectronic devices, nanoreservoirs for hydro- gen storage etc, it is very important to be able to predict reliably values of the ground-state parameters, especially, the molar binding energies and sizes of the nanotubes with given indexes and their relative stability. In present work, this task is solved for the most stable – achiral (zigzag and armchair) – single-walled forms.  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 227 Paper is organized as follows. In Section 1, we have introduced methods of synthesis of the boron nitride nanotubes and their technological applications. Section 2 is a brief summary of the structural and binding data available on nanotubular boron nitride. In Section 3, the theoretical approach based on the quasi-classical ap- proximation to the binding energy calculation and ge- ometries is presented. In Section 4, results of the per- formed calculations are presented in the form of curves “molar binding energy – structural parameter”. Section 5 is devoted to estimation of the nanotube lattice zero-point vibration energy. And finally, Section 6 discusses rela- tive stability of the boron nitride nanotubes of various radii and makes an attempt to generalize the obtained results. 2. Structural and Binding Data Let us start with a brief overview of the structural and binding data available on a boron nitride diatomic mole- cule, isolated sheet, and nanotubular form. 2.1. Molecular Boron Nitride The diatomic molecule BN can be considered as a sim- plest (degenerated) form for boron nitride nanotubes. In general, electronic theory of substance considers a dia- tomic molecule as a special problem for its intermediate structural and, consequently, electronic properties be- tween mono- and polyatomic systems. Peculiarities are related mainly with the system axial symmetry and uniqueness of the structural parameter – interatomic dis- tance d. Unlike the solid state or nanoscale boron ni- trides, which are materials with a diversity of technical and industrial applications, BN molecule, which exists under the extreme conditions, is only of academic inter- est as a “building block” for two- and three-dimensional boron nitride structures. From the standard thermoche- mical data, the energy of B–N bond at the equilibrium length is known to be considerably higher compared with those of B–B and N–N bonds. In addition, any stable regular BN structure is a network of atomic rings with alternating atoms such that the nearest-neighbor envi- ronment of both B and N atoms consists of only B–N bonds. Therefore, the B–N bond length is a key intera- tomic distance in the analysis of boron-nitrogen binding. There are known some old first principles and semiempirical investigations for boron–nitrogen interac- tion (see [53,54]). Applying a self-consistent-field (SCF) procedure to the BN molecule in [55], it was calculated molecular orbitals (MOs) in order to minimize total en- ergy of the diatomic system. Then using the spectro- scopic data available for the corresponding ground state, the BN molecule dissociation energy value E was found to be 4.6 eV. According to the original theoretical approach of [56], the equilibrium interatomic distance in this molecule equals to 1.307 Å. At the same time, spec- troscopic parameters characterizing the calculated bo- ron–nitrogen interaction potential curve lead to the dis- sociation energy estimation of 5.05 eV. Nearly the same theoretical value for the bond length of 1.320 Å was suggested in [57]. In [58], a short-ranged classical-force- field (CFF) modeling of BN modifications was per- formed on the basis of experimental and first principles solid-state and diatomic-molecular data. In particular, assuming that CFF can be correctly determined by a sum of only two-body interaction terms, the B−N potential energy had been expressed analytically via Morse poten- tial, which gave 1.32521d Å and 5.0007EeV. However, it was noted [25] that standard forms of the pair interatomic potentials, such as the Morse, Mee–Grüneisen, Buckingham, and other potentials, con- verge slowly and, therefore, a cutoff procedure should be used. But, in such a case a non-physical jump on the po- tential radial function can arise. In order to eliminate this problem, based on the embedded atom method, a new B−N interatomic potential was designed which fulfills the conditions for smooth end: the potential function and its derivative (i.e., the interatomic force) vanish at the cutoff radius. The equilibrium bond length of 1.4457 Å and binding energy of 4.00 eV were found to reproduce correctly relative stabilities of the boron nitride layered structures. We also suggested [59] a theoretical, namely, quasi- classical method of calculation of the dependence the B–N interatomic binding energy E upon the bond length d. The constructed ()EEd curve was shown to be useful for estimations of BN crystalline structures cohesion parameters as well. This function reveals stan- dard behavior characteristic for the central pair potentials. (0)E , and () 0Ed if d is equal or greater than the sum of B and N quasi-classical atomic radii () 2.30r and () 1.70r Å, i.e., ()() 4.00dr r Å (note that quasi-classical B−N interatomic potential automatically fulfills the conditions for the smooth end at () () dr r ), while within the intermediate region () () 0dr r it is an oscillating function with sev- eral maxima. Among these maxima only one is available kinetically and, therefore, it corresponds to the equilib- rium. Analysis of the piece of the quasi-classical ()EEd curve for BN diatomic molecule, in the vicin- ity of this maximum, yields the values of bond length of 1.55 Å and binding energy of 4.51 eV. Same dependence determined earlier [60,61] within the frames of another quasi-classical parameterization scheme (for this purpose the screening factor of the potential affecting the given electron in interacting atom was approximated by the  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 228 radial polynomial, not by the constant) is relatively flat and leads to the estimations of 1.58 Å and 4.79 eV. Thus, the spread in theoretical and semiempirical val- ues for BN molecule binding energy is (4.0-5.5) eV, which overlaps with the recommended [62] experimental dissociation energy value of (4.0 ± 0.5) eV. The available first principles and semiempirical calculations and ther- mochemical experimental data lead to the binding energy values of about (4-7) eV per B−N bond for various, dif- ferently coordinated, BN modifications (for sheet and nanotubular structures see below). Such kind of estima- tions may be considered to be in qualitative agreement with the quasi-classically calculated B–N bond energy as the ground state energetic parameters are quite sensitive to the atomic coordination. For this reason, we focus our attention on the differences in the bond length values between BN molecular and crystalline phases. The quasi-classical values for the isolated B−N bond length and other relevant theoretical and semiempirical data, which lie over the range (1.307-1.580) Å, are overes- timated in comparison with 1.281 Å measured in 11B14N molecule [63]. An explanation may be that BN molecular spectra [64] verify triplet ground state, but at the same time reveal a low-lying singlet state with longer bond. Quasi-classically calculated interatomic vibration en- ergy in a B−N diatomic system of 0.178 eV/mole (the corresponding vibration quantum equals to 1435 cm1) was found by fitting the quasi-classical B–N potential curve with parabola [60,65]. This value is in good agree- ment (accurate within 5 %) with the values experimen- tally obtained for a neutral BN molecule of 0.187 (1514.6) [63] and 0.188 eV/mole ((1519.0 ± 0.2) cm1, from the absorption spectra Fourier analysis for laser-induced molecular fluorescence) [64]. According to the SCF theoretical method of [56], the ground state vibration energy in molecular BN estimated as 0.179 eV/mole (1446 cm1), which is almost the quasi- classical result. In [57], it was suggested the higher theoretical value of 0.217 (1750), what is close with 0.216 eV/mole (1740 cm1) measured in ionized molecule BN+ [66]. Studies of more complex molecular clusters of B and N are also interesting to get deeper insight into the defect formation processes in boron nitride nanotubes. High- temperature Knudsen cell mass spectrometry was used to study the equilibria involving the B2N molecule [67]. The thermal functions necessary to evaluate the mass spectrometric equilibrium data had been calculated from available experimental and theoretical molecular pa- rameters. In particular, in some B2N formation reactions changes in enthalpy have been measured. Room tem- perature atomization and formation enthalpies were de- termined to be 10.84 and 5.71 eV, respectively. At the same time, first-principles calculations were performed to estimate the electronic parameters of B2N, such as ionization energy and electron affinity. Mixed clusters of B and N atoms – B2N, BN2, B3N, B4N, B2N2, and B3N2 – can be produced by sputtering of a solid state BN [68]. Atom ordering in assumed linear species had been derived from measurements of the mass distribution of both the positive and the negative prod- ucts from the fragmentation of the anionic clusters in a gas target. As for neutral configurations, they were cal- culated. A tendency was found that a structure with the highest number of B−N bonds is most stable both in neu- tral and anionic species (an exception is the BN2 mole- cule). In contrast to this, the species with the highest number of adjacent same atoms (except for triatomic chains) had the largest electron affinity. 