 Open Journal of Cell Biology, 2012, 2, 21-31 doi:10.4236/ojcb.2012.22002 Published Online December 2012 (http://www.SciRP.org/journal/ojcb) Helicobacter pylori Induction in Gastric Mucosal Prostaglandin and Nitric Oxide Generation Is Dependent on MAPK/ERK-Mediated Activation of IKK-β and cPLA2: Modulatory Effect of Ghrelin Bronislaw L. Slomiany, Amalia Slomiany Research Center, University of Medicine and Dentistry of New Jersey, Newark, USA Email: slomiabr@umdnj.edu Received July 17, 2012; revised August 25, 2012; accepted September 9, 2012 ABSTRACT Among the key factors defining the extent of gastric mucosal inflammatory involvement in response to H. pylori is the excessive generation of prostaglandin (PGE2) and nitric oxide (NO), caused by the overexpression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), and triggered by the activation of MAPK/JNK, p38 and ERK, and nuclear translocation of the cognate transcription factors. In this study, we report on the role of MAPK/ERK in the regulation of H. pylori LPS-induced gastric mucosal expression of COX-2 and iNOS. We show that ERK activation by the LPS leads to phosphorylation of the inhibitory κB kinase-β (IKK-β) and cytosolic phospholipase A2 (cPLA2), and is reflected in the upsurge in NF-κB nuclear translocation, induction in COX-2 and iNOS expression, and up-regulation in cPLA2 activity. The modulatory effect of peptide hormone, ghrelin, on the LPS-induced changes, although associated with further enhancement in ERK, IKK-β and cPLA2 phosphorylation, was reflected in the suppression of IKK-β and cPLA2 activity through S-nitrosylation. While the effect of ghrelin on S-nitrosylation was susceptible to suppression by the inhibitors of Src/Akt pathway, the inhibition of ERK activation caused the blockage in IKK-β and cPLA2 phos- phorylation as well as S-nitrosylation. Taken together, our data show that H. pylori-induced ERK activation plays a critical role in up-regulation of gastric mucosal PGE2 and NO generation at the level of IKK-β and cPLA2 activation, and that ghrelin counters these proinflammatory consequences of the LPS through Src/Akt-dependent S-nitrosylation. Keywords: H. pylori; Gastric Mucosa; Ghrelin; PGE2; NO; COX-2; iNOS; ERK; IKKβ; cPLA2; S-Nitrosylation 1. Introduction Infection with Helicobacter pylori is well-establish risk factor in etiology of gastric disease, and the excessive generation of prostaglandin (PGE2) and nitric oxide (NO) caused by the overexpression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) are consid- ered of primary importance in defining the extent of gas- tric mucosal inflammatory involvement [1-6]. The sig- naling events underlying the up-regulation in PGE2 and NO generation indicate that H. pylori cell wall lipopoly- saccharide (LPS), like that of other Gram-negative bacteria, is capable of triggering the stimulation of Toll-like re- ceptor-4 (TLR-4), which then through a series of down- stream effectors causes the activation of transcriptional factors that exert control over iNOS and COX-2 gene expression [2,7-9]. While the induction of iNOS gene by LPS has been convincingly linked to transcriptional factor NF-κB acti- vation [9-11], the nature of factors involved in transcrip- tional regulation of COX-2 expression is less apparent [7,12-14]. Depending on the cell type, at least four cen- tral response elements have been implicated in the regu- lation of COX-2 expression. These include, transcrip- tional factor, NF-κB, activator protein-1(AP-1), cAMP response element-biding protein (CREB), and CCATT/ enhancer-binding proteins (C/EBP) β and δ [7,13-17]. Moreover, LPS is a potent activator of mitogen-activated protein kinase (MAPK) cascade, including extracellular signal regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 kinase, which in turn exert their control over transcription factors activation through phosphory- lation [12,14-16]. Indeed, we have shown recently that stimulation of gastric mucosal cells with H. pylori LPS elicits the activation of all three MAPK subtypes (JNK, p38, and ERK), and have linked the involvement of JNK/p38 in the transcription factor AP-1 activation [17]. Studies into the role of LPS-elicited ERK activation in the regulation of factors linked to the induction of COX-2 and iNOS expression indicate the involvement of MAPK/ Copyright © 2012 SciRes. OJCB  B. L. SLOMIANY, A. SLOMIANY 22 ERK in the activation of transcriptional factors, C/EBP and CREB, implicated in the control of COX-2 expres- sion, as well as the processes associated with the en- hancement in NF-κB nuclear translocation and the induc- tion of iNOS expression [14,15,18-20]. Moreover, H. pylori LPS-induced ERK activation plays an essential role in the phosphorylation cPLA2, that facilitates the enzyme translocation form cytosol to membrane to gain access to phospholipid substrates for the increase in AA release [21]. The literature data, furthermore, point to similarities in the transcriptional regulation of COX-2 and cPLA2 expression; the gene locus of cPLA2 is located on chromosome I in the proximity of COX-2 gene, and the increased level of AA affects the COX-2 expression [22-24]. A substantial body of information also suggests that the systems involved in transcription factors activation are regulated through S-nitrosylation [9,25-27]. Indeed, S- nitrosylation has been linked to the processes of COX-2 and cPLA2 activation, as well as the regulation of inhibi- tory κB kinase-β (IKK-β) activity responsible for IκB-α degradation and NF-κB nuclear translocation [26-29]. Moreover, we have recently shown that S-nitrosylation of IKK-β plays a role in the modulatory influence of pep- tide hormone, ghrelin, on the gastric mucosal inflamma- tory responses to H. pylori [11,17,30]. As gastric mucosal responses to H. pylori are associ- ated with MAPK/JNK, p38 and ERK activation, while the modulatory influence of ghrelin is reflected in the inhibition of JNK and p38, but not the ERK, in this study we investigated the influence of H. pylori LPS and ghre- lin on the processes affected by MAPK/ERK activation. Our results demonstrate that the LPS-induced ERK acti- vation is of critical significance to up-regulation in gas- tric mucosal PGE2 and NO generation at the level of IKK-β and cPLA2 activation, and that ghrelin counters these proinflammatory consequences of the LPS through Src/Akt-dependent S-nitrosylation. 