 Open Journal of Cell Biology, 2011, 1, 1-10 doi:10.4236/ojcb.2011.11001 Published Online December 2011 (http://www.SciRP.org/journal/ojcb) Copyright © 2011 SciRes. OJCB Ghrelin Suppression of Helicobacter pylori-Induced Gastric Mucosal Expression of iNOS is Mediated through the Inhibition of IKK-β Activation by cNOS-Dependent S-Nitrosylation Bronislaw L. Slomiany*, Amalia Slomiany University of Medicine and Dentistry of New Jersey, Newark, USA E-mail: *slomiabr@umdnj.edu Received November 7, 2011; revised December 3, 2011; accepted December 12, 2011 Abstract Excessive nitric oxide generation, caused by the disturbances in nitric oxide synthase (NOS) isozyme system, plays a key role in defining the extent of gastric mucosal inflammatory response to H. pylori infection. Here, we report that H. pylori LPS-induced enhancement in gastric mucosal inducible (i) iNOS expression and the impairment in constitutive (c) cNOS activity was associated with up-regulation in the inhibitory kB kinase-β (IKKβ) activation through phosphorylation, rise in IκB-α degradation, and the increase in the transcriptional factor, NF-κB, nuclear translocation. Further, we show that the countering effect of peptide hormone, ghrelin, on the LPS-induced disturbances in NOS isozyme system was reflected in the increase in Src/Akt-dependent cNOS activation through phosphorylation and the suppression of IKK-β activity through cNOSmediated IKK-β protein S-nitrosylation. As a consequence, ghrelin exerted the inhibitory effect on the LPS-induced rise in IκB-α degradation and NF-κB nuclear translocation, thus leading to iNOS gene suppression and the repression of iNOS induction. These results point to a central role of cNOS activation in controlling the sig- naling pathways of the LPS-triggered iNOS gene induction. Moreover, our findings suggest a molecular mechanism by which ghrelin suppresses the gastric mucosal proinflammatory consequences of H. pylori in- fection. Keywords: H. Pylori, Gastric Mucosa, Ghrelin, cNOS Activation, IKK-β S-Nitrosylation 1. Introduction Cell-wall lipopolysaccharide (LPS) of H. pylori, a Gram- negative bacterium colonizing the gastric mucosa of over 50% of the human population, is recognized as a potent endotoxin capable of eliciting mucosal inflammatory changes that characterize gastritis and duodenal ulcer [1-3]. The stimulation of gastric mucosa with H. pylori LPS is known to provoke a marked up-regulation in pro- inflammatory cytokine production, massive rise in epi- thelial cell apoptosis, and the excessive nitric oxide (NO) generation caused by the disturbances in nitric oxide synthase (NOS) system [4-6]. A growing body of evi- dence, moreover, points to the disturbances in NO pro- duction associated with H. pylori colonization, and re- flected in continual activation of iNOS and the suppres- sion of cNOS isozyme system, as the event defining the extent of gastric mucosal inflammatory involvement. [4, 7-9]. Moreover, studies indicate that the expression of proinflammatory mediators induced by H. pylori infec- tion is regulated by the activity of the nuclear transcrip- tion factor, NF-κB [6,10-12]. Indeed, NF-κB is ubiquitously present in all types of mammalian cells, where it controls a variety of genes involved in immune, inflammatory, and proliferative res- ponses, including those of the NOS isozyme system that are responsible for NO production [12-14]. While the factor is known to consist of five homo- and heter- odimers of the Rel protein family, the most characterized NF-κB complex is a p50/p65 hetrodimer, which is se- questered in the cytoplasm of resting cells bound to a family of inhibitory IκB proteins [14-16]. Activation of NF-κB in response to such stimuli as TNF-α or LPS oc- curs through a rapid IκB-α phosphorylation by an IκB  