2.2. Boron Nitride Sheet The facts that boron nitride layered crystals and nano- tubes may be prepared suggest the necessity of analyzing the hypothetic isolated infinite hexagonal layer, i.e., the BN sheet. Corresponding two-dimensional BN crystal is represented as a planar layer composed of regular hexa- gons with vertexes alternately occupied by B and N at- oms. Classification and discussion of the BN haeckelite sheet structures, consisting of not only hexagonal atomic rings but also other even-membered rings, one can find in [69]. For the first time, the truncated crystal approach in the form of two semiempirical (standard and extended itera- tive Hückel) methods was applied to a two-dimensional hexagonal boron nitride structure [70]. The bond length was found to be 1.48 or 1.50 Å. However, when semiem- pirical calculations were performed on a two-dimensional periodic small cluster of the h-BN layer the equilibrium B−N distance was computed as 1.441 Å [71]. In [72], the 3-coordinated B12N12 network of 6-membered atomic rings was examined theoretically. Namely, the total en- ergy calculated using Hartree–Fock (HF) approach and DFT in local and gradient-corrected forms was mini- mized with respect to the B−N bond length. But, “graph- itic” isomer B12N12 is only a fragment of the BN sheet and its geometry appears to be somewhat distorted be- cause of finite sizes. As is to be expected, slight devia- tions of the bonds’ angles from the ideal value of 120 were observed for the bonds of atoms forming the central hexagon: −(2.52-2.65) for B atoms and +(2.52-2.65) for N atoms. There were also obtained number of unequal bond lengths: (1.266-1.283), (1.371-1.378), (1.427-1.442), (1.434-1.444), (1.520-1.536), and (1.553-1.576) Å. The finiteness of the quasi-classical atomic radii allowed us to obtain the B−N bond length for an infinite boron ni- tride sheet within the initial quasi-classical approxima-  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 229 tion [73]. The calculated dependence of the molar bind- ing energy on the lattice constant exhibits a maximum of 23.0 eV at 2.64 Å, which should correspond to the equi- librium state for an isolated hexagonal layer (analytical optimization [74] of the lattice parameter using the bind- ing energy calculated in quasi-classical approximation, which is possible only by neglecting the vibration energy, yields slightly different values: 23.2 eV and 2.66 Å, re- spectively). The lattice constant of 2.64 Å implies B−N bond length of 1.52 Å. Correction in energy introduced by zero-point vibrations was estimated as 0.242 eV/mole [65,73]. The quasi-classical bond length of 1.52 Å in an iso- lated BN sheet is in reasonable agreement (accurate within 4.6 %) with the bond length of 1.45 Å observed in layers of real h-BN crystals. At first glance, the surpris- ing thing is that the theoretical result for the isolated layer is in better agreement (with the accuracy of (2.6-3.8) %) with the bond lengths in tetrahedrally coordinated modifications c-BN (1.57 Å) and w-BN (1.56 and 1.58 Å). However, it is worth noting that to a certain extent two-dimensional boron nitride looks like three-dimen- sional crystals c-BN and w-BN: these three structures do not contain weak interlayer bonds, which occur in the h-BN layered modification. The lengths of (1.52-1.54) and (1.55-1.58) Å obtained in [72] for the bonds of atoms forming the central (almost undistorted) hexagon in B12N12 plane-fragment are also in good agreement with the quasi-classical result found for an idealized infinite BN sheet. Another quasi-classical approach using a different scheme of parameterization, employed to calculate h-BN binding and zero-point vibration energies, slightly un- derestimates the intralayer bond length [75]. The plausi- ble reason may be that the crystalline equilibrium con- figuration was selected to maximize its static binding energy with respect only to the layer lattice parameter, while the interlayer distance was fixed. Summarizing other theoretical and semiempirical re- sults concerning intralayer bond lengths in h-BN (and r-BN), one can state that all of them are in agreement with the experimental value of 1.446 Å [76]. For instance, in [77] the total energy of h-BN crystal as a function of unit cell volume V had been calculated using orthogo- nalized linear-combinations-of-atomic-orbitals (LCAOs) method within the local-density-approximation (LDA). The equilibrium was found at exp / 0.998VV , where exp V is the experimental value of V. Such result corre- sponds to the intralayer B−N distance of 1.438 Å. The calculations of [78] were also based on DFT within LDA, but PW expansion was used both for the pseudo- potential (PP) and the wave-function. The computed total energies and, consequently, the intralayer bond lengths in h-BN and r-BN were nearly the same: 1.441 and 1.439 Å, respectively. The short-ranged CFF modeling of boron nitrides leads to exactly the same intralayer B–N bond lengths in both layered structures: 1.454 Å [58]. The re- sults presented show satisfactory accuracy for the quasi- classically determined boron-nitrogen binding character- istics: accuracies of quasi-classical approach to deter- mine isolated B−N bond length and length of bonds in solid state structure amount a few percents, 7.2 and 5.1 %, respectively. Thus, the quasi-classically obtained B–N binding curve and its parameters mentioned above (namely, equilibrium bond length, binding energy, and vibration frequency) would be useful for investigations of compounds containing B−N bonds and, especially, BN nanosystems. As for the BN sheet binding and vibration energies, it is also reasonable to analyze correctness of the given predictions by comparing them with data available on the cohesion characteristics of h-BN layered crystals. As follows from standard thermochemical data, the binding energy of h-BN equals to 13.0 eV/mole [79]. The binding energies of 14.5, 16.0, and 14.4 eV/mole were deter- mined from semiempirical calculations performed using two variants of the semiempirical LCAOs method and an approach based on a periodic small-sized cluster [70,71]. Within the CFF potential model, the lower semiempirical estimate of 11.5 eV/mole was obtained [58]. In the framework of DFT, optimization of the structural pa- rameters led to the theoretical binding energy of 12.5 eV/mole [78]. Therefore, it can be expected that the mo- lar binding energy for h-BN layered crystal lies in the range from 11.5 to 16.0 eV. The binding energy of 23.0 eV/mole found by the quasi-classical method for the iso- lated layer is considerably higher. However, when com- paring these energies, one should take into account that interlayer bonds are substantially weaker than intralayer ones and that each atom in layered BN structures is in- volved in the formation of 5 bonds, of which only 3 are intralayer bonds. Consequently, if the interlayer energy is ignored as compared to the intralayer energy, we can assume that the binding energy per B−N bond of similar modifications is equal to 3/5 of the molar binding energy of the isolated layer. Making use of the result 23.0 eV/mole for layer, we find the molar binding energy of 3/5 23.0 eV = 13.8 eV. Indeed, this energy is close to the midpoint (13.75 eV) of the aforementioned energy range. On the other hand, the vibration energies of the isolated layer and layered crystals can be directly com- pared because the atoms of the low-dimensional system can execute vibrations in three independent directions in physical space. The quasi-classical result of 0.242 for two-dimensional BN agrees well with analogous calcula- tions of 0.266 [65], with the semiempirical estimate of 0.225 for zero-point vibrations energy in h-BN [58], and  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 230 coincides in order of magnitude with the estimate of 0.350 eV/mole from the theoretical phonon spectrum [78]. A few words about the defects in BN layer. Using PP and expanded unit cell methods, it was found that N- vacancies in BN sheet, as well as di- and trivacancional clusters including neighbor defects in BN layer, are characterized by small binding energies [80]. Calculated spectra and oscillator strengths allow to interpret local bands of the optical absorption in pyrolitic h-BN crystals before and after irradiation by fast-neutrons, protons, and C-ions. By first-principles calculations the 20 structures BxCyNz, derived from a hexagonal layer by placing B, N, or C atoms on each site, were considered [81] to investi- gate their relative stabilities. First-principles simulations of the interaction of molecular hydrogen H2 with the na- tive and substitutional defects of a single hexagonal BN sheet were performed in [36]. The adsorption of H2 on structures found to be endothermic with respect to disso- ciation. Vacancies reduce the barriers for H2 dissociation. The geometries of haeckelite BN sheets were con- structed by DFT [82]. Their molar energy of cohesion is found to be higher (by ~0.6 eV/mole) than that of regular one. 2.3. Nanotubular Boron Nitride The elementary form of a BN nanotube is a wrapped closed hexagonal surface inscribed in the cylinder. Such BN nanotubes can be found in regular – achiral, i.e., zig- zag (n,0) or armchair (n,n), and also in chiral (n,m) forms, 0mn. Here n and m are the tube indexes. Their symmetry operators have been identified in [83]: each type belongs to different family of the non-symmorphic rod groups; armchair tubes with even n are found to be centro-symmetric. The types and structures of the non- carbon, in particular, BN nanotubes were reviewed in [84]. In addition, the deformed regular or haeckelite nanotubes can exist. Concerning the haeckelite structures of BN tubes, a variety of chiral angles, including zigzag and armchair types, were observed. Depending on the structure formation kinetics characteristic for a given technology, BN nanotubes quite often take the bamboo- like morphology, form of a nanoarch (i.e., half-tube at the ends closed by planes) etc. Real nanotubular struc- tures are not infinite in length: they are definitely trun- cated. The three main different possible morphologies of the cylindrical tube closing with flat [8], conical, and amor- phous ends, as observed in experiments, were shown [85] to be directly related to the tube chirality. There are also possible rectangular BN nanotubes with linear defects on edges and with tips in the form of triangular flags. Such kinds of morphologies suggest the presence of energeti- cally unfavorable odd-membered atomic rings (i.e., pen- tagons and heptagons) in addition to favorable even-mem- bered rings (e.g. squares). As the growth of BN nanotubes cannot be directly ob- served and, consequently, the underlying microscopic mechanism is a controversial subject, in [86] first-prin- ciples MD simulation of the single-walled nanotube edges was performed. The behavior of growing BN nanotubes was found to strongly depend on the nanotube network chirality. In particular, open-ended zigzag tubes close rapidly into an amorphous tip, preventing further growth. In the case of armchair tubes, formation of squares traps the tip into a flat cap presenting a large central even- membered ring. This structure is meta-stable and is able to revert to a growing hexagonal framework by incorpo- rating incoming atoms. These findings are directly re- lated to frustration effects, namely that B−N bonds are energetically favored over B–B and N–N bonds. The expressions of radii, (,0)n R and (,)nn R, of the zig- zag and armchair, (n,0) and (n,n), BN nanotubes in