2. Materials and Methods 2.1. Gastric Mucosal Cell Incubation The gastric mucosal cells, collected form freshly dissected rat stomachs by scraping the mucosa with a blunt spatula, were suspended in five volumes of ice-cold Dulbecco’s modified (Gibco) Eagle’s minimal essential medium (DMEM), supplemented with fungizone (50 µg/ml), peni- cillin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf serum. After gentle trituration with a syringe, the dis- persed cells were settled by centrifugation, and resuspended in the medium to a concentration of 2 × 107 cell/ml [31]. Cell aliquots (1 ml) were then transferred to DMEM in culture dishes and incubated under 95% O2 - 5% CO2 atmosphere at 37˚C for up to8 h in the presence of 0 - 100 ng/ml of H. pylori LPS [6]. H. pylori used for LPS preparation was cultured from clinical isolates obtained from ATCC No. 4350 [6]. In the experiments evaluating the effect of ghrelin (rat, Sigma), JNK inhibitor, SP600125, p38 MAPK inhibitor, SB202190, ERK1/2 inhibitor, PD98059, Akt inhibitor, SH-5, Src inhibitor, PP2, Raf-1 kinase inhibitor, NF-κB inhibitor, PPM-18 (Calbiochem), and ascorbate (Sigma), the cells were first preincubated for 30 min with the indicated dose of the agent or vehicle before the addition of the LPS. The viability of cell preparations before and during the experimentation, as- sessed by Trypan blue dye exclusion assay [6], was greater than 97%. 2.2. COX-2 and iNOS Activity Assay For measurements of COX-2 activity, the gastric mucosal cells from the control and various experimental treatments were settled by centrifugation, rinsed with phosphate- buffered saline, and homogenized in 0.3 ml of cold sam- ple buffer containing 0.1 M Tris-HCl, pH 7.8, and 1 mM EDTA, centrifuged at 12,000 × g for 10 min at 4˚C, and the supernatant collected [5]. The COX-2 activity in 40 µl sample aliquots of the resulting supernatant was meas- ured using COX activity assay kit (Cayman) in the ab- sence and the presence of COX-1 inhibition (SC-560), by monitoring the appearance of oxidized TMPD at 590 nm. The activity of iNOS was measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline using NOS-detect kit (Stratagene). The gastric mucosal cells from the control and experimental treatments were ho- mogenized in a sample buffer containing 10 mM EDTA and centrifuged. The aliquots of the resulting supernatant were incubated for 30 min at 25˚C in the presence of 50 µCi/ml of L-[3H] arginine, 10 mM NAPDH, 5 µM tetra- hydrobiopterin, and 50 mM Tris-HCl buffer, pH 7.4, in a final volume of 250 µl. Following addition of stop buffer and Dowex-50 W (Na+) resin, the mixtures were trans- ferred to spin cups, centrifuged and the formed L-[3H] citrulline contained in the flow through was quantified by scintillation counting [6]. 2.3. cPLA2 Activity Assay The cPLA2 activity measurements were carried out using cPLA2 assay kit (Cayman) with thioarachidonoylphos- phatidylcholine as substrate. The gastric mucosal cells from the control and experimental treatments were set- tled by centrifugation, homogenized in 1 ml of 50 mM HEPES buffer, pH 7.4, containing 1 mM EDTA, and centrifuged at 10,000 × g for 15 min at 4˚C [29]. The su- pernatants were then filtered through an Amicon YM30 filter concentrators (m.w. cut-off 30 KDa) to remove any contamination with secretory PLA2, followed by 15 min incubation with 5 µM calcium-independent PLA2 inhibi- Copyright © 2012 SciRes. OJCB  B. L. SLOMIANY, A. SLOMIANY 23 tor, bromoenol lactone, and the aliquots (10 µl) of such prepared cell lysates were subjected to cPLA2 assay ac- cording to manufacturer’s instruction. 2.4. IκB Kinase Activity Assay To measure the IKK-β activity we utilized the ELISA- based detection kit, K-LISA (Calbiochem). The GST- IκB-α 50-amino acid peptide that includes the Ser32 and Ser36 of IκB-α phosphorylation sites was used as a sub- strate [32]. The gastric mucosal cell cytosolic extracts were incubated a glutathione-coated 96-well plate with GST-tagged IκB-α at room temperature for 30 min, and the phosphorylated GST-IκB-α substrate was detected using anti-phospho IκB-α (Ser32/Ser36) as first antibody, followed by horseradish peroxidase-conjugated secon- dary antibody. Following washing the retained complex was probed TMB reagent for spectrophotometric quanti- fication [11]. 2.5. cPLA2 and IKK-β Protein S-Nitrosylation Assay Detection of cPLA2 and IKK-β protein S-nitrosylation was carried out utilizing a biotin switch method for pro- tein S-nitrosylation [33,34]. The gastric mucosal cells were treated with ghrelin (0.5 µg/ml), or ERK inhibitor, PD 98059 (30 µM) + ghrelin, or Akt inhibitor, SH-5 (20 µM) + ghrelin, and incubated for 1h in the presence of 100ng/ml of H. pylori LPS. The cells were collected by centrifugation at 500 × g for 5 min, lysed in 0.2 ml of HEN lysis buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM neocuprin, pH 7.7), and the unnitrosylated thiol groups were blocked with S-methyl methanethiosul- fonate reagent at 50˚C for 20 min [34]. The proteins were precipitated with acetone, resuspended in 0.2 ml of HEN buffer containing 1% SDS, and subjected to targeted ni- trothiol group reduction with sodium ascorbate (100 mM). The free thiols were then labeled with biotin and the biotinylated proteins were recovered on streptavidin beads. The formed streptavidin bead-protein complex was washed with neutralization buffer, and the bound proteins were dissociated from streptavidin beads with 50 µl of elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, pH 7.7) containing 1% 2-mercaptoethanol [34]. The obtained proteins were then analyzed by Western blotting. 