B. L. SLOMIANY ET AL. 2 kinase (IKK) complex, that targets IκB-α for degradation through the ubiquitin-proteasomal pathway, thus allow- ing nuclear translocation of NF-κB, its binding to pro- moter response elements, and activation of the transcrip- tion of genes implicated in immunoregulation, inflam- mation and cellular proliferation [14-17]. The IKK complex consists of two catalytic subunits, IKK-α and IKK-β, and one regulatory subunit, IKK-γ, also known as NEMO (NF-κB essential modulator) [12, 14,15,17]. While the activation of IKK complex requires phosphorylation of IKK-α and IKK-β on the specific conserved serine residues in the activation loops, the IKK-γ regulatory subunit appears to be responsible for IKK complex stabilization and the interaction with up- stream regulatory proteins [15,17]. Furthermore, al- though IKK-α and IKK-β show a high degree of homol- ogy in their structural domains, the available literature indicates that IKK-β displays up to 50 fold higher level of kinase activity towards IκB proteins, and the gene deletion studies demonstrated that it is IKK-β and not IKK-α which is responsible for NF-κB activation in re- sponse to stimulation by TNF-α or bacterial LPS [18- 20]. In addition, recent reports indicate that the activity of IKK complex may also be regulated through S-nitrosy- lation of a specific cysteine residue within the activation loop of IKK-β, by endogenous NO donors, leading to suppression of IκB-α degradation and the inhibition of NF-κB nuclear translocation, thus blocking the transcrip- tion of NF-κB-regulated gene products [17,21,22]. As peptide hormone, ghrelin, is recognized as an im- portant modulator of gastric mucosal inflammatory re- sponses to H. pylori through the regulation of NOS sys- tem [4,9,23-26], in this study we investigated the effect of H. pylori LPS on the activation of NF-κB signaling pathway in gastric mucosal cells, and the mechanism of ghrelin modulatory action. Our results demonstrate that the LPS elicit induction in iNOS expression via up- regulation in IKK-β activation through phosphorylation. We further show that ghrelin inhibits the LPS-induced activation of IKK-β through cNOS-mediated IKK-β pro- tein S-nitrosylation, thus leading to suppression of IκB-α phosphorylation and degradation, inhibition of NF-κB nuclear translocation, and the repression of iNOS gene induction. 2. Materials and Methods 2.1 Gastric Mucosal Cell Incubation The cells were collected by scraping the mucosa of freshly dissected rat stomachs with a blunt spatula, and suspended in five volumes of ice-cold Dulbecco’s modi- fied (Gibco) Eagle’s minimal essential medium (DMEM), supplemented with fungizone (50 µg/ml), penicillin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf se- rum. The cells were then gently dispersed by trituration with a syringe, settled by centrifugation, and following rinsing resuspended in the medium to a concentration of 2 × 107 cell/ml [9]. Cell aliquots (1 ml) were then trans- ferred to DMEM in culture dishes and incubated under 95% O2 - 5% CO2 atmosphere at 37˚C for 16 h in the presence of 0 - 100 ng/ml of H. pylori LPS [25]. H. py- lori used for LPS preparation was cultured from clinical isolates obtained from ATCC No. 4350 [5]. In the ex- periments evaluating the effect of ghrelin (rat, Sigma), cNOS inhibitor, L-NAME, iNOS inhibitor, 1400W, Akt inhibitor, SH-5, Src inhibitor, PP2 (Calbiochem), and ascorbate (Sigma), the cells were first preincubated for 30 min with the indicated dose of the agent or vehicle before the addition of the LPS. The viability of cell pre- parations before and during the experimentation, asse- ssed by Trypan blue dye exclusion assay [9], was greater than 97%. 