terms of the index and the structure parameter a were ob- tained [87]. The parameter a corresponds to the lattice constant of the boron nitride layered crystals, i.e., intra- layer B–B or N–N bonds lengths. Therefore, the B–N bond length d equals to /3a. The nanotube index 1,2,3,...n determines the number of atoms as nano- tube unit cell consists of 2n formula units BN. The estimations of radii of the single-walled BN nanotubes, for their part, can be used for predicting their most prob- able combinations in multi-walled structures. Analyzing this problem, it is necessary to take into account that actually the question involves the average radii. A detailed study using the generalized tight-binding MD method has revealed [85] that, as a result of the dy- namical relaxation, the structure acquires a wave-like or “rippled” surface in which B atoms are displaced inward, while N atoms are displaced outward. This relaxation is similar to the reconstruction occurring at clean surfaces of III-V type crystalline semiconductors. However a general feature of BN nanotubular systems is that stronger surface potentials are associated with regions of higher curvature [88]. Thus, the interlayer interaction in BN nanotubes differs from bonding in three-dimensional layered crystals. However, most probably, these distinc- tions for nanotubular BN are weak enough to change essentially the equilibrium interlayer distances which are observed in h-BN and r-BN crystals. This conclusion is also confirmed by the results of an experimental study of the multi-walled nanotubes by high-resolution electronic microscopy [89]. In these structures, like in three-dimen- sional layered BN crystals, hexagonal and rhombohedral stacking sequences can freely coexist in nanotube wall- assembly. There are also possible some different cross-  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 231 section flattening, as well as ordering of layers in non- spiral zigzag. According to first-principles total-energy calculations [90], the most favorable double-walled BN nanotubes are structures in which the inter-wall distances are about 3 Å, i.e., as interlayer distances in layered BN crystals. Therefore, due to the weakness of the interlayer van der Waals forces, various types of multi-walled BN nanotubes can exist. Consequently, it is more probable the formation of such multi-walled BN nanotubes in which the difference between the radii of adjacent regu- lar nanotubes is close to the interlayer distance in a lay- ered h-BN crystal, i.e., to half of the height of the hex- agonal unit cell 6.6612c Å [91]. Thus, the similarity between types of the adjacent nanotubes has no crucial importance. In view of these factors, from the calculated single- walled nanotubes [87] pairs most suitable for the forma- tion of the stable double-walled BN nanotubes have been chosen. Remaining small divergences in sizes of the neighboring regular nanotubes can be compensated by defects and small chiral distortions. Such transformations of the zigzag and armchair nanotubes into chiral one will be accompanied, respectively, by the increase and de- crease in their radii. If the difference in radius between regular nanotubes is more (less) than /2c, the realiza- tion of structure in which the internal wall will be zigzag (armchair) and external – armchair (zigzag) is more probable. Hence, based on estimations of sizes of the single-walled BN nanotubes, it is possible to predict successfully the most stable double-walled forms. But, how can be solved the same problem for multi-walled nanotubes? A few words on the task. In this case, all over, it will be necessary to calculate radii of nanotubes with high indexes to choose sequences of single-walled nano- tubes, whose radii are close to terms of arithmetic pro- gression with common difference of /2c. However, now only geometrical considerations will be insufficient. The point is that unlike double-walled nanotubes in multi-walled ones there are also medial layers. For this reason, the choice of the most stable multi-walled struc- ture should be based on the comparison between the gains in energy, which are caused by the deviation from the equilibrium interlayer distance, on the one hand, and by chiral distortions, on the other hand. We can mention some theoretical results available on binding properties and stabilities of BN nanotubes. Sta- bilities of the boron nitride nanotubular structures were studied by means of non-orthogonal tight-binding for- malism [92]. The radii and energies of the BN nanotubes also were estimated by MD simulation [25] within the embedded atom model in which parameter d took the experimental value 1.4457 Å of the intralayer B–N bond length in real h-BN crystals. In [69], the binding energy of the regular BN nanotubes has been calculated within the DFT in generalized gradient approximation (GGA). Seeking equilibrium values of B–N bond length and radii, the geometry of the tubular 32-atom supercell was opti- mized. For (8,0), (10,0) and (4,4) tubes, it has been found 1.46d Å, and for (5,5) tube, 1.45d Å. Within the frame of semiempirical calculations of the nanotubular piezoelectric characteristics performed by the method of modified neglecting of diatomic overlapping (MNDO) [50], their radii also were determined. In this case the dependence of energies on the bond length was calcu- lated for the molecular fragments containing 3 or 4 ele- mentary layers (presumably, in this work for d the empirical value known for h-BN crystals was fixed as an equilibrium value). The possible contribution of ionicity of bonds in boron nitride structures is important to explain the binding dif- ferences between BN tubes and similar C tubes [1]. In order to facilitate understanding and prediction of nano- tube interactions in a multi-walled structure, the electro- static potentials on both outer and inner surfaces of some single-walled BN nanotubes have been calculated at a HF Slater-type-orbital level [88]. Structures were opti- mized computationally. Fictitious hydrogen atoms were introduced at the ends of the open tubes to satisfy the unfulfilled valences. It was found that BN tubes have stronger and more variable surface potentials than graph- itic ones. There are characteristic patterns of positive and negative sites on the outer lateral surfaces, while the in- ner ones are markedly positive. The binding and vibrations in small-radius single- walled BN nanotubes in [93] were studied by DFT using LDA. The results show that the chirality preference ob- served in experiments may be explained from the relative stability of the corresponding BN strips: the zigzag strips have larger binding energies and thus may be more easily formed. The smallest stable BN nanotube is found to be the (5,0) zigzag nanotube. The dependence of the tube deformation energy on its radius R was approximated by the formula E [eV/mole] 2.09 5.82 /R [Å]. The phonon dispersions of BN nanotubes were calculated and the frequency of the radial breathing mode was found to be inversely proportional to the nanotube radius. The geometries of the BN nanotubes were also constructed in DFT [82]. Based on DFT calculations [69], it was found that the energies of haeckelite BN nanotubes exceed by ~ 0.6 eV/mole those of corresponding hexagonal nanotubes. They are less stable in comparison with corresponding haeckelite sheets as well. However, still they are stable and can be synthesized. Energy of deformation (i.e., en- ergy needed to wrap nanotube from its sheet prototype) for large haeckelite BN tubes extrapolated by the formula ~/CR , where R is the tube radius, with different  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 232 parameters C and 2 for different structures. Using the symmetry properties in [83], it was deter- mined the numbers of Raman- and infrared (IR)-active vibrations in single-walled BN nanotubes. In contrast to the regular carbon nanotubes, zigzag boron nitride tubes possess almost twice as many vibrations as armchair ones. An extensive first principles study of the phonons in BN nanotubes using perturbation DFT in the LDA was performed in [94], where, based on the non-symmorphic rod group symmetry of tubes, the Raman- and IR-active modes at the point of the one-dimensional Brillouin zone were evaluated. For zigzag and chiral nanotubes, the set of IR-active modes is a subset of the Raman-active modes. In particular, the radial breathing mode is not only Raman-, but also IR-active. However, for armchair tubes, the sets of IR- and Raman-active modes are dis- joint. The frequencies of the active modes of zigzag, chiral, and armchair tubes were presented as a function of the tube diameter. They were compared with the fre- quencies obtained by the zone-folding method (i.e., by rolling of a BN sheet into a tube). Except for the high- frequency tangential modes, the zone-folding results are in very good agreement with the first principles calcula- tions. The radial breathing mode frequency can be de- rived by folding a sheet of finite width. Finally, the ef- fects of bundling on the phonon frequencies are shown to be small. First principles calculations of the nonresonant Raman spectra of zigzag and armchair BN nanotubes were presented in [95]. In comparison, a generalized bond-polarizability model, where the parameters are ex- tracted from first principles calculations of the polariza- bility tensor of a BN sheet, was implemented. For light polarized parallel to the tube axis, the agreement between model and first principles spectra is almost perfect, but for perpendicular polarization, depolarization effects have to be included in the model in order to reproduce Raman intensities. The possible dislocation dipoles as defect nuclei under tension in BN nanotubes were identified by dislocation theory and MD simulations [96]. Formation energies of the dipoles evaluated by gradient-corrected DFT are high and remain positive at large strains, thus suggesting great yield resistance of BN nanotubes. The dipole appears to be more favorable in spite of its homoelemental B−B and N−N bonds. The resonant photoabsorption and vibration spectroscopy combined with scanning tunneling micros- copy unambiguously identify the presence of Stone–Wales defects in BN nanotubes [97]. Based on extensive time- dependent DFT calculations, it was proposed to reso- nantly photoexcite such defects in the IR and UV re- gimes as a means of their identification. Intrinsic defects in zigzag BN nanotubes, including single vacancy, diva- cancy, and Stone–Wales defects, were systematically investigated using DFT calculation in [98]. It was found