2.6. Nuclear Protein Extraction The aliquots of gastric mucosal cell suspension from the control and various experimental conditions were settled by centrifugation at 500 × g for 5 min, rinsed with phos- phate-buffered saline, and lysed by incubation for 10 min on ice in the lysis buffer, containing 10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5% Nonidet P-40, 1 mM dithiothreitol, and 0.5 mM PMSF [35]. Following centrifugation at 12,000 × g for 10 min at 4˚C, the super- natant was subjected to centrifugation at 100,000 × g for 1h and the obtained soluble fraction was used as source of cytosolic extract [11]. The pellets, from 12,000 × g centrifugation, containing crude nuclei were suspended for 20 min at 4˚C in the extraction buffer, containing 20 mM HEPES, pH 7.9, 25% glycerol, 400 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM PMSF. The samples were centrifuged at 15,000 × g for 10 min at 4˚C, and the supernatants containing nu- clear extracts were collected and stored at –70˚C until use. 2.7. Immunoblot Analysis The gastric mucosal cells from the control and experi- mental treatments were collected by centrifugation and resuspended for 30 min in ice-cold lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Tri- ton X-100, 2 mM EDTA, 1 mM sodium orthovanadate, 4 mM sodium pyrophosphate, 1 mM PMSF, and 1 mM NaF), containing 1 µg/ml leupeptin and 1 µg/ml pepstatin [5]. Following brief sonication, the lysates were centri- fuged at 12,000 × g for 10 min, and the supernatants were collected and normalized with respect to protein concen- tration using BCA protein assay kit (Pierce). The samples, including those subjected to biotin switch procedure, were then resuspended in loading buffer, boiled for 5 min, and subjected to SDS-PAGE using 40 µg protein/lane. The separated proteins were transferred onto nitrocellu- lose membranes, blocked for 1 h with 5% skim milk in Tris-buffered Tween (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and probed with specific anti- bodies directed against IκΒ-α, COX-2 and iNOS (Cal- biochem), NF-κB p65 and IKK-β (EMD Millipore), and phospho-IKK-β, cPLA2 and phospho-cPLA2 (Cell Sig- naling) Antibodies directed against ERK1/2 and phos- pho-ERK1/2 MAPK were from Calbiochem, whereas ati-β-actin from Sigma. 2.8. Data Analysis All experiments were carried out using duplicate sampling, and the results are expressed as means ± SD. Analysis of variance (ANOVA) and nonparametric Kruskal-Wallis tests were used to determine significance. Any difference detected was evaluated by means of post hoc Bonferroni test, and the significance level was set at P < 0.05. 3. Results In our previous study, we reported that ghrelin modulates H. pylori LPS-elicited induction in gastric mucosal COX-2 and iNOS expression by affecting p38 MAPK/ATF-2 and IKK-β/NF-κB activation pathways [17]. Indeed, as shown in Figure 1, exposure of rat gastric mucosal cells Copyright © 2012 SciRes. OJCB 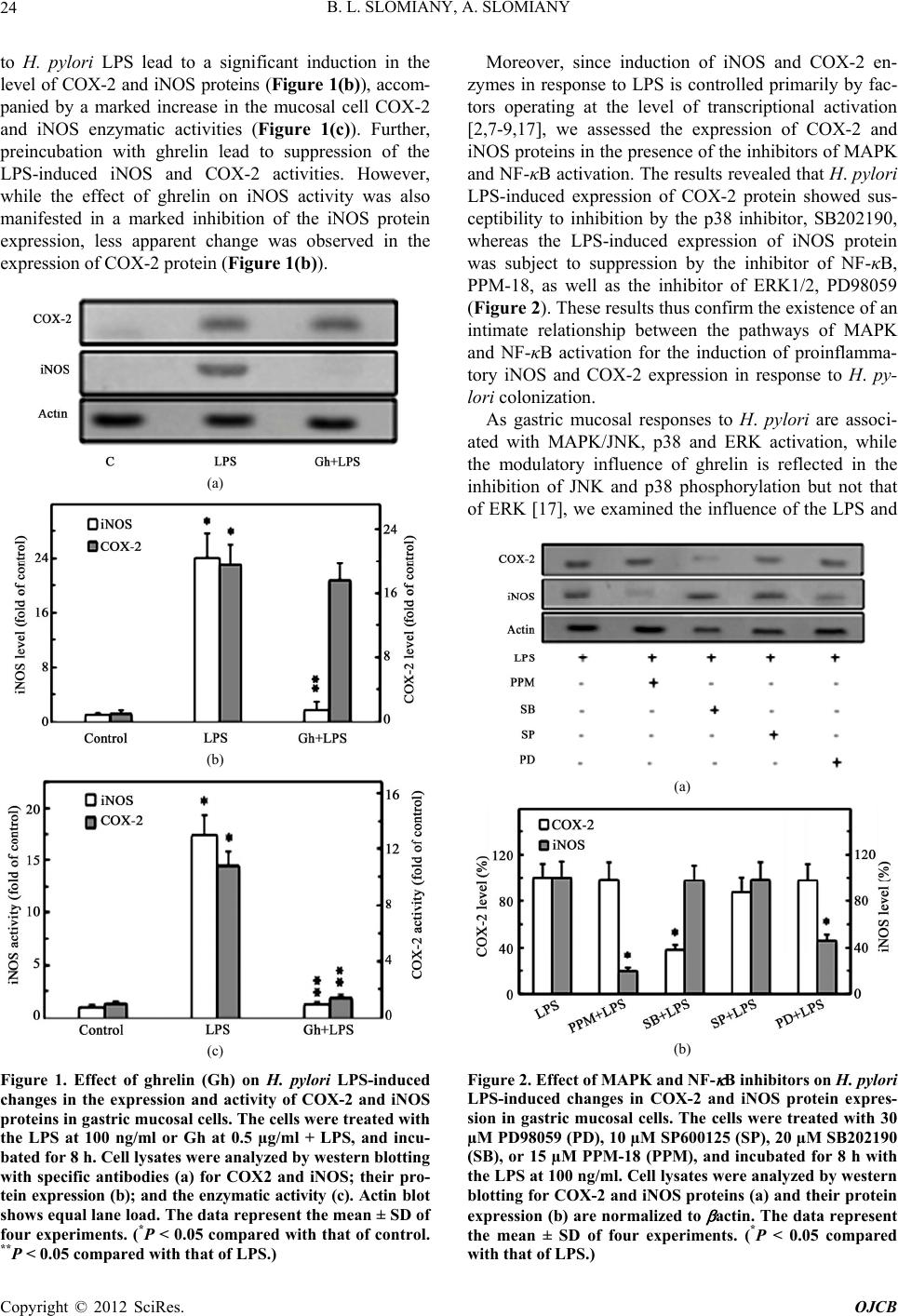 B. L. SLOMIANY, A. SLOMIANY 24 to H. pylori LPS lead to a significant induction in the level of COX-2 and iNOS proteins (Figure 1(b)), accom- panied by a marked increase in the mucosal cell COX-2 and iNOS enzymatic activities (Figure 1(c)). Further, preincubation with ghrelin lead to suppression of the LPS-induced iNOS and COX-2 activities. However, while the effect of ghrelin on iNOS activity was also manifested in a marked inhibition of the iNOS protein expression, less apparent change was observed in the expression of COX-2 protein (Figure 1(b)). (a) (b) (c) Figure 1. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in the expression and activity of COX-2 and iNOS proteins in gastric mucosal cells. The cells were treated with the LPS at 100 ng/ml or Gh at 0.5 µg/ml + LPS, and incu- bated for 8 h. Cell lysates were analyzed by western blotting with specific antibodies (a) for COX2 and iNOS; their pro- tein expression (b); and the enzymatic activity (c). Actin bl ot shows equal lane load. The data repre sent the mean ± SD of four experiments. (*P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS.) Moreover, since induction of iNOS and COX-2 en- zymes in response to LPS is controlled primarily by fac- tors operating at the level of transcriptional activation [2,7-9,17], we assessed the expression of COX-2 and iNOS proteins in the presence of the inhibitors of MAPK and NF-κB activation. The results revealed that H. pylori LPS-induced expression of COX-2 protein showed sus- ceptibility to inhibition by the p38 inhibitor, SB202190, whereas the LPS-induced expression of iNOS protein was subject to suppression by the inhibitor of NF-κB, PPM-18, as well as the inhibitor of ERK1/2, PD98059 (Figure 2). These results thus confirm the existence of an intimate relationship between the pathways of MAPK and NF-κB activation for the induction of proinflamma- tory iNOS and COX-2 expression in response to H. py- lori colonization. As gastric mucosal responses to H. pylori are associ- ated with MAPK/JNK, p38 and ERK activation, while the modulatory influence of ghrelin is reflected in the inhibition of JNK and p38 phosphorylation but not that of ERK [17], we examined the influence of the LPS and (a) (b) Figure 2. Effect of MAPK and NF- B inhibitors on H. pylori LPS-induced changes in COX-2 and iNOS protein expres- sion in gastric mucosal cells. The cells were treated with 30 µM PD98059 (PD), 10 µM SP600125 (SP), 20 µM SB202190 (SB), or 15 µM PPM-18 (PPM), and incubated for 8 h with the LPS at 100 ng/ml. Cell lysates were analyzed by western blotting for COX-2 and iNOS proteins (a) and their protein expression (b) are normalized to actin. The data represent the mean ± SD of four experiments. (*P < 0.05 compared with that of LPS.) Copyright © 2012 SciRes. OJCB 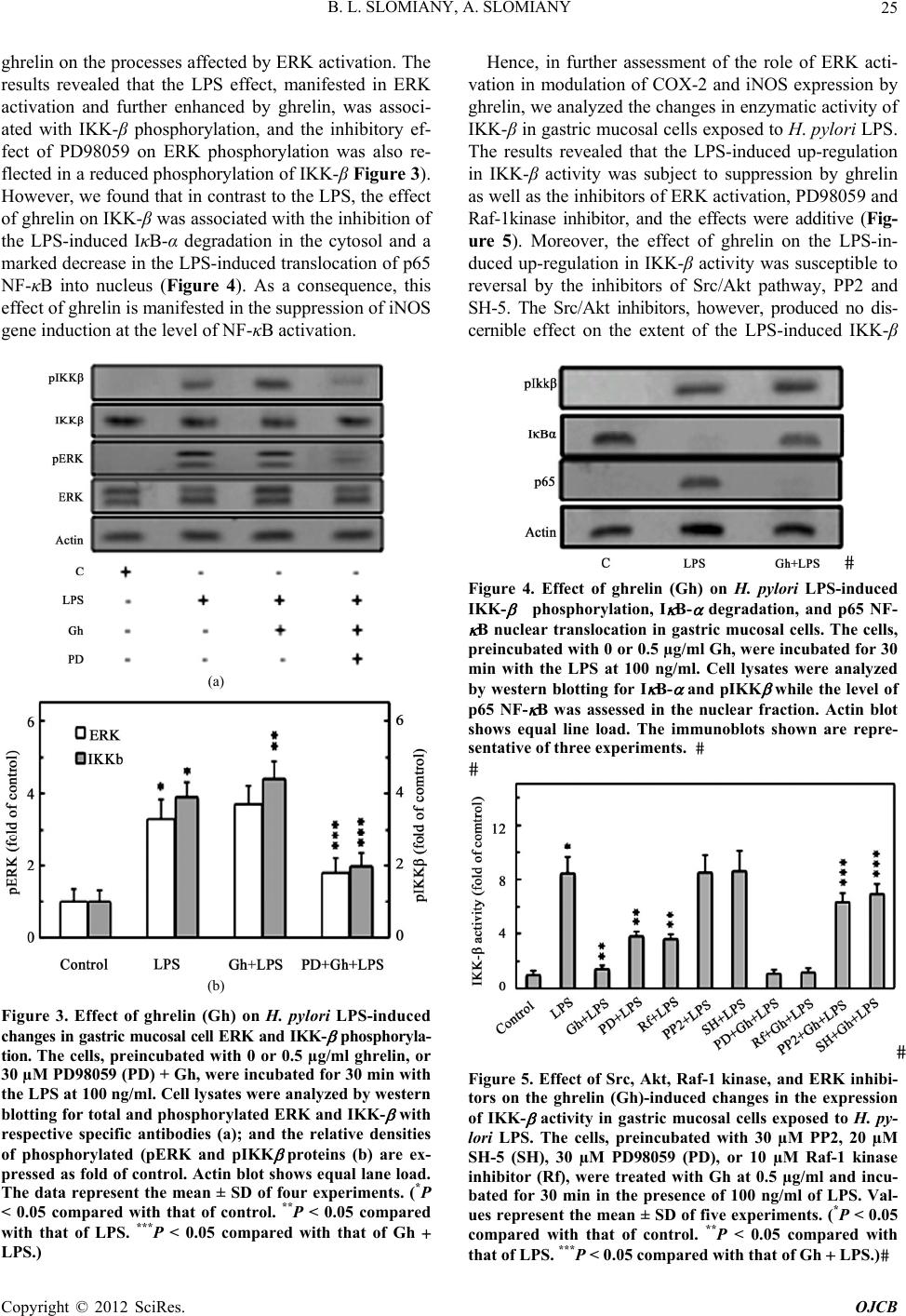 B. L. SLOMIANY, A. SLOMIANY 25 ghrelin on the processes affected by ERK activation. The results revealed that the LPS effect, manifested in ERK activation and further enhanced by ghrelin, was associ- ated with IKK-β phosphorylation, and the inhibitory ef- fect of PD98059 on ERK phosphorylation was also re- flected in a reduced phosphorylation of IKK-β Figure 3). However, we found that in contrast to the LPS, the effect of ghrelin on IKK-β was associated with the inhibition of the LPS-induced IκB-α degradation in the cytosol and a marked decrease in the LPS-induced translocation of p65 NF-κB into nucleus (Figure 4). As a consequence, this effect of ghrelin is manifested in the suppression of iNOS gene induction at the level of NF-κB activation. (a) (b) Figure 3. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in gastri c mucosal cell ERK and IKK- phosphoryla- tion. The cells, preincubated with 0 or 0.5 µg/ml ghrelin, or 30 µM PD98059 (PD) + Gh, were incubated for 30 min with the LPS at 100 ng/ml. Cell lysates were analyzed by western blotting for total and phosphorylated ERK and IKK- with respective specific antibodies (a); and the relative densities of phosphorylated (pERK and pIKK proteins (b) are ex- pressed as fold of control. Actin blot show s equal lane load. The data represent the mean ± SD of four experiments. (*P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS. ***P < 0.05 compared with that of Gh LPS.) Hence, in further assessment of the role of ERK acti- vation in modulation of COX-2 and iNOS expression by ghrelin, we analyzed the changes in enzymatic activity of IKK-β in gastric mucosal cells exposed to H. pylori LPS. The results revealed that the LPS-induced up-regulation in IKK-β activity was subject to suppression by ghrelin as well as the inhibitors of ERK activation, PD98059 and Raf-1kinase inhibitor, and the effects were additive (Fig- ure 5). Moreover, the effect of ghrelin on the LPS-in- duced up-regulation in IKK-β activity was susceptible to reversal by the inhibitors of Src/Akt pathway, PP2 and SH-5. The Src/Akt inhibitors, however, produced no dis- cernible effect on the extent of the LPS-induced IKK-β Figure 4. Effect of ghrelin (Gh) on H. pylori LPS-induced IKK- phosphorylation, I B- degradation, and p65 NF- B nuclear translocation in gastric mucosal cells. The cells, preincubated with 0 or 0.5 µg/ml Gh, were incubated for 30 min with the LPS at 100 ng/ml. Cell lysates were analyzed by western blotting for I B- and pIKK while the level of p65 NF- B was assessed in the nuclear fraction. Actin blot shows equal line load. The immunoblots shown are repre- sentative of three experiments. Figure 5. Effect of Src, Akt, Raf-1 kinase, and ERK inhibi- tors on the ghrelin (Gh)-induced changes in the expression of IKK- activity in gastric mucosal cells exposed to H. py- lori LPS. The cells, preincubated with 30 µM PP2, 20 µM SH-5 (SH), 30 µM PD98059 (PD), or 10 µM Raf-1 kinase inhibitor (Rf), were treated with Gh at 0.5 µg/ml and incu- bated for 30 min in the presence of 100 ng/ml of LPS. Val- ues represent the mean ± SD of five experiments. (*P < 0.05 compared with that of control. **P < 0.05 compared with *** that of LPS. P < 0.05 compared with that of Gh LPS.) Copyright © 2012 SciRes. OJCB 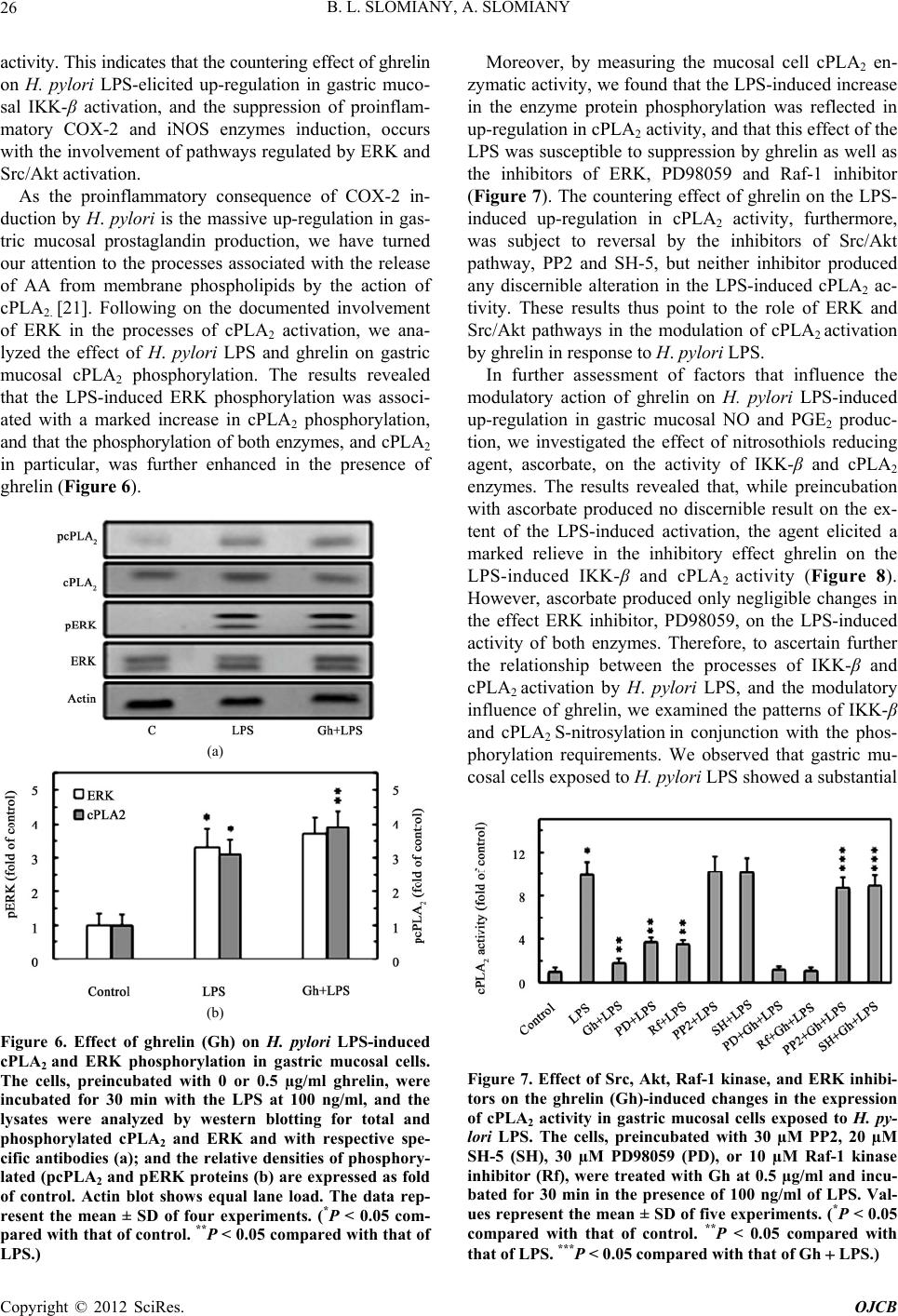 B. L. SLOMIANY, A. SLOMIANY 26 activity. This indicates that the countering effect of ghrelin matory consequence of COX-2 in- du on H. pylori LPS-elicited up-regulation in gastric muco- sal IKK-β activation, and the suppression of proinflam- matory COX-2 and iNOS enzymes induction, occurs with the involvement of pathways regulated by ERK and Src/Akt activation. As the proinflam ction by H. pylori is the massive up-regulation in gas- tric mucosal prostaglandin production, we have turned our attention to the processes associated with the release of AA from membrane phospholipids by the action of cPLA2. [21]. Following on the documented involvement of ERK in the processes of cPLA2 activation, we ana- lyzed the effect of H. pylori LPS and ghrelin on gastric mucosal cPLA2 phosphorylation. The results revealed that the LPS-induced ERK phosphorylation was associ- ated with a marked increase in cPLA2 phosphorylation, and that the phosphorylation of both enzymes, and cPLA2 in particular, was further enhanced in the presence of ghrelin (Figure 6). (a) (b) Figure 6. Effect of ghrelin H. pylori LPS-induced LPS.) r (Gh) on cPLA2 and ERK phosphorylation in gastric mucosal cells. The cells, preincubated with 0 or 0.5 µg/ml ghrelin, were incubated for 30 min with the LPS at 100 ng/ml, and the lysates were analyzed by western blotting for total and phosphorylated cPLA2 and ERK and with respective spe- cific antibodies (a); and the relative densities of phosphory- lated (pcPLA2 and pERK proteins (b) are expressed as fold of control. Actin blot shows equal lane load. The data rep- resent the mean ± SD of four experiments. (*P < 0.05 com- pared with that of control. **P < 0.05 compared with that of Moreover, by measuring the mucosal cell cPLA2 en- zymatic activity, we found that the LPS-induced incease in the enzyme protein phosphorylation was reflected in up-regulation in cPLA2 activity, and that this effect of the LPS was susceptible to suppression by ghrelin as well as the inhibitors of ERK, PD98059 and Raf-1 inhibitor (Figure 7). The countering effect of ghrelin on the LPS- induced up-regulation in cPLA2 activity, furthermore, was subject to reversal by the inhibitors of Src/Akt pathway, PP2 and SH-5, but neither inhibitor produced any discernible alteration in the LPS-induced cPLA2 ac- tivity. These results thus point to the role of ERK and Src/Akt pathways in the modulation of cPLA2 activation by ghrelin in response to H. pylori LPS. In further assessment of factors that influence the modulatory action of ghrelin on H. pylori LPS-induced up-regulation in gastric mucosal NO and PGE2 produc- tion, we investigated the effect of nitrosothiols reducing agent, ascorbate, on the activity of IKK-β and cPLA2 enzymes. The results revealed that, while preincubation with ascorbate produced no discernible result on the ex- tent of the LPS-induced activation, the