2.2. cNOS and iNOS Activity Assay Nitric oxide synthase activities of cNOS and iNOS en- zymes in the gastric mucosal cells were measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline using NOS-detect kit (Stratagene). The cells from the control and experimental treatments were ho- mogenized in a sample buffer containing either 10 mM EDTA (for Ca2+-independent iNOS) or 6 mM CaCl2 (for Ca2+-depenedent cNOS), and centrifuged. The ali- quots of the resulting supernatant were incubated for 30 min at 25˚C in the presence of 50 µCi/ml of L-[3H] ar- ginine, 10 mM NAPDH, 5 µM tetrahydrobiopterin, and 50 mM Tis-HCl buffer, pH 7.4, in a final volume of 250 µl. Following addition of stop buffer and Dowex-50 W (Na+) resin, the mixtures were transferred to spin cups, centrifuged and the formed L-[3H] citrulline contained in the flow through was quantified by scintillation counting. 2.3. Nuclear Protein Extraction The aliquots of gastric mucosal cell suspension from the control and various experimental conditions were settled by centrifugation at 1500 xg for 5 min, rinsed with phos- phate-buffered saline, and lysed by incubation for 10 min on ice in the lysis buffer, containing 10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5% Nonidet P-40, 1 mM dithiothreitol, and 0.5 mM PMSF [13]. Following centrifugation at 12,000 xg for 10 min at 4˚C, the super- natant was subjected to centrifugation at 100,000 xg for 1h and the obtained soluble fraction was used as source of cytosolic extract [27]. The pellets, from 12,000 xg Copyright © 2011 SciRes. OJCB  B. L. SLOMIANY ET AL.3 centrifugation, containing crude nuclei were suspended for 20 min at 4˚C in the extraction buffer, containing 20 mM HEPES, pH 7.9, 25% glycerol, 400 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM PMSF. The samples were centrifuged at 15,000 xg for 10 min at 4˚C, and the supernatants containing nu- clear extracts were collected and stored at −70˚C until use. 2.4. IκB Kinase Activity Assay To measure the IKK-β activity we utilized the ELISA- based detection kit, K-LISATM (Calbiochem). The GST- IkB-a 50-amino acid peptide that includes the Ser32 and Ser36 of IkB-α phosphorylation sites was used as a sub- strate [28]. The gastric mucosal cell cytosolic extracts were incubated a glutathione-coated 96-well plate with GST-tagged IκB-α at room temperature for 30 min, and the phosphorylated GST-IκB-α substrate was detected using anti-phospho IkB-α (Ser32/Ser36) as first antibody, followed by horseradish peroxidase-conjugated secon- dary antibody. Following washing the retained complex was probed TMB reagent for spectrophotometric quanti- fication at 450 nm. 2.5. IKK-β Protein S-Nitrosylation Assay A biotin switch procedure was employed to assess IKK-β protein S-nitrosylation [29,30]. The gastric mucosal cells were treated with ghrelin (0.5 µg/ml), or Akt inhibitor, SH-5 (20 µM) + ghrelin (0.5 µg/ml), and incubated for 1h in the presence of 100ng/ml of H. pylori LPS. Fol- lowing centrifugation at 500 xg for 5 min, the recovered cells were lysed in 0.2 ml of HEN lysis buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM neocuprin, pH 7.7), and the unnitrosylated thiol groups were blocked with S- methyl methanethiosulfonate reagent at 50˚C for 20 min [30]. The proteins were precipitated with acetone, resus- pended in 0.2 ml of HEN buffer containing 1% SDS, and subjected to targeted nitrothiol group reduction with so- dium ascorbate (100 mM). The free thiols were then la- beled with biotin and the biotinylated proteins were re- covered on streptavidin beads. The formed streptavidin bead-protein complex was washed with neutralization buffer, and the bound proteins were dissociated from streptavidin beads with 50 µl of elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, pH 7.7) contain- ing 1% 2-mercaptoethanol [30]. The obtained proteins were then analyzed by Western blotting. 