that the structural configurations and formation energies of the topological defects are dependent on tube diameter. The results demonstrate that such properties are origi- nated from the strong curvature effect in BN nanotubes. The scanning tunneling microscope images of intrinsic defects in the BN nanotubes also were predicted. The defected BN tubes with C-substitutions were considered in [50]. The theoretical studies of the elastic properties of sin- gle-walled BN nanotubes, carried out using the total -energy non-orthogonal tight-binding parameterization, were reported in [99]. Tubes of different diameters, ranging from 0.5 to 2 nm, were examined. The study found that in the limit of large diameters the mechanical properties of nanotubes approach those of the graph- ite-like sheet. The stiffness and plasticity of BN nano- tubes was investigated [100] using generalized tight- binding MD and first principles total-energy methods. Due to the B−N bond rotation effect, the compressed zigzag nanotubes were found to undergo anisotropic strain release followed by anisotropic plastic buckling. The strain is preferentially released toward N atoms in the rotated B−N bonds. The tubes buckle anisotropically toward only one end when uniaxially compressed from both ends. Based on these results, a skin-effect-model of smart nanocomposite materials is proposed, which local- izes the structural damage toward the surface side of the material. B−N bond-rotation mode of plastic yield in BN nanotubes in [101] was investigated combining first principles computations with a probabilistic rate approa- ch to predict the kinetic and thermodynamic strength. BN nanotubes yield defects have low activation, but high formation energies. In [50], elastic characteristics of BN nanotubes also were calculated applying MNDO method. 3. Theoretical Basis Our calculations are based on the quasi-classical expres- sion for binding molar energy of a substance, on the one hand, and on the geometric characterization of nano- tubular boron nitride, on the other hand. 3.1. Quasi-Classical Binding Energy of Substance Under the term ‘substance’ we imply polyatomic struc- tures at the ground state, i.e., molecules, various clusters, and crystals. Consequently, any substance is considered as a non-relativistic electron system affected by the static external field of nuclei, which are fixed at the sites in structure, and the averaged SCF of electrons. Because of singularities at the points, where the nuclei are located, and electron shell effects as well the inner potential of  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 233 substance does not satisfy the standard Wentzel–Kramers– Brillouin (WKB) quasi-classical condition of spatial smoothness. Nevertheless, beginning from Bohr’s fun- damental work ‘On the constitution of atoms and mole- cules’ up to the present the semi-classical analysis of the electronic spectrum has been widely used for light atoms and their small complexes. Besides, heavy atoms, large molecules, and crystals can be treated within the LDA using the total energy functional in the form of quasi- classical expansion. Success of quasi-classical approa- ches can be attributed to the diffuseness of atomic poten- tials. The expression for bounded electron states energies obtained by Maslov yields that precise and quasi-classi- cal spectra are close to each other if the characteristic values of potential 0 and the radius of its action 0 R meet requirement 2 00 21R (here and below all rela- tions are given in atomic units). For atomic potential 0~/ Z R and 0~RR where 1 Z is the atomic number and R is the radius of electron cloud. Therefore, in case of atoms it is required that 21 Z R . Even for light atoms their radii are several times larger than Bohr radius, 1R. Thus atoms and all polyatomic struc- tures indeed are quasi-classical electron systems and their structural and electronic characteristics can be calculated based on the quasi-classically parameterized electric charge density and electrical field potential distributions in atoms. The values of i-th electron classical turning point ra- dii i r and i r , ii rr , are obtained by solving the equations 2 (1) () 1,2,3,..., 2 ii ii ll Eri Z r , where r denotes the distance from the center of atom, () ir is the potential affecting the given electron, 0 i E and i l are its energy and orbital quantum num- ber, respectively. In the ground state the inner classical turning point for relative motion of atomic nucleus and electron cloud coincides with the center of system. As for the corre- sponding outer classical turning point radius r , it is obtained by solving the equation ()EZ r , where 0E denotes the energy associated with rela- tive motion and 2 1 1 () () 1 iZ i i Z rr Zr is the electron cloud potential affecting the nucleus. In particular, using the quasi-classical parameteriza- tion based on the Coulomb-like atomic potentials () ir i Z r we are able to get exact formulas 2(1) iii ii i i nnn ll rZ , 2(1) iiiii i i nnnll rZ , 2 22 1 2( 1) iZ i i Z r ZZ Z , 2 32 1 2 2( 1) iZ i i ZZ Z EZ . Here 2 ii i Z nE is the effective charge of the screened nucleus and i n is the principal quantum num- ber of i-th electron. The numerical values of i Z , E , i r , i r and r can be found by fitting the quasi-classical energy levels i E to ab initio (for instance HF) ones. Quasi-classical limit implies the truncation of electron states charge densities outside the classical turning points and space-averaging within the range between them. In this case i-th electron partial charge density is approxi- mated by the piecewise-constant radial function 33 ()0 3 4( ) 0 ii ii ii i rrr rrr rr rr . As for the nucleus charge density, it should be aver- aged inside the r -sphere: 3 3 () 0 4 0 Z rrr r rr . Consequently, the full atomic charge density is ex- pressed by the step-like radial function 1 () ()() iZ ik i rr r 1 1,2,3,..., kk rrrk q , where k r and k denote known constants which de- pend on parameters i r , i r and r , 012 0q rrr r , 2qZ is the number of different homogenous-charge-density spherical layers in atom. Using the Poisson equation, the radial dependence of the full atomic potential also can be approximated by the  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 234 step-like function, 22 55 11 33 33 11 3()3() () 2( )5( ) kk kkk k kk kk kk ar rbrr rc rr rr 1 1,2,3,..., kk rrrk q , 33 3 111 1 4( )4 33 ik ii ikk k i rrr a , 2 3 k k b , 22 2 1 1 2( )2 iq kiiikk ik crrr , if it is substituted by the space-averaged values inside each of the 1kk rrr intervals. In the region q rr, both the charge density and po- tential vanish identically, () 0r and () 0r . Thus finite parameter q r acquires a meaning of the quasi- classical atomic radius. Based on the presented step-like parameterization of the charge density and potential distributions in an atom, its quasi-classical total energy can be expressed in the following form: 33 () 1 1 ()0 3 iq Atomk kkk k Err . Note that it includes the non-physical energy of self- action, () 0 Atom SelfAction E which arises from substitut- ing the charge density for the probability density. Its value can be easily calculated in the quasi-classical appro- ximation and then excluded from the total energy. When the molecular or crystalline charge densities and potentials are expressed by the superposition of the step- like atomic charge densities and potentials, respectively, the molar (i.e., per chemical formula unit of the sub- stance) ground state static energy and its zero-point vi- bration correction are calculated as ()( ) ()( ) ()( )()() () 1 ()111 1()0 4 ik jq lq iNkN Staticijk likjlik t ti kj l EVr , ()( ) ()( )()()()()( )( ) / () 1() 111() () () () 30. 22 ik jq lq iN kNijk lk lijikjlikt Vibration itkjl iik tik t Vr EMr r Here the primed summation symbol denotes the elimination of the terms with 0t and ik ; in- dexes in parentheses ()i and ()k denote the atoms in the molecule or crystal unit cell, N is the full number of atoms, ()i M is the mass of i-th atom, t is the crystal translational vector – in case of a mole- cule 0t , ()ik t r is a distance between atomic sites and ()()( )()1( )1()( )1()1( ) ()()() () () ()(,,)(,,)(,,)(,,) ikjli jkli jkli jkli jkl ik tik tik tik tik t VrVrrrVrrrVr rrVrrr . We have introduced an universal geometric function 12 12 (, ,)VR RD which expresses the volume of the inter- section of two spheres as a function of their radii 1 R and 2 R, and the inter-center distance 12 D. 12 12 (, ,)VR RD and its partial derivative 1 21212 (, ,)/VR RDD both are continuous piecewise algebraic functions as follows: 3 1 12 12 4 (, ,)3 R VRR D 122 1 DRR , 3 2 4 3 R 1212 DRR , 2222 121212 121 122 12 ()(()4( )) 12 RRD RRDRRRR D 1212 12 ||RR DRR , 0 12 12 RRD ; 12 12 12 (,,) 0 VRR D D 122 1 DRR , 0 1212 DRR , 2222 1212 1212 2 12 (())(()) 4 RRDD RR D 1212 12 ||RR DRR , 0 12 12 RR D . In the lowest quasi-classical approximation, the equi- librium structure of substance is obtained by maximizing the molar binding energy of expected structures. () () () () 1 () ()0 iN Bindingii SelfAction i StaticSelf ActionVibration EEE EE E  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 235 with respect to their structural parameters. However, neglecting the insignificant redistribution of valence electrons arisen from association of atoms into a molecular or crystalline structure, the quasi-classical self- action energy of the substance is approximated by the sum of self-action energies of constituent atoms: () () () 1 iN SelfActioni SelfAaction i EE . And consequently, the binding energy approximately can be calculated without excluding of the self-action terms in advance: () () () 1 0 iN BindingiStatic Vibration i EEEE . The expected errors of the quasi-classical approach can be estimated for the model inner potential in the form of the analytical solution of the Thomas–Fermi (TF) equ- ation for the semi-classical atomic potential: structural and energy parameters of the electronic system deter- mined within the initial quasi-classical approximation are shown to differ from their exact values by factors 1/3 (10 / 3)1.02~ 1 and 2/3 (3/10)0.96 ~ 1 , respe- ctively. Thus the expected errors of the quasi-classical approach amount to a few percents. Even more, within the initial quasi-classical approximation there are no un- controllable calculation errors due the finiteness of quasi- classical atomic radii − the pair interactions without se- ries termination are truncated at the distances exceeded the sums of atomic radii. A complete quasi-classical theory of substance in- cluding calculation schemes for structural and binding, as well as for electronic spectrum characteristics, one can find in [102,103]. These schemes have been applied suc- cessfully for Na molecular and crystalline structures [104], various diatomic molecules [60,61], boron nano- tubes [105,106], and mainly for one-, two- and three- dimensional structural modifications of boron nitride – diatomic molecule, isolated plane sheet, hexagonal h-BN, cubic c-BN, and wurtzite-like w-BN crystals [59,60,64, 73-75,107,108]. 