agent elicited a marked relieve in the inhibitory effect ghrelin on the LPS-induced IKK-β and cPLA2 activity (Figure 8). However, ascorbate produced only negligible changes in the effect ERK inhibitor, PD98059, on the LPS-induced activity of both enzymes. Therefore, to ascertain further the relationship between the processes of IKK-β and cPLA2 activation by H. pylori LPS, and the modulatory influence of ghrelin, we examined the patterns of IKK-β and cPLA2 S-nitros ylation in conjunction with the phos- phorylation requirements. We observed that gastric mu- cosal cells exposed to H. pylori LPS showed a substantial Figure 7. Effect of Src, Akt, Raf-1 kinase, and ERK inhib tors on the ghrelin (Gh)-induced changes in the expressioi- n of cPLA2 activity in gastric mucosal cells exposed to H. py- lori LPS. The cells, preincubated with 30 µM PP2, 20 µM SH-5 (SH), 30 µM PD98059 (PD), or 10 µM Raf-1 kinase inhibitor (Rf), were treated with Gh at 0.5 µg/ml and incu- bated for 30 min in the presence of 100 ng/ml of LPS. Val- ues represent the mean ± SD of five experiments. (*P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS. ***P < 0.05 compared with that of Gh LPS.) Copyright © 2012 SciRes. OJCB 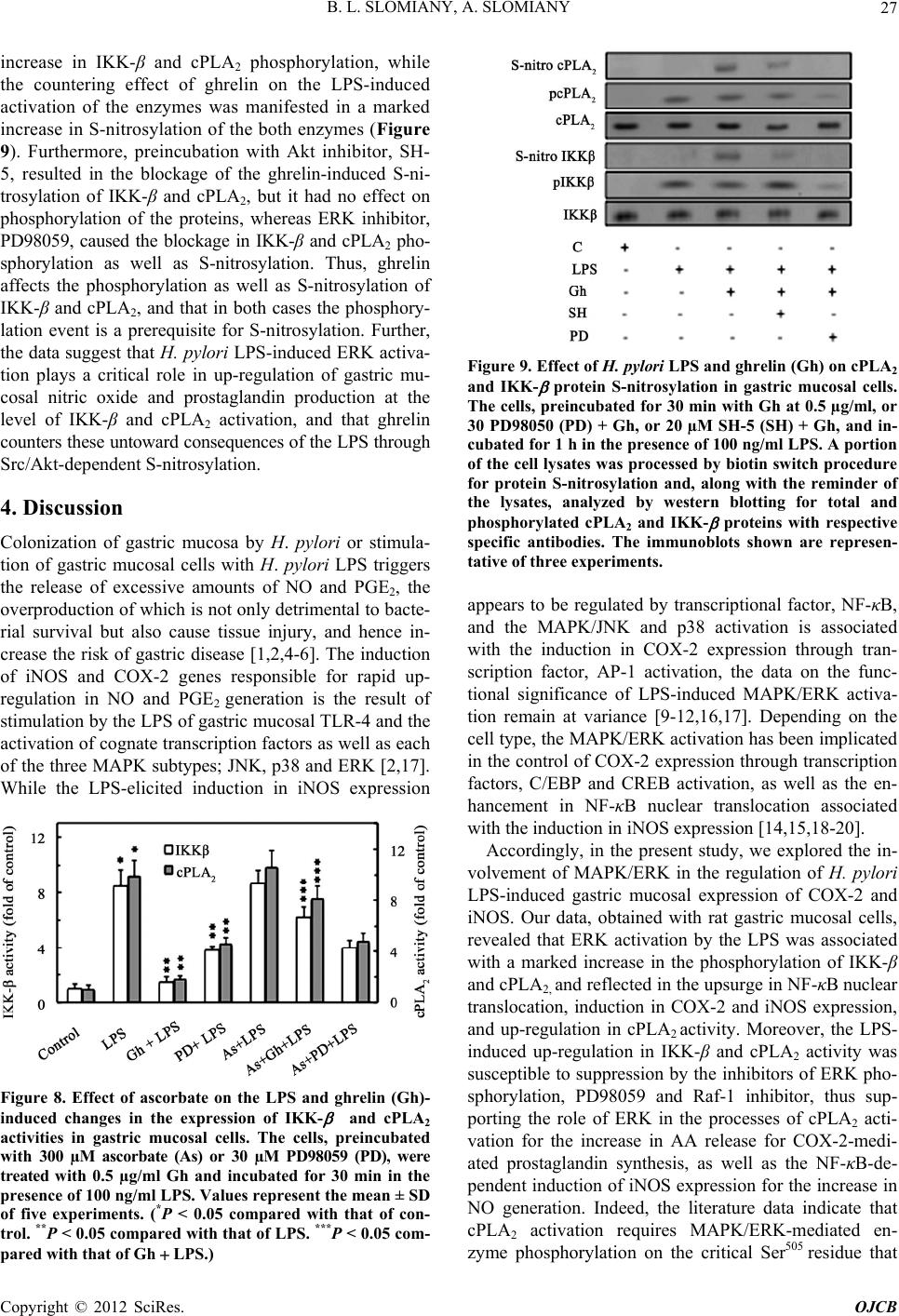 B. L. SLOMIANY, A. SLOMIANY 27 increase in IKK-β and cPLA2 phosphorylation, while the countering effect of ghrelin on the LPS-induced astric mucosa by H. pylori or stimula- cosal cells with Hggers activation of the enzymes was manifested in a marked increase in S-nitrosylation of the both enzymes (Figure 9). Furthermore, preincubation with Akt inhibitor, SH- 5, resulted in the blockage of the ghrelin-induced S-ni- trosylation of IKK-β and cPLA2, but it had no effect on phosphorylation of the proteins, whereas ERK inhibitor, PD98059, caused the blockage in IKK-β and cPLA2 pho- sphorylation as well as S-nitrosylation. Thus, ghrelin affects the phosphorylation as well as S-nitrosylation of IKK-β and cPLA2, and that in both cases the phosphory- lation event is a prerequisite for S-nitrosylation. Further, the data suggest that H. pylori LPS-induced ERK activa- tion plays a critical role in up-regulation of gastric mu- cosal nitric oxide and prostaglandin production at the level of IKK-β and cPLA2 activation, and that ghrelin counters these untoward consequences of the LPS through Src/Akt-dependent S-nitrosylation. 4. Discussion Colonization of g tion of gastric mu. pylori LPS tri the release of excessive amounts of NO and PGE2, the overproduction of which is not only detrimental to bacte- rial survival but also cause tissue injury, and hence in- crease the risk of gastric disease [1,2,4-6]. The induction of iNOS and COX-2 genes responsible for rapid up- regulation in NO and PGE2 generation is the result of stimulation by the LPS of gastric mucosal TLR-4 and the activation of cognate transcription factors as well as each of the three MAPK subtypes; JNK, p38 and ERK [2,17]. While the LPS-elicited induction in iNOS expression Figure 8. Effect of ascorbate on the LPS and ghrelin (Gh) induced changes in the expression of IKK- and cPLA - 2 activities in gastric mucosal cells. The cells, preincubated with 300 µM ascorbate (As) or 30 µM PD98059 (PD), were treated with 0.5 µg/ml Gh and incubated for 30 min in the presence of 100 ng/ml LPS. Values represent the mean ± SD of five experiments. (*P < 0.05 compared with that of con- trol. **P < 0.05 compared with that of LPS. ***P < 0.05 com- pared with that of Gh LPS.) Figure 9. Effect of H. pylori LPS and ghrelin (Gh) on LA2 and IKK- protein S-nitrosylation in gastric mucosal cells. ranscriptional factor, NF-κB, nd the MAPK/JNK and p38 activation is associated ori LP cP The cells, preincubated for 30 min with Gh at 0.5 µg/ml, or 30 PD98050 (PD) + Gh, or 20 µM SH-5 (SH) + Gh, and in- cubated for 1 h in the presence of 100 ng/ml LPS. A portion of the cell lysates was processed by biotin switch procedure for protein S-nitrosylation and, along with the reminder of the lysates, analyzed by western blotting for total and phosphorylated cPLA2 and IKK- proteins with respective specific antibodies. The immunoblots shown are represen- tative of three experiments. appears to be regulated by t a with the induction in COX-2 expression through tran- scription factor, AP-1 activation, the data on the func- tional significance of LPS-induced MAPK/ERK activa- tion remain at variance [9-12,16,17]. Depending on the cell type, the MAPK/ERK activation has been implicated in the control of COX-2 expression through transcription factors, C/EBP and CREB activation, as well as the en- hancement in NF-κB nuclear translocation associated with the induction in iNOS expression [14,15,18-20]. Accordingly, in the present study, we explored the in- volvement of MAPK/ERK in the regulation of H. pyl S-induced gastric mucosal expression of COX-2 and iNOS. Our data, obtained with rat gastric mucosal cells, revealed that ERK activation by the LPS was associated with a marked increase in the phosphorylation of IKK-β and cPLA2, and reflected in the upsurge in NF-κB nuclear translocation, induction in COX-2 and iNOS expression, and up-regulation in cPLA2 activity. Moreover, the LPS- induced up-regulation in IKK-β and cPLA2 activity was susceptible to suppression by the inhibitors of ERK pho- sphorylation, PD98059 and Raf-1 inhibitor, thus sup- porting the role of ERK in the processes of cPLA2 acti- vation for the increase in AA release for COX-2-medi- ated prostaglandin synthesis, as well as the NF-κB-de- pendent induction of iNOS expression for the increase in NO generation. Indeed, the literature data indicate that cPLA2 activation requires MAPK/ERK-mediated en- zyme phosphorylation on the critical Ser505 residue that Copyright © 2012 SciRes. OJCB  B. L. SLOMIANY, A. SLOMIANY 28 plays a crucial role in Ca2+-dependent translocation of cPLA2 form cytosol to membrane to gain access to pho- spholipid substrates [21,36]. Likewise, the ability of ERK to exert a stimulatory effect on IKK/NF-κB activation