2.6. Immunoblotting Analysis The mucosal cells from the control and experimental treatments were collected by centrifugation and resus- pended for 30 min in ice-cold lysis buffer (20 mM Tris- HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X- 100, 2 mM EDTA, 1 mM sodium orthovanadate, 4 mM sodium pyrophosphate, 1 mM PMSF, and 1 mM NaF), containing 1 µg/ml leupeptin and 1 µg/ml pepstatin [25]. Following brief sonication, the lysates were centrifuged at 12,000 g for 10 min, and the supernatants were col- lected and normalized with respect to protein concentra- tion using BCA protein assay kit (Pierce). The samples, including those subjected to biotin switch procedure, were then resuspended in loading buffer, boiled for 5 min, and subjected to SDS-PAGE using 40 µg protein/ lane. The separated proteins were transferred onto nitro- cellulose membranes, blocked for 1 h with 5% skim milk in Tris-buffered Tween (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and probed with specific polyclonal rabbit antibodies directed against IκB-α and cNOS (Calbiochem), phospho-IκB-α and phospho-IKK- β (Cell Signaling), NF-κB p65 and IKK-β (EMD Milli- pore). The phosphorylated cNOS (pcNOS) was analyzed using specific antibody (Calbiochem) directed against phospho-cNOS (mouse anti-eNOS, pSer1179). 2.7. Data Analysis All experiments were carried out using duplicate samp- ling, and the results are expressed as means ± SD. Ana- lysis of variance (ANOVA) and nonparametric Kruskal- Wallis tests were used to determine significance. Any difference detected was evaluated by means of post hoc Bonferroni test, and the significance level was set at P < 0.05. 3. Results We have reported that the modulatory effect of ghrelin on gastric mucosal inflammatory responses to H. pylori LPS, characterized by the disturbances in NO production, was reflected in the cNOS activation through phos- phorylation and the suppression of iNOS protein expres- sion [9,25]. Indeed, as illustrated in Figure 1, the LPS at 100 ng/ml elicited a 19.8-flold increase in the mucosal cell iNOS activity, whereas the cNOS activity showed a 4.3-fold decrease. Moreover, preincubation with ghrelin at 0.5 µg/ml lead to a 90.2% reduction in the LPS-in- duced iNOS activity, while the activity of cNOS in- creased by a 77%. Since iNOS expression is controlled primarily by the transcription factor, NF-κB [10-12], we further investigated the effect on ghrelin on the LPS- induced NF-κB activation. As NF-κB activation requires phosphorylation and degradation of the inhibitory protein IκB-α, we exposed Copyright © 2011 SciRes. OJCB 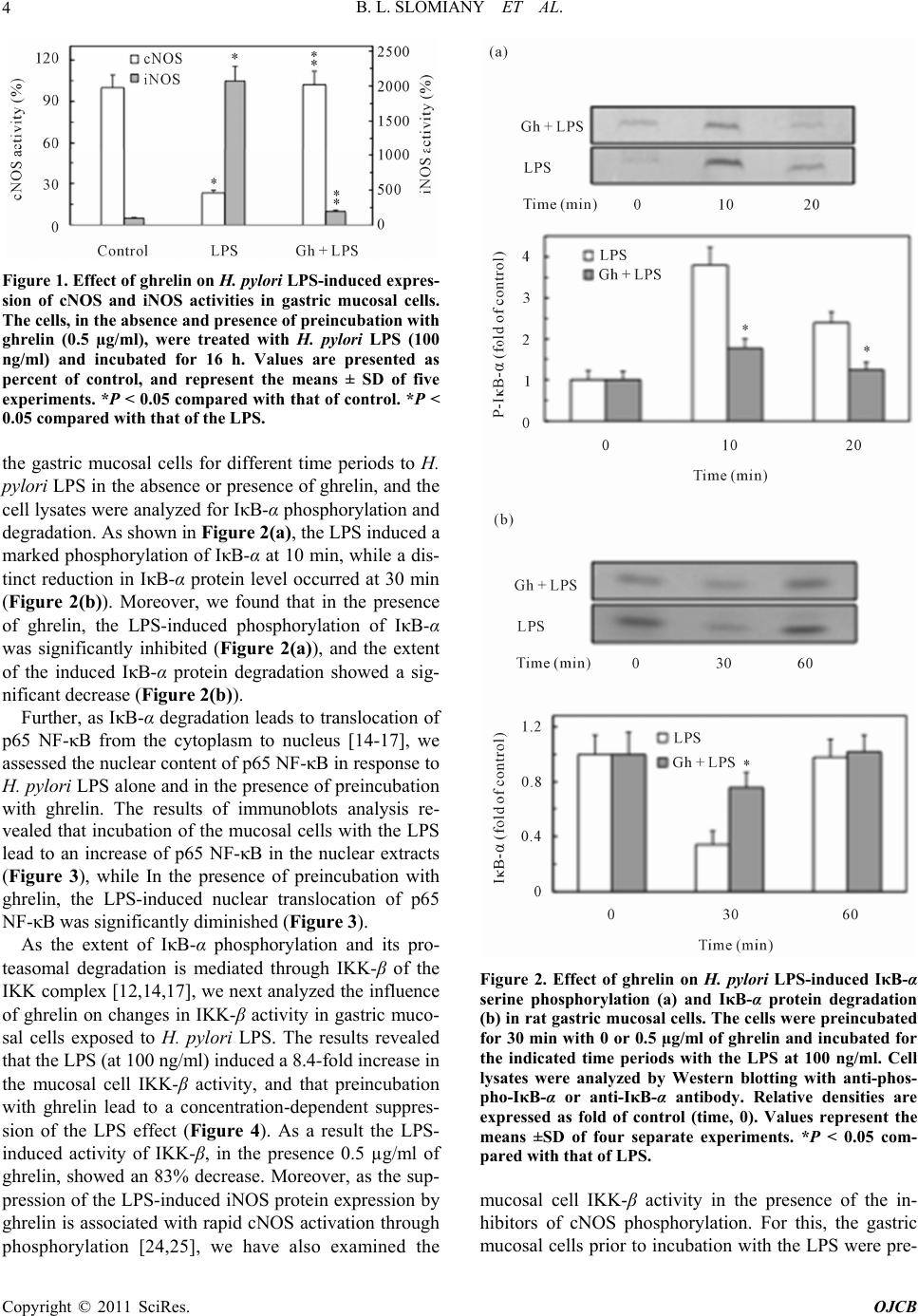 B. L. SLOMIANY ET AL. 4 Figure 1. Effect of ghrelin on H. pylori LPS-induced expres- sion of cNOS and iNOS activities in gastric mucosal cells. The cells, in the absence and presence of preincubation with ghrelin (0.5 µg/ml), were treated with H. pylori LPS (100 ng/ml) and incubated for 16 h. Values are presented as percent of control, and represent the means ± SD of five experiments. *P < 0.05 compared with that of control. *P < 0.05 compared with that of the LPS. the gastric mucosal cells for different time periods to H. pylori LPS in the absence or presence of ghrelin, and the cell lysates were analyzed for IκB- α phosphorylation and degradation. As shown in Figure 2(a), the LPS induced a marked phosphorylation of IκB-α at 10 min, while a dis- tinct reduction in IκB-α protein level occurred at 30 min (Figure 2(b)). Moreover, we found that in the presence of ghrelin, the LPS-induced phosphorylation of IκB-α was significantly inhibited (Figure 2(a)), and the extent of the induced IκB-α protein degradation showed a sig- nificant decrease (Figure 2(b)). Further, as IκB-α degradation leads to translocation of p65 NF-κB from