3.2. Geometries of the Boron Nitride Regular Single-Walled Nanotubes Summarizing previous subsection, one can conclude that equilibrium structural and binding parameters of the bo- ron nitride nanotubes can be calculated quasi-classically based on analytical expressions describing their geome- tries. This task has been solved in [87,108,109] for regu- lar (achiral), i.e., zigzag (n,0) and armchair (n,n) BN nanotubes. A model of regular nanotubes used here as- sumes that all atomic sites are located on cylindrical sur- face at the vertexes of regular hexagons broken along B–N or B–B and N–N diagonals, i.e., the expected small differences in bond length distinguished by their orienta- tion toward the tube axis are neglected. Namely, radii (,0)n R and (,)nn R of the zigzag and armchair nanotubes have been obtained [87,108] in terms of the nanotube index 1,2,3,... n and the structure parameter a: (,0) 4sin /2 n a Rn , (,) 54cos/2 43sin /2 nn na Rn . As it was mentioned, the parameter a corresponds to a lattice constant of the boron nitride layered crystals, i.e., to an intralayer B–B or N–N bond lengths. Therefore, the B–N bond length d equals to /3a. The nanotube index n determines the number of atoms, as a nanotube unit cell consists of 2n formula units, B2nN2n. Detailed regular geometries of zigzag and armchair BN nanotubes have been described in [108,109] using cylindrical coordinates (,,)z , which are useful for calculating binding energy. A unit cell of zigzag nanotubes consists of 4 atomic rings in parallel planes perpendicular to the axis. There are 2 pairs of rings, each consisting of 2 planes with n boron or n nitrogen atoms. Obviously, the cylindrical coordinate for all atomic sites equals to the tube ra- dius: (,0)n R . As for the coordinates and z in the first and sec- ond pairs of atomic rings, they equal to 2/ln , (61)/ 23zma , (61)/ 23zma , and (21) /ln , (31) /3zma , (31) /3zma , respectively. Here 0,1, 2,...,1ln and 0,1,2,...m number atomic pairs in a given pair of the atomic rings and these rings themselves, respectively. The unit cell of armchair nanotubes consists of 2 ato- mic rings in parallel planes perpendicular to the tube axis. For its part, each ring consists of n boron and n ni- trogen atoms. The coordinate for all atomic sites again equals to the tube radius: (,)nn R ,  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 236 while the rest cylindrical coordinates in the first and second atomic rings equal to 12/ln , 12/ln , zzma , and 12 22/ln , 12 22/ln , (21)/ 2zz ma , respectively. Here 1 2sin /2 sin 54cos/2 n n , 2 sin/2 sin 54cos/2 n n , and 0,1,2,...,1ln and 0, 1,2,...m number B or N atoms in atomic rings and these rings themselves. Based on the above discussion, the distances between a given atomic site and the sites in the so-called central atomic pairs (with 0 lm) in zigzag and armchair nanotubes have been found. For zigzag (0 lm: 0 ,B/2 3za, and N/23za ) tubes 00 22 (,0) (,0)2 22 (1 1)sin/3 4sin /2 lm nn ln m an , 00 222 (,0) (,0) 22 (2 1) sin(21)/23(21) 4 4sin /2 lm nn lnm an , 00 222 (,0)(,0) 22 (1 1) sin/(3 1) 3 4sin /2 lm nn lnm an , 00 222 ( ,0)( ,0) 22 (2 1) sin(21)/ 2(61) 12 4sin /2 lm nn lnm an , 00 222 (,0) (,0) 22 (1 1)sin/(3 1) 3 4sin /2 lm nn lnm an , 00 222 ( ,0)( ,0) 22 (2 1) sin(21)/ 2(61) 12 4sin /2 lm nn lnm an , 00 22 (,0)(,0)2 22 (1 1) sin /3 4sin /2 lm nn ln m an , 00 222 (,0) (,0) 22 (2 1) sin(21)/23(21) 4 4sin /2 lm nn lnm an . For armchair (0 lm:1 ,1 , and 0zz ) tubes, 00 22 (,) (,)2 22 (1 1) (54cos/2)sin/ 12sin/ 2 lm nn nnnln m an , 00 2 (,) (,) 2 22 2 (2 1) (54cos/2)sin (21)/2(21) 4 12sin/ 2 lm nn nn a nl nm n . 00 2 (,) (,) 2 2 2 2 (1 1) (2sin(21)/ 2sin/) 12sin/ 2 lm nn nn a lnln m n , 00 2 (,) (,) 2 22 2 (2 1) (sin(21)/22sin/)(21) 4 12sin/ 2 lm nn nn a lnlnm n , 00 2 (,) (,) 2 2 2 2 (1 1) (2sin(21)/ 2sin/) 12sin/ 2 lm nn nn a lnln m n , 00 2 (,) (,) 2 22 2 (2 1) (sin(21)/ 22sin/)(21) 4 12sin/ 2 lm nn nn a lnlnm n , 00 22 (,) (,)2 22 (1 1) (54cos/ 2)sin/ 12sin/ 2 lm nn nnnln m an , 00 2 (,) (,) 2 22 2 (2 1) (54cos/ 2)sin(21)/ 2(21) 4 12sin/ 2 lm nn nn a nl nm n . 4. Binding Energies in Dependence on Structural Parameter At first, based on the above stated relations and HF val- ues of the atomic electron energy-levels tabulated in [110], the required quasi-classical parameters k r, k , and k for constituent atoms B and N have been calcu- lated. They are given in Tables 1 and 2, respectively. Here the values are shown in atomic units with 7 sig- nificant digits in accordance with the accuracy of input data (HF energies). Such high accuracy is useful in in- terim calculations. As for the final results, they should be expressed in rounded figures with 3 or 4 significant digits (in Å or eV for structure or energy parameters, respec- tively) because it corresponds to the usual experimental errors when determining structure and energy parameters of a substance, and the relative errors of the semi-classical calculations aimed at finding theoretically these parame- ters for polyatomic systems amount to a few percents.  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 237 Table 1. Quasi-classical parameters of step-like radial distributions of electron-charge-density and electric-field-potential in boron atom (in a.u.). k 1 2 3 4 5 k r 2.758476E − 02 5.098016E − 01 7.441219E − 01 4.021346E + 00 4.337060E + 00 k 5.686514E + 04 −3.610951E + 00 −7.342212E − 03 −1.028341E − 02 −2.941197E − 03 k 2.105468E + 02 8.882329E + 00 3.652920E + 00 2.060720E − 01 6.135348E − 04 Table 2. Quasi-classical parameters of step-like radial distributions of electron-charge-density and electric-field-potential in nitrogen atom (in a.u.). k 1 2 3 4 5 k r 9.446222E − 03 3.577244E − 01 5.498034E − 01 2.909074E + 00 3.204489E + 00 k 1.982589E + 06 −1.044967E + 01 −1.939444E − 02 −4.126981E − 02 −2.187537E − 02 k 8.784581E + 02 2.022523E + 01 8.464698E + 00 5.096684E − 01 3.993358E − 03 Using these parameters and expressions for the com- ponents of the quasi-classical molar binding energy and squared interatomic distances in zigzag and armchair BN nanotubes (Figures 1 and 2), their binding energies were calculated versus the structural parameter a with spac- ing of 0.001 a.u., i.e., within the accuracy of 4 significant digits. The vibration energy is assumed to be zero when radicand in its formula becomes negative. In order to carry out these massive calculations, a spe- cial computer code has been designed. Calculations were performed within the range of the structure parameter a which varied from 1 to 13 a.u. with a step 0.001a a.u. (that is quite enough to cover values having any physical sense). The nanotube indexes varied from 1n , covering zigzag and armchair nanotubes up to (18,0) and (10,10), respectively. The radii of the largest calculated species are approximately equal. They are sufficiently large for the tube molar binding energy to almost reach the “saturation” value, which is given by the binding energy of the planar hexagonal BN sheet. In order to make sure that such “saturation” indeed takes place, test species with very large indexes (45,0) and (26,26) (again with approximately equal radii) have also been calcu- lated. Figures 3-6 show () Binding Ea curves (and their trends near the peaks) for some zigzag and armchair BN nano- tubes, respectively. One can see that at sufficiently small interatomic distances binding energy might take a large negative value that implies that the structure is unstable, while at sufficiently large interatomic distances the binding energy always equals to zero which reveals atomization of a structure. As for the intermediate distances, the mo- lar binding energy is positive that is a signature of struc- tural stability. In this case, general trend in binding en- ergy value is decreasing. However, () Binding Ea curves are not monotonous, but with several extremes. Such kind of oscillatory behavior of the molar biding energy of Figure 1. Structure of a zigzag BN nanotube. Figure 2. Structure of an armchair BN nanotube.  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 238 (a) (b) Figure 3. Molar binding energy of zigzag BN nanotubes vs. structural parameter a for different nanotube indexes n. any atomic structure against the inter-atomic distances reflects electron-shell-structure of the constituent atoms (note that the interaction between particles of matter with forces non-monotonously decreasing with distance was foreseen as early as in 18th century by Boscovich [111], whose atomic theory was based only on abstract philoso- phical speculations). Figures 7 and 8 show the binding energies for the two types of achiral nanotubes with the same index n (na- mely (3,0) and (3,3) nanotubes). Let us discuss which of the peaks in these figures cor- respond to the equilibrium structure. The first peak from the right is lower than the successive one. This second peak for all tubes located at 5.085a a.u. (2.691 Å) seems to correspond to the realized stable BN nanotubu- lar structures (the detailed behavior of () Binding Ea curves in its vicinity is shown in Figures 4, 6 and 8). The next peak, even being higher than this, can not been reached kinetically in standard laboratory conditions because they correspond to lower interatomic distances and these two peaks are separated by very deep and sufficiently wide (a) (b) Figure 4. Molar binding energies of zigzag BN nanotubes with different indexes n near the peak a = 2.691 Å. Figure 5. Molar binding energy of armchair BN nanotubes vs structural parameter a for different nanotube indexes n. minima, i.e., by high and wide potential barriers which can be overcome only at ultrahigh temperatures or tun- neled only at ultrahigh pressures. The obtained equilibrium binding energies of BN nano- tubes of both achiral types are summarized in Table 3.  