through Raf-1/MEK/ERK signaling cascade has been implicated in the regulation of adenosine A1 receptor mediated activation of NF-κB in human lymphocytic cells [19]. Further, we found that, while the LPS-induced phos- phorylation of ERK, IKK-β and cPLAwas enhanced by pr ty of substrates, and the signaling through Src/ A and cPLA activation by th n as to the critical role of M 2 eincubation with gastric hormone, ghrelin, the modu- latory effect of the hormone was reflected in the suppres- sion of IKK-β and cPLA2 activity, as well as that of iNOS and COX-2. The effect of ghrelin on iNOS was manifested in a marked decline of the enzyme protein expression, and associated with the inhibition of the LPS-induced IκB-α degradation and a decrease in NF-κB nuclear translocation, while the effect on COX-2 was primarily manifested in the suppression of the LPS-in- duced up-regulation in COX-2 activity. Interestingly, NO stimulation through iNOS induction has been linked to COX-2 activation through S-nitrosylation and the in- crease in PGE2 production [9,26], and we have recently shown that ghrelin suppression of H. pylori LPS-induced COX-2 S-nitrosylation results in the inhibition of PGE2 generation [5]. Moreover, following our earlier leads as to the involvement of cNOS in the mechanism of ghrelin action [6,11], we revealed that the countering effect of ghrelin on the LPS-induced up-regulation in the activity of IKK-βand cLPA2 enzymes was susceptible to sup- pression by the inhibitors of cNOS activation through phosphorylation, an Akt inhibitor, SH-5 and Src inhibitor, PP2. The Src/Akt inhibitors, however, produced no dis- cernible alteration in the LPS-induced activity of IKK-β and cPLA2 enzymes. These data and the finding that the LPS-induced up-regulation in the activities of IKK-β and cPLA2 was also susceptible to suppression by the inhibi- tors of ERK activation, PD98059 and Raf-1, provide strong indication as to the role of ERK and Src/Akt pathways in the mediation of modulatory influence of ghrelin on the processes associated with up-regulation in gastric mucosal PGE2 and NO generation in response to H. pylori. While as an upstream kinase, cSrc phosphorylates a wide varie kt pathway is known to occupy a central stage in the receptor (GHS-R)-mediated responses to ghrelin stimula- tion [6,11,37-39], it is becoming increasingly apparent that the hormone is also capable of exerting its modula- tory influence through the process of protein S-nitrosy- lation. Indeed, the induction IKK-β S-nitrosylation by ghrelin exerts the inhibitory effect on the extent of IκB- α degradation, causing suppression in NF-κB nuclear translocation and resulting in the repression of iNOS gene induction [11,31]. Moreover, ghrelin has been im- plicated in the modulation of S-nitrosylation-dependent activation COX-2 and cPLA2 enzymes, thus affecting the processes of PGE2 generation [11,27,29,30]. Therefore, in further assessment of the modulatory action of ghrelin on H. pylori LPS-induced up-regulation in gastric muco- sal generation of PGE2 and NO, we evaluated the effect of nitrosothiol reducing agent, ascorbate on the activity of IKK-β and cPLA2. We found that while ascorbate elic- ited a marked relieve in the inhibitory effect of ghrelin on the LPS-induced IKK-β and cPLA2 activation, the agent produced only negligible changes in the effect of ERK inhibitor, PD98059, on the LPS-induced activity of both enzymes. Hence, consistent with the above findings and considering the fact that the changes in activity of both enzymes in the presence of ghrelin was susceptible to suppression by the inhibitors of cNOS activation, we concluded that the countering effect of ghrelin on H. py- lori LPS-induced activation of IKK-β and cPLA2 are in- timately linked to the events of cNOS-dependent S-ni- trosylation of these enzymes. Consequently, to ascertain further the relationship be- tween the processes of IKK-β2 e LPS, and the modulatory action of ghrelin, we exam- ined the patterns of IKK-β and cPLA2 S-nitrosylation in conjunction with the phosphorylation requirements in the presence of the inhibitors of cNOS and ERK. We ob- served that the mucosal cells exposed to the LPS showed a marked increase in IKK-β and cPLA2 phosphorylation, while the countering effect of ghrelin was associated with the increased phosphorylation as well as S-nitrosy- lation of the enzymes. Furthermore, preincubation with the inhibitor of cNOS activation, SH-5, lead to the blockage in ghrelin-induced S-nitrosylation of IKK-β and cPLA2, but had no effect on phosphorylation of the en- zymes, whereas ERK inhibitor, PD98059, caused the blockage in IKK-β and cPLA2 phosphorylation as well as S-nitrosylation. This indicated that the phosphorylation event is a prerequisite for the IKK-β and cPLA2 protein S-nitrosylation. Together, these findings suggest that the activation MAPK/ERK by H. pylori LPS plays a pivotal role in up-regulation in gastric mucosal PGE2 and NO generation at the level of IKK-β and cPLA2 activation through phosphorylation, and that ghrelin counters these proinflammatory consequences of the LPS through Src/ Akt-mediated and cNOS-dependent S-nitrosylation of the IKK-β and cPLA2 proteins. In conclusion, the data present in this report add fur- ther support to our assertio APK/ERK/JNK and p38 signaling cascades in media- tion of gastric mucosal inflammatory responses to H. pylori LPS While the LPS-elicited induction in COX-2 expression relays primarily on JNK/p38-dependent acti- vation of transcription factor AP-1 [17], the ERK Copyright © 2012 SciRes. OJCB 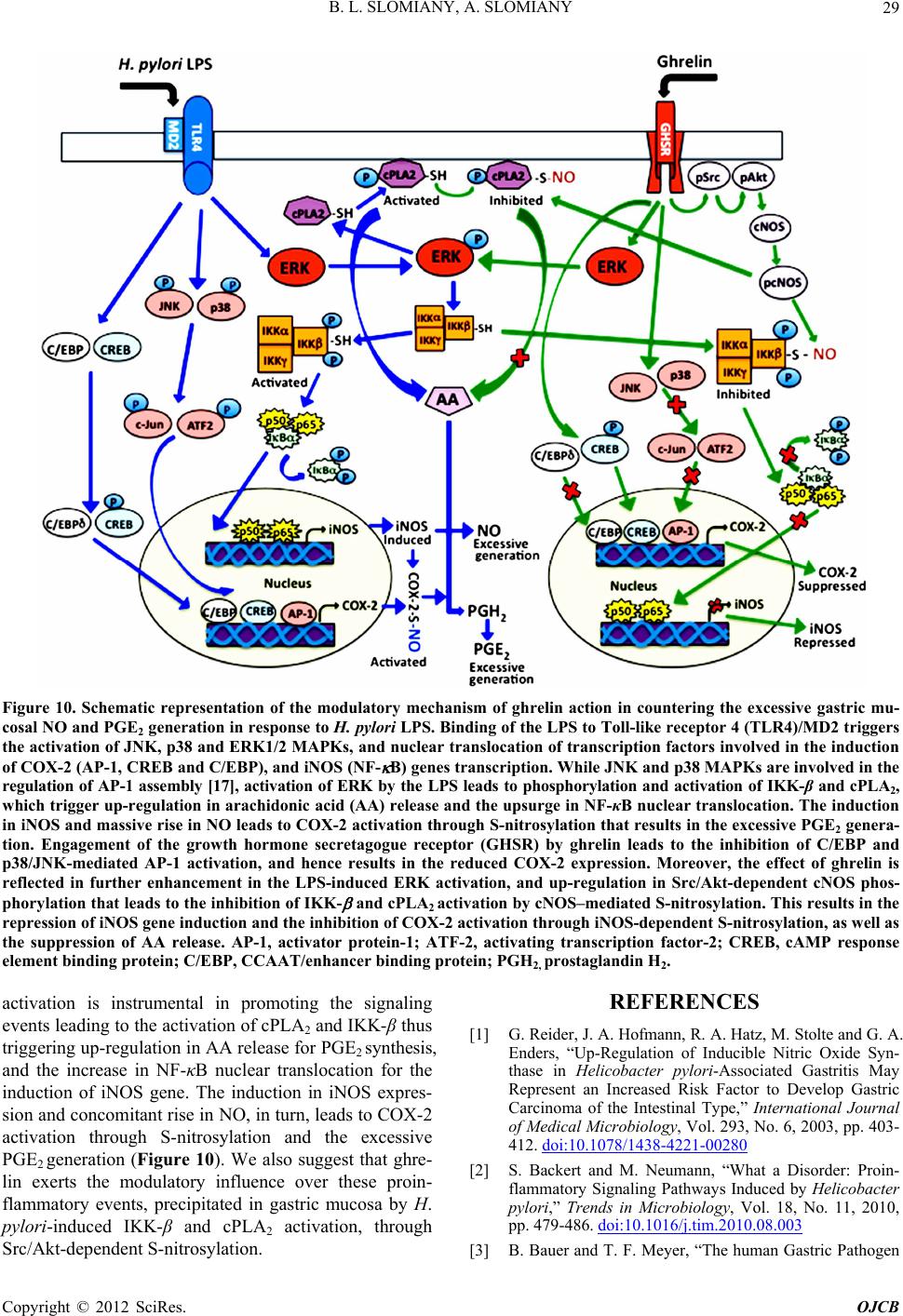 B. L. SLOMIANY, A. SLOMIANY Copyright © 2012 SciRes. OJCB 29 Figure 10. Schematic representation of the modulatory mechanism of ghrelin action in countering the excessive gastric mu- cosal NO and PGE2 generation in response to H. pylori LPS. Binding of the LPS to Toll-like receptor 4 (TLR4)/MD2 tri vents leading to the activation of cPLA and IKK-β thus ENCES ated Gastritis May Represent an o Develop Gastric ggers the activation of JNK, p38 and ERK1/2 MAPKs, and nuclear translocation of transcription factors involved in the inductio n of COX-2 (AP-1, CREB and C/EBP), and iNOS (NF- B) genes transcription. While JNK and p38 MAPKs are involved in the regulation of AP-1 assembly [17], activation of ERK by the LPS leads to phosphorylation and activation of IKK-β and cPLA2, which trigger up-regulation in arachidonic acid (AA) re lease and the upsurge in NF-κB nuclear translocation. The induction in iNOS and massive rise in NO leads to COX-2 activation through S-nitrosylation that results in the excessive PGE2 genera- tion. Engagement of the growth hormone secretagogue receptor (GHSR) by ghrelin leads to the inhibition of C/EBP and p38/JNK-mediated AP-1 activation, and hence results in the reduced COX-2 expression. Moreover, the effect of ghrelin is reflected in further enhancement in the LPS-induced ERK activation, and up-regulation in Src/Akt-dependent cNOS phos- phorylation that leads to the inhibition of IKK- and cPLA2 activation by cNOS–mediated S-nitrosy lation. This results in the repression of iNOS gene induction and the inhibition of COX-2 activation through iNOS-dependent S-nitr osy lation, as well as the suppression of AA release. AP-1, activator protein-1; ATF-2, activating transcription factor-2; CREB, cAMP response element binding protein; C/EBP, CCAAT/enhancer binding protein; PGH2, prostaglandin H2. activation is instrumental in promoting the signaling REFER e2 triggering up-regulation in AA release for PGE2 synthesis, and the increase in NF-κB nuclear translocation for the induction of iNOS gene. The induction in iNOS expres- sion and concomitant rise in NO, in turn, leads to COX-2 activation through S-nitrosylation and the excessive PGE2 generation (Figure 10). We also suggest that ghre- lin exerts the modulatory influence over these proin- flammatory events, precipitated in gastric mucosa by H. pylori-induced IKK-β and cPLA2 activation, through Src/Akt-dependent S-nitrosylation. [1] G. Reider, J. A. Hofmann, R. A. Hatz, M. Stolte and G. A. Enders, “Up-Regulation of Inducible Nitric Oxide Syn- thase in Helicobacter pylori-Associ Increased Risk Factor t Carcinoma of the Intestinal Type,” International Journal of Medical Microbiology, Vol. 293, No. 6, 2003, pp. 403- 412. doi:10.1078/1438-4221-00280 [2] S. Backert and M. Neumann, “What a Disorder: Proin- flammatory Signaling Pathways Induced by Helicobacter pylori,” Trends in Microbiology, Vol. 18, No. 11, 2010, pp. 479-486. doi:10.1016/j.tim.2010.08.003 [3] B. Bauer and T. F. Meyer, “The human Gastric Pathogen  B. L. SLOMIANY, A. SLOMIANY 30 Helicobacter pylori and Its Association with Gastric Cancer and Ulcer Disease,” Ulcers, Vol. 2011, 2011, 23 Pages, Article ID 340157. doi:10.1155/2011/340157 [4] L. A. Wroblewski, R. M. Peek and K. T. Wilson, “Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk,” Clinical Microbiology Reviews, Vol. 23, No. 4, 2010, pp. 713-739. doi:10.1128/CMR.00011-10 [5] B. L. Slomiany and A. Slomiany, “Role of Constitutive Nitric Oxide Synthase in Regulation of Helicobacter py- lori-Induced Gastric Mucosal Cyclooxygenase-2 Activa- tion through S-Nitrosylation: Mecha tion,” Open Journal of Gastr nism of Ghrelin Ac- oenterology, Vol. 1, No. 2, 2011, pp. 13-22. doi:10.4236/ojgas.2011.12003 [6] B. L. Slomiany and A. Slomiany, “Role of Ghrelin In- duced cSrc activation in Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori,” In- flammopharmacology, Vol. 19, No. 4, 2011, pp. 197-204. doi:10.1007/s10787-011-0083-7 [7] M. Joo, J. G. Wright, N. N. Hu, et al., “Yin Yang 1 En- hances Cyclooxygenase-2 Gene Expression in Macro- phages,” American Journal of Physiology Lung and Cell Molecular Physiology, Vol. 292, No. 5, 2007, pp. L1219- L1226. doi:10.1152/ajplung.00474.2006 hysiology [8] B. D. Lamon, R. K. Upmacis, R. S. Deeb, H. Koyuncu and D. Haijar, “Inducible Nitric Oxide Synthase Gene Deletion Exaggerates MAPK-Mediated Cyclooxygenase- 2 Induction by Inflammatory Stimuli,” American Journal of Physiology Heart and Circulatory P, Vol. 299, No. 3, 2010, pp. H613-H623. doi:10.1152/ajpheart.00144.2010 [9] Y. Ye, J. D. Martinez, R. J. Perez-Polo, Y. Lin, B. F. Uretsky and Y. Birnbaum, “The Role of eNOS, iNOS, and NF-B in Upregulation and Activation of Cyclooxy- genase-2 and Infarct Size Reduct American Journal of Physiology Heart and ion by Atorvastin,” Circulatory Physiology, Vol. 295, No. 1, 2008, pp. H343-H351. doi:10.1152/ajpheart.01350.2007 [10] S. Cuzzocrea and D. Salvemini, “Molecular Mechanisms Involved in the Reciprocal Regulation of Cyclooxygenase and Nitric Oxide Synthase Enzymes,” Kidney Interna- tional, Vol. 71, No. 4, 2007, pp. 290-297. doi:10.1038/sj.ki.5002058 [11] B. L. Slomiany and A. Slomiany, “Ghrelin Suppression of Helicobacter Pylori-Induced Gastric Mucosal iNOS Is Mediated through the Inhibition of IKK-β Activation by cNOS-Dependent S-Nitrosylation,” Open Jo Biology Vol. 1, No. 1, 2011 urnal of Cel , pp. 1-10. doi:10.4236/ojcb l 1, 2009, pp. 86-99. [12] A. V. Grishin, J. Wang, D. A. Potoka, et al., “Lipopoly- saccharide Induces Cyclooxygenase-2 in Intestinal Epi- thelium via a Noncanonical p38 MAPK Pathway,” Jour- nal of Immunology, Vol. 176, No. 1, 2006, pp. 580-588. Y. J. Kang, B. A. Wingerd, T. Arakawa and W. L. Smith,[13] “Cyclooxygenase-2 Gene Transcription in a Macrophage Model of Inflammation,” Journal of Immunology, Vol. 177, No. 11, 2006, pp. 8111-8122. I Cho and S. G. Kim, “A novel Mitogen-Activated Pro- [14] tein Kinase Phosphatase-1 and Glucocorticoid Re- ceptor (GR) Interacting Protein-1-Dependent Combinato- rial Mechanism of Gene Transrepression by GR,” Mo- lecular Endocrinology, Vol. 23, No. doi:10.1210/me.2008-0257 [15] M. Caivano, B. Gorgoni, P. Cohen and V. Poli, “The Induction of Cyclooxygenase-2 mRNA in Macrophages Is Biphasic and Requires Both CCAAT Enhancer-Bind- ing Protein β (C/EBPβ) and C/EBPδTranscription Fac- tors,” Journal of Biological Chemistry, Vol. 276, No. 52, 2001, pp. 