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 239 Figure 6. Molar binding energies of armchair BN nanotubes with different indexes n near the peak a = 2.691 Å. Figure 7. Dependence of molar binding energies of zigzag and armchair BN nanotubes with the nanotube index n = 3 on the structural parameter a. together with their radii calculated from formulas for equilibrium value of the structural parameter 2.691a Å. Thus, the molar binding energies of small-sized nano- tubes, both zigzag and armchair have peaks at (3,0) and (2,2), respectively. Then for large indexes the binding energies decrease toward the same constant value (see Figures 9 and 10). Figure 11 presents the dependence of the molar bind- ing energy of achiral BN nanotubes on their radii R. It reveals pairs of minima at (1,0) and (2,0), and maxima at (1,1) and (3,0), i.e., all the extremes are located in low- radii-region. At higher radii, the molar binding energy slowly decreases to the value of 23.26 eV, which, appar- ently, corresponds to that of the plane hexagonal BN sheet. The obtained dependence () Binding Ea in its domain of monotonicity seems to be quite smooth. It allows us to extrapolate this curve also to chiral BN nanotubes (Figure 12) because the radius of a chiral tube (, )nm R Figure 8. Molar binding energies of zigzag and armchair BN nanotubes with the index n = 3 near the peak a = 2.691 Å. Figure 9. Molar binding energy of zigzag BN nanotubes for different nanotube indexes n at the peak a = 2.691 Å. Figure 10. Molar binding energy of armchair BN nanotubes for different nanotube indexes n at the peak a = 2.691 Å. (0mn ) and radii of the corresponding achiral tubes always meet the condition (,0) (,) (,)nnmnn RR R . The results of the carried out calculations are pre-  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 240 Table 3. Quasi-classically calculated radii and binding en- ergies of BN nanotubes. Nanotube Radius, Å Binding Energy, eV/mole (1,0) 0.673 12.26 (1,1) 0.868 26.08 (2,0) 0.951 23.17 (3,0) 1.345. 29.72 (2,2) 1.537 27.59 (4,0) 1.758 26.40 (5,0) 2.177 24.74 (3,3) 2.260 24.62 (6,0) 2.599 24.19 (4,4) 2.993 23.94 (7,0) 3.023 23.92 (8,0) 3.448 23.76 (5,5) 3.729 23.68 (9,0) 3.874 23.64 (10,0) 4.300 23.57 (6,6) 4.668 23.54 (11,0) 4.727 23.51 (12,0) 5.154 23.47 (7,7) 5.207 23.46 (13,0) 5.581 23.43 (8,8) 5.947 23.41 (14,0) 6.008 23.41 (15,0) 6.436 23.39 (9,9) 6.687 23.37 (16,0) 6.863 23.37 (17,0) 7.291 23.35 (10,10) 7.428 23.35 … … … (45,0) 19.276 23.26 (26,26) 19.290 23.26 sented in Figures 13 and 14 in the form of surface plots where the molar binding energy (,) Binding Ean of zigzag and armchair BN nanotubes is shown as a function of the structural parameter a and the nanotube index n. 5. Zero-Point Vibration Energies First, let us emphasize some features characteristic for the quasi-classical procedure of estimation of the zero-point vibration energies. Figure 11. Molar binding energy of achiral BN nanotubes vs the nanotube radius R. Figure 12. Structure of a chiral BN nanotube. On the one hand, within the above formulated quasi- classical approach, all binding energy maxima are related with the onset of overlapping between certain regions of homogeneity of electric charge density and electric field potential in interacting atoms, constituents of the struc- ture under the consideration. Namely, equilibrium point at 5.085a a.u. corresponds to the B–N bond length of 2.937d a.u. which is a sum of radii B 10.028r a.u. and N 42.909r a.u. (Tables 1 and 2). On the other hand, quasi-classical expression of the vibration energy is based on the parabolic approximation of the () Binding Ea curve and formula for the volume of the intersection of two spheres, 12 12 (, ,)VR RD, which is a continuously differentiable function of the inter-central distance 12 D. However, one can see readily from ex- pression of its first (continuous) derivative that second derivative is not a continuous function. That is the reason why the parabolic approximation of the () Binding Ea curve in the immediate vicinity of the 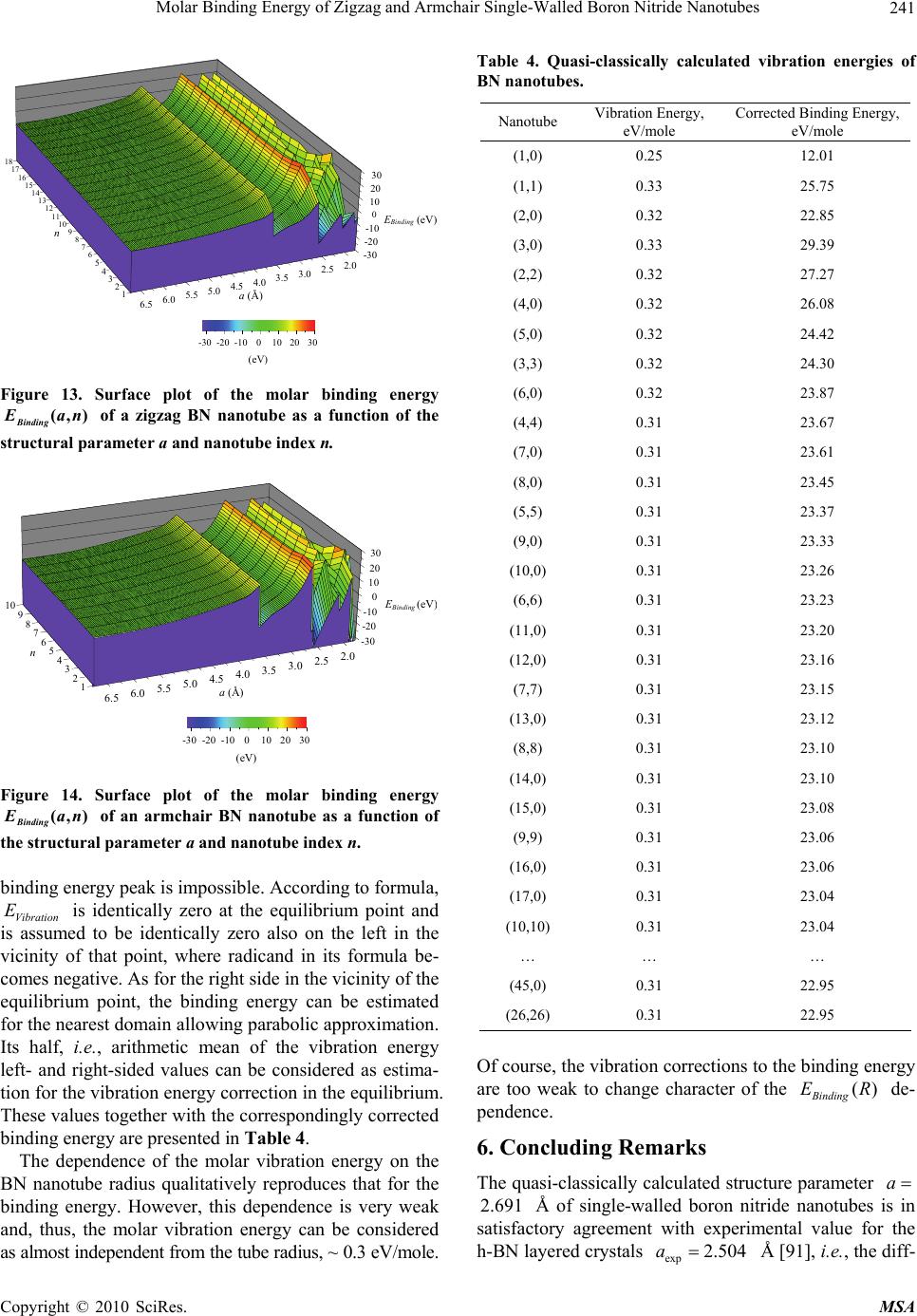 Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 241 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 n 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 a (Å) -30 -20 -10 0 10 20 30 EBind ing (eV ) -30 -20 -10 0 10 20 30 ( e V ) Figure 13. Surface plot of the molar binding energy (,) Binding E an of a zigzag BN nanotube as a function of the structural parameter a and nanotube index n. 1 2 3 4 5 6 7 8 9 10 n 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 a (Å) -30 -20 -10 0 10 20 30 E Binding (eV ) -30 -20 -100 10 20 30 (eV) Figure 14. Surface plot of the molar binding energy (,) Binding E an of an armchair BN nanotube as a function of the structural parameter a and nanotube index n. binding energy peak is impossible. According to formula, Vibration E is identically zero at the equilibrium point and is assumed to be identically zero also on the left in the vicinity of that point, where radicand in its formula be- comes negative. As for the right side in the vicinity of the equilibrium point, the binding energy can be estimated for the nearest domain allowing parabolic approximation. Its half, i.e., arithmetic mean of the vibration energy left- and right-sided values can be considered as estima- tion for the vibration energy correction in the equilibrium. These values together with the correspondingly corrected binding energy are presented in Table 4. The dependence of the molar vibration energy on the BN nanotube radius qualitatively reproduces that for the binding energy. However, this dependence is very weak and, thus, the molar vibration energy can be considered as almost independent from the tube radius, ~ 0.3 eV/mole. Table 4. Quasi-classically calculated vibration energies of BN nanotubes. Nanotube Vibration Energy, eV/mole Corrected Binding Energy, eV/mole (1,0) 0.25 12.01 (1,1) 0.33 25.75 (2,0) 0.32 22.85 (3,0) 0.33 29.39 (2,2) 0.32 27.27 (4,0) 0.32 26.08 (5,0) 0.32 24.42 (3,3) 0.32 24.30 (6,0) 0.32 23.87 (4,4) 0.31 23.67 (7,0) 0.31 23.61 (8,0) 0.31 23.45 (5,5) 0.31 23.37 (9,0) 0.31 23.33 (10,0) 0.31 23.26 (6,6) 0.31 23.23 (11,0) 0.31 23.20 (12,0) 0.31 23.16 (7,7) 0.31 23.15 (13,0) 0.31 23.12 (8,8) 0.31 23.10 (14,0) 0.31 23.10 (15,0) 0.31 23.08 (9,9) 0.31 23.06 (16,0) 0.31 23.06 (17,0) 0.31 23.04 (10,10) 0.31 23.04 … … … (45,0) 0.31 22.95 (26,26) 0.31 22.95 Of course, the vibration corrections to the binding energy are too weak to change character of the () Binding ER de- pendence. 6. Concluding Remarks The quasi-classically calculated structure parameter a 2.691 Å of single-walled boron nitride nanotubes is in satisfactory agreement with experimental value for the h-BN layered crystals exp 2.504a Å [91], i.e., the diff-  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 242 erence is about 7%. As is mentioned above the overesti- mations in the structural parameter is characteristic for the quasi-classical approach. However, at least partially, this overestimation seems to be related with expansion of lattice of the single hexagonal layer (plane or cylindrical) if compared with that of the three-dimensional layered crystal. It is also instructive to analyze the obtained spread of the molar zero-point vibration energy of BN nanotubes (0.25-0.33) eV and, in particular, its limit for ultra-large- radius tubes 0.31 eV using the data available on the vi- bration characteristics of h-BN layered crystal. The vi- bration energies of an isolated tubular layer and layered crystals can be directly compared as the atoms of the low-dimensional system can execute vibrations in three independent directions in physical space. Our quasi-classical estimations made for BN nanotubes agree well with ana- logous calculations (but in tight-binding approximation) for BN plane sheet of 0.27, the semiempirical estimate of 0.23 for zero-point vibrations energy in h-BN, and the estimate of 0.35 eV/mole from the theoretical phonon spectrum (see Subsection 2.2). We have found that the binding energies of BN sin- gle-walled nanotubes corrected with zero-point vibration energies lies within the interval (12.01-29.39) eV. In par- ticular, the calculated corrected binding energy of the ultra-large-radius tube is predicted as 22.95 eV. Previous quasi-classical calculations (but in tight-binding appro- ximation) performed for BN isolated plane sheet have given the binding energy 23.00 eV/mole, which coin- cides in order of magnitude with this interval and agrees very well with present result obtained for large tubes. As it was demonstrated in Subsection 2.2, for its part the binding energy ~23 eV/mole for single-layer boron ni- tride structures should be in good agreement with bind- ing energy data available for BN multi-layered structures. Summarizing the obtained results, it should be empha- sized that a complex dependence of the BN nanotube molar binding energy on its radius is found out, though all the binding energy values are found to be positive, i.e., all tubes should be stable. However, they have rather different degrees of stability. On the one hand, ultra-small-radius BN nanotubes (1,0) and (2,0) seem to be meta-stable, though their molar binding energies are positive, they are less than that for isolated hexagonal boron nitride layer. Especially the smallest (1,0) tube structure degenerated in zigzag ato- mic strip should be meta-stable because its binding en- ergy is only about half of this value. Such a structure can be realized only as an inner wall in a multi-walled tube. On the other hand, the formation probabilities for BN tubes with indexes (1,1), (3,0), and (4,0) should exceed that for isolated sheet. Among them the (3,0) tube is well pronounced, formation of which is predicted to be ener- getically most preferable than the layer growth. Molar binding energies for other BN nanotubes slightly exceed that of sheet and their formation probabilities should be almost same as for layered crystal growth. Finally, it should be noted that, in addition to the en- ergy considerations concerning the relative stability of tubular structure, it is also necessary to take into account features characteristic to BN nanosystems, in view of the general equations derived for energy fluctuations of small completely open (incompressible) systems [112]. They show that the fluctuations should be unusually large because there are no constraints on the size of a system and, in addition, the fluctuations of the total or partial number of atoms in binary systems indirectly con- tribute to the fluctuations in their energy. 7. Acknowledgements L. Chkhartishvili acknowledges the financial support from the Georgia National Science Foundation (GNSF) under the Project # GNSF/ST 08/4-411-Geometry of the boron nitride nanostructures. REFERENCES [1] A. Rubio, J. L. Corkill and M. L. Cohen, “Theory of Graphitic Boron Nitride Nanotubes,” Physical Review B, Vol. 49, No. 7, 1994, pp. 5081-5084. [2] Z. Weng-Sieh, K. Cherrey, N. G. Chopra, X. Blasé, Y. Miyamoto, A. Rubio, M. L. Cohen, S. G. Louie, A. Zettl and R. Gronsky, “Synthesis of BxCyNz Nanotubules,” Physical Review B, Vol. 51, No. 16, 1995, pp. 11229- 11232. [3] N. G. Chopra, R. J. Luyken, K. Cherrey, V. H. Crespi, M. L. Cohen, S. G. Louie and A. Zettl, “Boron-Nitride Nano- tubes,” Science, Vol. 269, No. 5226, 1995, pp. 966-967. [4] K. Suenaga, C. Colliex, N. Demoncy, A. Loieseau, H. Pascard and F. Willaime, “Synthesis of Nanoparticles and Nanotubes with Well-Separated Layers of Boron Nitride and Carbon,” Science, Vol. 278, No. 5338, 1997, pp. 653- 655. [5] K. Suenaga, F. Willaime, A. Loieseau and C. Colliex. “Organization of Carbon and Boron Nitride Layers in Mixed Nanoparticles and Nanotubes Synthesized by Arc Discharge,” Applied Physics A, Vol. 68, No. 3, 1999, pp. 301-308. [6] A. Loiseau, F. Willaime, N. Demoncy, G. Hug and H. Pascard, “Boron Nitride Nanotubes with Reduced Num- bers of Layers Synthesized by Arc Discharge,” Physical Review Letters, Vol. 76, No. 25, 1996, pp. 4737-4740. [7] Y. Saito, M. Maida and T. Matsumoto, “Structures of Boron Nitride Nanotubes with Single-Layer and Multi- Layers Produced by Arc Discharge,” Japanese Journal of Applied Physics, Vol. 38, No. 1A, 1999, pp. 159-163. [8] Y. Saito and M. Maida, “Square, Pentagon, and Heptagon  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 243 rings at BN Nanotube Tips,” Journal of Physical Chem- istry A, Vol. 103, No. 10, 1999, pp. 1291-1293. [9] D. Golberg, Y. Bando, M. Eremets, K. Takemura, K. Kurashima and H. Yusa “Nanotubes in Boron Nitride Laser Heated at High Pressure,” Applied Physics Letters, Vol. 69, No. 14, 1996, pp. 2045-2047. [10] R. Sen, B. C. Satishkumar, A. Govindaraj, K. R. Hariku- mar, G. Raina, J.-P. Zhang, A. K. Cheetham and C. N. R. Rao, “B-C-N, C-N and B–N Nanotubes Produced by the Pyrolysis of Precursor Molecules over Co Catalysts,” Chemical Physics Letters, Vol. 287, No. 5-6, 1998, pp. 671-676. [11] D. Golberg, Y. Bando, W. Han, K. Kurashima and T. Sato, “Single-Walled B-Doped Carbon, B/N-Doped Car- bon, and BN Nanotubes Synthesized from Single-Walled Carbon Nanotubes through a Substitution Reaction,” Chemical Physics Letters, Vol. 308, No. 3-4, 1999, pp. 337-342. [12] D. Golberg, W. Han, Y. Bando, L. Bourgeois, K. Kura- shima and T. Sato, “Fine Structure of Boron Nitride Nanotubes Produced from Carbon Nanotubes by a Sub- stitution Reaction,” Journal of Applied Physics, Vol. 86, No. 4, 1999, pp. 2364-2366. [13] Y. Chen, L. T. Chadderton, J. F. Gerald and J. S. Wil- liams, “A Solid-State Process for Formation of Boron Ni- tride Nanotubes,” Applied Physics Letters, Vol. 74, No. 20, 1999, pp. 2960-2962. [14] T. S. Bartnitskaya, G. S. Oleinik, V. V. Pokropivnyi, N. V. Danilenko, V. M. Vershchaka and A. V. Kotko, “Na- notubes from Graphite-Like BN,” Superhard Materials, No. 6, 1998, pp. 71-74. [15] T. S. Bartnitskaya, G. S. Oleinik, A. V. Pokropivnyi and V. V. Pokropivnyi, “Synthesis, Structure, and Formation Mechanism of Boron Nitride Nanotubes,” JEPT Letters, Vol. 69, No. 2, 1999, pp. 163-168. [16] C. Colazo-Davila, E. Bengu, L. D. Marks and M. Kirk, “Nucleation of Cubic Boron Nitride Thin Films,” Dia- mond Related Materials, Vol. 8, No. 6, 1999, pp. 1091- 1100. [17] T. Oku, T. Hirano, M. Kuno, T. Kusunose, K. Niihara and K. Suganuma, “Synthesis, Atomic Structures, and Proper- ties of Carbon and Boron Nitride Fullerene Materials,” Materials Science and Engineering B, Vol. 74, No. 1-3, 2000, pp. 206-217. [18] J. Wang, V. K. Kayastha, Y. K. Yap, Z. Fan, J. G. Lu, Z. Pan, I. N. Ivanov, A. A. Puretzky and D. B. Geohegan, “Low Temperature Growth of Boron Nitride Nanotubes on Substrates,” Nano Letters, Vol. 5, No. 12, 2005, pp. 2528-2532. [19] L. L. Sartinska, S. Barchikovski, N. Wagenda, B. M. Rud’ and I. I. Timofeeva, “Laser Induced Modification of Surface Structures,” Applied Surface Science, Vol. 253, No. 9, 2007, pp. 4295-4299. [20] L. L. Sartinska, A. A. Frolov, A. Yu. Koval’, N. A. Danilenko, I. I. Timofeeva and B. M. Rud’, “Transforma- tion of Fine-Grained Graphite-Like Boron Nitride In- duced by Concentrated Light Energy,” Materials Chem- istry and Physics, Vol. 109, No. 1, 2008, pp. 20-25. [21] M. V. P. Altoe, J. P. Sprunck, J.-C. P. Gabriel and K. Bradley, “Nanococoon Seeds for BN Nanotube Growth,” Journal of Materials Science, Vol. 38, No. 24, 2003, pp. 4805-4810. [22] M. W. Smith, K. C. Jordan, C. Park, J.-W. Kim, P. T. Lillehei, R. Crooks and J. S. Harrison, “Very Long Sin- gle- and Few-Walled Boron Nitride Nanotubes via the Pressurized Vapor/Condenser Method,” Nanotechnology, Vol. 20, No. 50, 2009, pp. 505604-505610. [23] X. Blasé, J.-C. Charlier, A. de Vita and R. Car, “Theory of Composite BxCyNz Nanotube Heterojunctions,” Ap- plied Physics Letters, Vol. 70, No. 2, 1997, pp. 197-199. [24] X. Blasé, J.-C. Charlier, A. de Vita and R. Car, “Struc- tural and Electrical Properties of Composite BxCyNz Nanotubes and Heterojunctions,” Applied Physics A, Vol. 68, No. 3, 1999, pp. 293-300. [25] V. V. Pokropivnyj, V. V. Skorokhod, G. S. Oleinik, A. V. Kurdyumov, T. S. Bartnitskaya, A. V. Pokropivnyj, A. G. Sisonyuk and D. M. Sheichenko, “Boron Nitride Analogs of Fullerenes (the Fulborenes), Nanotubes, and Fullerites (the Fulborenites),” Journal of Solid State Chemistry, Vol. 154, No. 1, 2000, pp. 214-215. [26] J. H. Lee, “A Study on a Boron-Nitride Nanotube as a Gigahertz Oscillator,” The Journal of the Korean Physi- cal Society, Vol. 49, No. 1, 2006, pp. 172-176. [27] V. Verma and K. Dharamvir, “BNNT in Contact with h-BN Sheet and Other BNNT and DW-BNNT as GHz Oscillator,” International Journal of Nanosystems, Vol. 1, No. 1, 2008, pp. 27-34. [28] B. Baumeier, P. Krüger and J. Pollmann, “Structural, Elastic, and Electronic Properties of SiC, BN, and BeO Nanotubes,” Physical Review B, Vol. 76, 2007, p. 085407. [29] Y.-H. Kim, K. J. Chang and S. G. Louie, “Electronic Structure of Radially Deformed BN and BC3 Nanotubes,” Physical Review B, Vol. 63, 2001, p. 205408. [30] G. Y. Guo and J. C. Lin, “Second-Harmonic Generation and Linear Electro-Optical Coefficients of BN Nano- tubes,” Physical Review B, Vol. 72, 2005, p. 075416. [31] G. Y. Guo and J. C. Lin, “Erratum: Second-Harmonic Generation and Linear Electro-Optical Coefficients of BN Nanotubes,” Physical Review B, Vol. 77, 2008, p. 049901 (E). [32] C. Zhi, Y. Bando, C. Tang and D. Golberg, “Engineering of Electronic Structure of Boron-Nitride Nanotubes by Covalent Functionalization,” Physical Review B, Vol. 74, 2006, p. 153413. [33] C. Zhi, Y. Bando, C. Tang and D. Golberg, “Publisher’s Note: Engineering of Electronic Structure of Boron-Ni- tride Nanotubes by Covalent Functionalization,” Physical Review B, Vol. 74, 2006, p. 199902 (E). [34] W.-Q. Han, C. W. Chang and A. Zettl, “Encapsulation of One-Dimensional Potassium Halide Crystals within BN Nanotubes,” Nano Letters, Vol. 4, No. 7, 2004, pp. 1355- 1357. [35] K. Yum and M.-F. Yu, “Measurement of Wetting Proper-  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 244 ties of Individual Boron Nitride Nanotubes with the Wil- helmy Method Using a Nanotube-Based Force Sensor,” Nano Letters, Vol. 6, No. 2, 2006, pp. 329-333. [36] S. A. Shevlin and Z. X. Guo, “Hydrogen Sorption in De- fective Hexagonal BN Sheets and BN Nanotubes,” Physi- cal Review B, Vol. 76, 2007, p. 024104. [37] E. Durgun, Y.-R. Jang and S. Ciraci, “Hydrogen Storage Capacity of Ti-Doped Boron-Nitride and B/Be-Substitut- ed Carbon Nanotubes,” Physical Review B, Vol. 76, 2007, p. 073413. [38] C. Tang, Y. Bando, X. Ding, S. Qi and D. Golberg, “Catalyzed Collapse and Enhanced Hydrogen Storage of BN Nanotubes,” Journal of the American Chemical Soci- ety, Vol. 124, No. 49, 2002, pp. 14550-14551. [39] C. Zhi, Y. Bando, C. Tang and D. Golberg. “Immobiliza- tion of Proteins on Boron Nitride Nanotubes,” Journal of the American Chemical Society, Vol. 127, No. 49, 2005, pp. 17144-17145. [40] C. Zhi, Y. Bando, C. Tang, R. Xie, T. Sekiguchi and D. Golberg, “Perfectly Dissolved Boron Nitride Nanotubes Due to Polymer Wrapping,” Journal of the American Chemical Society, Vol. 127, No. 46, 2005, pp. 15996- 15997. [41] M. Côté, P. D. Haynes and C. Molteni, “Boron Nitride Polymers: Building Blocks for Organic Electronic De- vices,” Physical Review B, Vol. 63, 2001, p. 125207. [42] M. Ishigami, J. D. Sau, Sh. Aloni, M. L. Cohen and A. Zettl, “Observation of the Giant Stark Effect in Bo- ron-Nitride Nanotubes,” Physical Review Letters, Vol. 94, 2005, p. 056804. [43] J. Cumings and A. Zettl, “Mass-Production of Boron Nitride Double-Wall Nanotubes and Nanococones,” Chemical Physics Letters, Vol. 316, No. 3-4, 2000, pp. 211-216. [44] Y. Chen, J. Zou, S. J. Campbell and G. le Caer, “Boron Nitride Nanotubes: Pronounced Resistance to Oxidation,” Applied Physics Letters, Vol. 84, No. 13, 2004, pp. 2430- 2432. [45] E. Bengu and L. D. Marks, “Single-Walled BN Nanos- tructures,” Physical Review Letters, Vol. 86, No. 11, 2001, pp. 2385-2387. [46] C. Tang, Y. Bando, Y. Huang, S. Yue, C. Gu, F. F. Xu and D. Golberg, “Fluorination and Electrical Conductivity of BN Nanotubes,” Journal of the American Chemical Society, Vol. 127, No. 18, 2005, pp. 6552-6553. [47] E. J. Mele and P. Král, “Electric Polarization of Heter- opolar Nanotubes as a Geometric Phase,” Physical Re- view Letters, Vol. 88, 2002, p. 056803. [48] N. Sai and E. J. Mele, “Microscopic Theory for Nanotube Piezoelectricity,” Physical Review B, Vol. 68, 2003, p. 241405. [49] S. M. Nakhmanson, A. Calzolari, V. Meunier, J. Bernholc and M. Buongiorno Nardelli, “Spontaneous Polarization and Piezoelectricity in Boron Nitride Nanotubes,” Physi- cal Review B, Vol. 67, 2003, p. 235406. [50] N. G. Lebedev and L. A. Chernozatonskiĭ, “Quantum- Chemical Calculations of the Piezoelectric Characteristics of Boron Nitride and Carbon Nanotubes,” Physics of the Solid State, Vol. 48, No. 10, 2006, pp. 2028-2034. [51] Y. Zhang, K. Suenaga, C. Colliex and S. Iijima, “Coaxial Nanocable: Silicon Carbide and Silicon Oxide Sheathed with Boron Nitride and Carbon,” Science, Vol. 281, No. 5379, 1998, pp. 973-975. [52] A. Zobelli, A. Gloter, C. P. Ewels, G. Seifert and C. Col- liex, “Electron Knock-on Cross Section of Carbon and Boron Nitride Nanotubes,” Physical Review B, Vol. 75, 2007, p. 245402. [53] I. V. Weiz, “Supplement II,” In: K.