48693-48701. doi:10.1074/jbc.M108282200 [16] D. X. Hou, S. Masuzaki, F. Hashimoto, et al., (2007) “Green Tea Proanthocyanidins Inhibit Cyclooxygenase-2 Expression in LPS-Activated Mouse Macrophages: Mo- lecular Mechanisms and Structure-Activity Relationship,” Archives of Biochemistry and Biophysics, Vol. 460, No. 1, 2007, pp. 67-74. doi:10.1016/j.abb.2007.01.009 [17] B. L. Slomiany and A. Slomiany, “Involvement of p38 MAPK-Dependent Activator Protein (AP-1) Activation in Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori by Ghrelin,” Inflammopharmacol- ogy, 2012. doi:10.1007/s10787-012-0141-9 [18] S. Akira and K. Takeda, “Toll-Like Receptor Signaling,” Nature Reviews Immunology, Vol. 4, No. 7, 2004, pp. 499- 511. doi:10.1038/nri1391 [19] A. M. F. Liu and Y. H. Wong, “G16-Mediated Activation of Nuclear Factor B by the Adenosine A R 1 vol eceptor In- 074/jbc.M410196200 ves c-Src, Protein Kinase C, and ERK Signaling,” Journal of Biological Chemistry, Vol. 279, No. 51, 2004, pp. 53196-53204. doi:10.1 [20] R. Medzhitov and T. Horng, “Transcriptional Control of the Inflammatory Response,” Nature Reviews Immunol- ogy, Vol. 9, No. 10, 2009, pp. 692-703. doi:10.1038/nri2634 [21] B. L. Slomiany and A. Slomiany, “Cytosolic Phospholi- pase A2 Activation in Helicobacter pylori Lipopolysac- charide-Induced Interference with Gastric Mucin Synthe- sis,” IUBMB Life, Vol. 58, No. 4, 2006, pp. 217-223. 600732021doi:10.1080/15216540 pp. doi:10.1093/carcin/bgi112 [22] S. P. Newman, J. D. Croxtall, Q. Choudhury and R. J. Flower, “The Coordinate Regulation of Lipocortin 1, COX-2 and cPLA2 by IL-1β in A549 Cells,” Advances in Experimental Medicine and Biology, Vol. 407, 1997, 249-253. [23] M. Hughes-Fulford, R. R. Yjandrawinata, C. F. Li and S. Sayyah, “Arachidonic Acid, an Omega-6 Fatty Acid, In- duces Cytoplasmic Phospholipase A2 in Prostate Carci- noma Cells,” Carcinogenesis Vol. 26, No. 9, 2005, pp. 1520-1526. vr/cvp069 [24] C. C. Lin, W. N. Lin, W. J. Wang, et al., “Functional Coupling of COX-2 and cPLA2 Induced by ATP in Rat Vascular Smooth Muscle Cells: Role of ERK1/2, p38 MAPK, and NF-κB,” Cardiovascular Research, Vol. 82, No. 3, 2009, pp. 522-531. doi:10.1093/c Nitric [25] N. L. Reynaert, K. Ckless, S. H. Korn, et al., “Nitric Ox- ide Represses Inhibitory κB Kinase through S-Nitrosy- lation,” Proceedings of the National Academy of Sciences of theUSA, Vol. 101, No. 24, 2004, pp. 8945-8950. [26] S. F. Kim, D. A. Huri and S. H. Snyder, “Inducible Oxide Synthase Binds, Snitrosylates, and Activates Cyclo- Copyright © 2012 SciRes. OJCB  B. L. SLOMIANY, A. SLOMIANY Copyright © 2012 SciRes. OJCB 31 oxygenase-2,” Science, Vol. 310, No. 5756, 2005, pp. 1966-1970. doi:10.1126/science.1119407 [27] L. Xu, C. Han and T. Wu, “Activation of Cytosolic Phos- pholipase A2 through Nitric Oxide-Induced S-Nitrosy- lation. Involvement of Inducible Nitric-Oxide Synthase and Cyclooxygenase-2,” Journal of Biological Chemistry, Vol. 283, No. 6, 2008, pp. 3077 -3087. doi:10.1074/jbc.M705709200 [28] N. D. Perkins, “Integrating Cell-Signalling Pathways with NF-κB and IKK Function,” Nature Reviews Molecular Cell Biology, Vol. 8, No. 1, 2007, pp. 49-62 doi:10.1038/nrm2083 [29] B. L. Slomiany and A. Slomiany, “Involvement of Con- Inflammo- , No. 5, 2009, pp. 245-253. stitutive Nitric Oxide Synthase in Ghrelin-Induced Cyto- solic Phospholipase A2 Activation in Gastric Mucosal Cell Protection against Ethanol Cytotoxicity,” pharmacology, Vol. 17 doi:10.1007/s10787-009-0013-0 [30] B. L. Slomiany and A. Slomiany, “Modulation of gastric Mucosal Inflammatory Responses to Helicobacter pylori by Ghrelin: Role of cNOS-Dependent IKK-β S-Nitrosyla- tion in the Regulation of COX-2 Activation,” Ame Journal of Molecular Biology, Vol. 2, No. 2, rican 2012, pp . 113-123. doi:10.4236/ajmb [31] B. L. Slomiany and A. Slomiany, “Helicobacter pylori Induces Disturbances in Gastric Mucosal Akt Activation through Inducible Nitric Oxide Synthase-Dependent S-Nitrosylation: Effect of Ghrelin,” ISRN Gastroenterol- ogy, 2011, Article ID: 308727. doi:10.5402/2011/308727 [32] S. M. Noha, A. G. Atanasov, D. Schuster, et al., “Dis- covery of a Novel IKK-β Inhibitor by Ligand-Based Vir- tual Screening Techniques,” Bioorganic & Medicinal Chemistry Letters, Vol. 21, No. [33] S. R. Jaffrey, H. Erdjument 1, 20011, pp. 577-583. -Bromage, D. Ferris, P. Tempst and S. H. Snyder, “Protein S-Nitrosylation: A Physio- logical Signal for Neuronal Nitric Acid,” Nature Cell Bi- ology, Vol. 3, No. 2, 2001, pp. 193-197. doi:10.1038/35055104 [34] M. T. Forrester, M. W. Foster and J. S. Stamler, “As- sessment and Application of the Biotin Sw for Examining Protein itch Technique S-Nitrosylation under Conditions of Pharmacologically Induced Oxidative Stress,” Journal of Biological Chemistry, Vol. 282, No. 19, 2007, pp. 13977- 13983. doi:10.1074/jbc.M609684200 [35] K. W. Kang, S. Y. Choi, M. K. Cho, C. C. Lee and S. G. Kim, “Thrombin Induces Nitric-Oxide Synthase via Ga12/13- Coupled Protein Kinase C-Dependent I-κBα and JNK-Mediated I-κBα Degradation,” Journal of Biological Chemistry, Vol. 278, No. 19, 2003, pp. 17368-17378. doi:10.1074/jbc.M300471200 [36] T. Hirabayashi and T. Shimizu, “Localization and Regu- lation of Cytosolic Phospholipase A2,” Biochimica et Bi physica Acta, Vol. 1488, No. 1- o- 2, 2000, pp. 124-138. doi:10.1016/S1388-1981(00)00115-3 [37] P. Lodeiro, M. Theodoropoulou, M. Pardo, F. F. Casa- nueva and J. P. Camina, “c-Src Regulates Akt Signali in Response to Ghrelin via b-Arres ng tin Signaling-Inde- pendent and Dependent Mechanism,” PLoS ONE, Vol. 4, No. 3, 2009, p. e4686. doi:10.1371 [38] W. Wu, Z. Sun, J. Wu, et al., “Trihydrophobin 1 Phos- phorylatio by c-Src Regulates MAPK/ERK Signaling and Cell Migration,” PLoS One, Vol. 7, No. 1, 2012, p. e29920. doi:10.1371/journal.pone.0029920 [39] X. Xu, B. S. Jhun, C. H. Ha and Z. G. Jin, (2008) “Mo- lecular Mechanisms of Ghrelin-Mediated Endothelial Ni- tric-Oxide Synthase Activation,” Endocrinology, Vol. 149, No. 8, 2008, pp. 4183-4192. doi:10.1210/en.2008-0255
|