-P. Huber and G. Herzberg, Constants of Diatomic Molecules, Part 2: Molecules N2–ZrO, Mir, Moscow, 1984, pp. 295-366. [54] O. I. Bukhtyarov, S. P. Kurlov and B. M. Lipenskikh, “Calculation of the Structure and Physicochemical Prop- erties of B-Based Systems by the Computer Modeling Method,” In: Abstracts of the 8th International Sympo- sium on Boron, Borides, Carbides, Nitrides, and Related Compounds, Tbilisi, October 1984, pp. 135-136. [55] J. L. Masse and M. Bärlocher, “Etude par la Méthode du Autocohérent (Self-Consistent) de la Molécule BN,” Helvetica Chimia Acta, Vol. 47, No. 1, 1964, pp. 314- 318. [56] Y. G. Khajt and V. I. Baranovskij, “Ab Initio Calcula- tions of the BN Molecule,” Journal of Structural Chem- istry, Vol. 21, No. 1, 1980, pp. 153-154. [57] H. Bredohl, J. Dubois, Y. Houbrechts and P. Nzo- habonayo, “The Singlet Bands of BN,” Journal of Phys- ics B, Vol. 17, No. 1, 1984, pp. 95-98. [58] C. M. Marian, M. Gastreich and J. D. Gale, “Empirical Two-Body Potential for Solid Silicon Nitride, Boron Ni- tride, and Borosilazane Modifications,” Physical Review B, Vol. 62, No. 5, 2000, pp. 3117-3124. [59] L. Chkhartishvili, “Quasi-Classical Analysis of Boron- Nitrogen Binding,” In: Proceedings of the 2nd Interna- tional Boron Symposium, Eskişehir, Turkey, September 2004, pp. 165-171. [60] L. Chkhartishvili, D. Lezhava, O. Tsagareishvili and D. Gulua, “Ground State Parameters of B2, BC, BN, and BO Diatomic Molecules,” Transactions of the AMIAG, Vol. 1, 1999, pp. 295-300. [61] L. Chkhartishvili, D. Lezhava and O. Tsagareishvili, “Quasi-Classical Determination of Electronic Energies and Vibration Frequencies in Boron Compounds,” Jour- nal of Solid State Chemistry, Vol. 154, No. 1, 2000, pp. 148-152. [62] A. Gaydon, “Dissociation Energies and Spectra of Dia- tomic Molecules,” Chapman & Hall, London, 1947. [63] K. P. Huber and G. Herzberg, “Molecular Spectra and Molecular Structure: IV. Constants of Diatomic Mole- cules,” van Nostrand Reinhold Co, New York, 1979. [64] M. Lorenz, J. Agreiter, A. M. Smith and V. E. Bondybey, “Electronic Structure of Diatomic Boron Nitride,” Jour- nal of Chemical Physics, Vol. 104, No. 8, 1996, pp. 3143- 3146.  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 245 [65] L. Chkhartishvili, “Zero-Point Vibration Energy within Quasi-Classical Approximation: Boron Nitrides,” Pro- ceedings of the TSU (Physics), Vol. 40, 2006, pp. 130-138. [66] V. I. Vedeneev, L. V. Gurvich, V. N. Kondrat’ev, V. A. Medvedev and E. A. Frankevich, “Chemical Bonds Breaking Energies, Ionization Potentials and Electron Af- finities,” Academy of Sciences of USSR, Moscow, 1962. [67] G. Meloni, M. Sai Baba and K. A. Gingerich, “Knudsen Cell Mass Spectrometric Investigation of the B2N Mole- cule,” Journal of Chemical Physics, Vol. 113, No. 20, 2000, pp. 8995-8999. [68] R. Vanderbosch, “Gas-Phase Anions Containing B and N,” Physical Review A, Vol. 67, 2003, p. 013203. [69] S. V. Lisenkov, G. A. Vinogradov, T. Y. Astakhova and N. G. Lebedev, “Non-Spiral ‘Haeckelite-Type’ BN-Nano- tubes,” JETP Letters, Vol. 81, No. 7, 2005, pp. 431-436. [70] A. Zunger, “A Molecular Calculation of Electronic Prop- erties of Layered Crystals: I. Truncated Crystal Approach for Hexagonal Boron Nitride,” Journal of Physics C, Vol. 7, No. 1, 1974, pp. 76-95. [71] A. Zunger, “A Molecular Calculation of Electronic Prop- erties of Layered Crystals: II. Periodic Small Cluster Calculation for Graphite and Boron Nitride,” Journal of Physics C, Vol. 7, No. 1, 1974, pp. 96-106. [72] D. L. Strout, “Structure and Stability of Boron Nitrides: Isomers of B12N12,” Journal of Physical Chemistry A, Vol. 104, No. 15, 2000, pp. 3364-3366. [73] L. S. Chkhartishvili, “Quasi-Classical Estimates of the Lattice Constant and Band Gap of a Crystal: Two-Di- mensional Boron Nitride,” Physics of the Solid State, Vol. 46, No. 11, 2004, pp. 2126-2133. [74] L. S. Chkhartishvili, “Analytical Optimization of the Lat- tice Parameter Using the Binding Energy Calculated in the Quasi-Classical Approximation,” Physics of the Solid State, Vol. 48, No. 5, 2006, pp. 846-853. [75] L. Chkhartishvili and D. Lezhava, “Zero-Point Vibration Effect on Crystal Binding Energy: Quasi-Classical Cal- culation for Layered Boron Nitride,” Transactions of the GTU, Vol. 439, 2001, pp. 87-90. [76] K.-H. Hellwege and O. von Madelung, Eds., “Boron Ni- tride (BN),” In: Landolt-Börnstein, Numerical Data and Functional Relationships in Science and Technology, New Series, Group III: Crystal and Solid State Physics, Volume 17: Semiconductors, Subvolume g: Physics of Non-Tetrahedrally Bonded Binary Compounds III, Springer-Verlag, Berlin, 1982. [77] Y.-N. Xu and W. Y. Ching, “Calculation of Ground-State and Optical Properties of Boron Nitrides in the Hexagonal, Cubic, and Wurtzite Structures,” Physical Review B, Vol. 44-I, No. 15, 1991, pp. 7784-7798. [78] K. Albe, “Theoretical Study of Boron Nitride Modifica- tions at Hydrostatic Pressures,” Physical Review B, Vol. 55-II, No. 10, 1997, pp. 6203-6210. [79] D. D. Wagmann, W. H. Evans, V. B. Parker, J. Halow, S. M. Bairly and R. H. Shumn, Eds., Selected Values of Chemical Thermodynamic Properties, National Bureau of Standards, Washington, 1968. [80] S. N. Grinyaev, F. V. Konusov and V. V. Lopatin, “Deep Levels of Nitrogen Vacancy Complexes in Graphite-Like Boron Nitride,” Physics of the Solid State, Vol. 44, No. 2, 2002, pp. 286-293. [81] M. S. C. Mazzoni, R. W. Nunes, S. Azevedo and H. Chacham, “Electronic Structure and Energetics of BxCyNz Layered Structures,” Physical Review B, Vol. 73, 2006, p. 073108. [82] S. V. Lisenkov, G. A. Vonogradov, T. Y. Astakhova and N. G. Lebedev, “Geometric Structure and Electronic Properties of Planar and Nanotubular BN Structures of the Haeckelite Type,” Physics of the Solid State, Vol. 48, No. 1, 2006, pp. 192-198. [83] O. E. Alon, “Symmetry Properties of Single-Walled Bo- ron Nitride Nanotubes,” Physical Review B, Vol. 64, 2001, p. 153408. [84] V. V. Pokropivnyi, “Non-Carbon Nanotubes (Review). 2. Types and Structure,” Powder Metallurgy and Metal Ce- ramics, Vol. 40, No. 11-12, 2002, pp. 582-594. [85] M. Menon and D. Srivastava, “Structure of Boron Nitride Nanotubes: Tube Closing Versus Chirality,” Chemical Physics Letters, Vol. 307, No. 5-6, 1999, pp. 407-412. [86] J.-C. Charlier, X. Blase, A. de Vita and R. Car, “Micro- scopic Growth Mechanisms for Carbon and Boron-Ni- tride Nanotubes,” Applied Physics A, Vol. 68, No. 3, 1999, pp. 267-273. [87] L. S. Chkhartishvili, “On Sizes of Boron Nitride Nano- tubes,” In: Proceedings of the 18th International Sympo- sium ‘Thin Films in Optics in Nanoelectronics’, Kharkiv, October 2006, pp. 367-373. [88] Z. Peralta–Inga, P. Lane, J. S. Murray, S. Boyd, M. E. Grice, C. J. O’Connor and P. Politzer, “Characterization of Surface Electrostatic Potentials of Some (5,5) and (n,1) Carbon and Boron/Nitrogen Model Nanotubes,” Nano Letters, Vol. 3, No. 1, 2003, pp. 21-28. [89] D. Golberg, Y. Bando, L. Bourgeois, K. Kurashima and T. Sato, “Insights into the Structure of BN Nanotubes,” Ap- plied Physics Letters, Vol. 77, No. 13, 2000, pp. 1979- 1981. [90] S. Okada, S. Saito and A. Oshiyama, “Inter-Wall Interac- tion and Electronic Structure of Double-Walled BN Nanotubes,” Physical Review B, Vol. 65, No. 16, 2002, p. 165410. [91] Y. B. Kuz’ma and N. F. Chaban, “Boron Containing Bi- nary and Ternary Systems: Handbook,” Metallurgiya, Moscow, 1990. [92] E. Hernández, C. Goze, P. Bernier and A. Rubio, “Elastic properties of C and BxCyNz Composite Nanotubes,” Physical Review Letters, Vol. 80, No. 20, 1998, pp. 4502- 4505. [93] H. J. Xiang, J. Yang, J. G. Hou and Q. Zhu, “First-Prin- ciples Study of Small-Radius Single-Walled BN Nano- tubes,” Physical Review B, Vol. 68, 2003, p. 035427. [94] L. Wirtz, A. Rubio, R. A. de la Concha and A. Loiseau, “Ab initio Calculations of the Lattice Dynamics of Boron  Molar Binding Energy of Zigzag and Armchair Single-Walled Boron Nitride Nanotubes Copyright © 2010 SciRes. MSA 246 Nitride Nanotubes,” Physical Review B, Vol. 68, 2003, p. 045425. [95] L. Wirtz, M. Lazzeri, F. Mauri and A. Rubio, “Raman Spectra of BN Nanotubes: Ab Initio and Bond-Polari- zability Model Calculations,” Physical Review B, Vol. 71, 2005, p. 241402 (R). [96] H. F. Bettinger, T. Dumitrică, G. E. Scuseria and B. I. Yakobson, “Mechanically Induced Defects and Strength of BN Nanotubes,” Physical Review B, Vol. 65, 2002, p. 041406. [97] Y. Miyamoto, A. Rubio, S. Berber, M. Yoon and D. To- mánek, “Spectroscopic Characterization of Stone–Wales Defects in Nanotubes,” Physical Review B, Vol. 69, 2004, p. 121413 (R). [98] G. Y. Gou, B. C. Pan and L. Shi, “Theoretical Study of Size-Dependent Properties of BN Nanotubes with Intrin- sic Defects,” Physical Review B, Vol. 76, 2007, p. 155414. [99] E. Hernandez, C. Goze, P. Bernier and A. Rubio, “Elastic Properties of Single-Wall Nanotubes,” Applied Physics A, Vol. 68, No. 3, 1999, pp. 287-292. [100] D. Srivastava, M. Menon and K.-J. Cho, “Anisotropic Nanomechanics of Boron Nitride Nanotubes: Nanostruc- tured “Skin” Effect,” Physical Review B, Vol. 63, 2001, p. 195413. [101] T. Dumitrică and B. I. Yakobson, “Rate Theory of Yield in Boron Nitride Nanotubes,” Physical Review B, Vol. 72, 2005, p. 035418. [102] L. Chkhartishvili, “Quasi-Classical Theory of Substance Ground State,” Technical University Press, Tbilisi, 2004. [103] L. Chkhartishvili, “Quasi-Classical Method of Calcula- tion of Substance Structural and Electronic Energy Spec- trum Parameters,” Tbilisi University Press, Tbilisi, 2006. [104] L. Chkhartishvili, “Selection of Equilibrium Configura- tions for Crystalline and Molecular Structures Based on Quasi-Classical Inter-Atomic Potential,” Transactions of the GTU, No. 427, 1999, pp. 13-19. [105] L. Chkhartishvili, “On Quasi-Classical Estimations of Boron Nanotubes Ground-State Parameters,” Journal of Physics: Conference Series, Vol. 176, 2009, p. 012013. [106] L. Chkhartishvili, “Molar Binding Energy of the Boron Nanosystems,” In: Proceedings of the 4th International Boron Symposium, Osmangazi University Press, Eskişe- hir, Turkey, 2009, pp. 153-160. [107] L. Chkhartishvili, “Ground State Parameters of Wurtzite Boron Nitride: Quasi-Classical Estimations,” In: Pro- ceedings of the 1st International Boron Symposium, Dumlupinar University Press, Kütahya, 2002, pp. 139- 143. [108] L. Chkhartishvili, “Boron Nitride Nanosystems of Regu- lar Geometry,” Journal of Physics: Conference Series, Vol. 176, 2009, p. 012014. [109] L. S. Chkhartishvili, “Equilibrium Geometry of Ultra- Small-Radius Boron Nitride Nanotubes,” Material Sci- ence of Nanostructures, No. 1, 2009, pp. 33-44. [110] Ch. Froese–Fischer, “The Hartree–Fock Method for At- oms: A Numerical Approach,” Wiley, New York, 1977. [111] R. J. Boscovich, “Philosophiae Naturalis Theoria Redacta ad Inicam Legem Vitium in Natura Existentiam,” Colle- gio Romano, Milano, 1758. [112] T. L Hill and R. V. Chamberlin, “Fluctuations in Energy in Completely Open Small Systems,” Nano Letters, Vol. 2, No. 6, 2002, pp. 609-613. |