the cytoplasm to nucleus [14-17], we assessed the nuclear content of p65 NF-κB in response to H. pylori LPS alone and in the presence of preincubation with ghrelin. The results of immunoblots analysis re- vealed that incubation of the mucosal cells with the LPS lead to an increase of p65 NF-κB in the nuclear extracts (Figure 3), while In the presence of preincubation with ghrelin, the LPS-induced nuclear translocation of p65 NF-κB was significantly diminished (Figure 3). As the extent of IκB-α phosphorylation and its pro- teasomal degradation is mediated through IKK-β of the IKK complex [12,14,17], we next analyzed the influence of ghrelin on changes in IKK-β activity in gastric muco- sal cells exposed to H. pylori LPS. The results revealed that the LPS (at 100 ng/ml) induced a 8.4-fold increase in the mucosal cell IKK-β activity, and that preincubation with ghrelin lead to a concentration-dependent suppres- sion of the LPS effect (Figure 4). As a result the LPS- induced activity of IKK-β, in the presence 0.5 µg/ml of ghrelin, showed an 83% decrease. Moreover, as the sup- pression of the LPS-induced iNOS protein expression by ghrelin is associated with rapid cNOS activation through phosphorylation [24,25], we have also examined the Figure 2. Effect of ghrelin on H. pylori LPS-induced IκB-α serine phosphorylation (a) and IκB-α protein degradation (b) in rat gastric mucosal cells. The cells were preincubated for 30 min with 0 or 0.5 µg/ml of ghrelin and incubated for the indicated time periods with the LPS at 100 ng/ml. Cell lysates were analyzed by Western blotting with anti-phos- pho-IκB-α or anti-IκB-α antibody. Relative densities are expressed as fold of control (time, 0). Values represent the means ±SD of four separate experiments. *P < 0.05 com- pared with that of LPS. mucosal cell IKK-β activity in the presence of the in- hibitors of cNOS phosphorylation. For this, the gastric mucosal cells prior to incubation with the LPS were pre- Copyright © 2011 SciRes. OJCB  B. L. SLOMIANY ET AL.5 Figure 3. Effect of ghrelin on H. pylori LPS-induced nuclear translocation of p65 NF-κB in rat gastric mucosal cells. The cells were preincubated for 30 min with 0 or 0.5 µg/ml of ghrelin and incubated for 30 min with the LPS at 100 ng/ml. Cells were lysed and the level of p65 NF-κB protein in the nuclear fraction was analyzed immunochemically. Relative densities are expressed as fold of control. The data repre- sent the means ± SD of four separate experiments. *P < 0.05 compared with that of LPS. Figure 4. Effect of ghrelin on H. pylori LPS-induced changes in IKK-β activity in gastric mucosal cells. The cells, prein- cubated for 30 min with the indicated concentrations of ghrelin, were treated with the LPS at 100 ng/ml and incu- bated for 30 min. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. treated with the inhibitors of Src/Akt pathway in the ab- sence or presence of ghrelin, and assayed for IKK-β ac- tivity. We found that the countering effect of ghrelin on H. pylori LPS-induced up-regulation in the mucosal cell IKK-β activity was subject to suppression by both, the inhibitor of Src, PP2, as well as Akt inhibitor, SH-5 (Figure 5). A significant decrease in the effectiveness of ghrelin on the LPS-induced IKK-β activation, further- more, was observed in the presence of cNOS inhibitor, L-NAME, whereas the inhibitor of iNOS, 1400 W, had no effect (Figure 5). Further, examination of the effect of H. pylori LPS and ghrelin on IKK-β and cNOS phos- Figure 5. Effect of Src inhibitor, PP2, Akt inhibitor, SH-5, cNOS inhibitor, L-NAME, and iNOS inhibitor, 1400 W, on the ghrelin (Gh)-induced changes in IKK-β activity in gas- tric mucosal cells exposed to H. pylori LPS. The cells, pre- incubated with 30 µM PP2, 20 µM SH-5 (SH), 200 µM L-NAME (LN), or 40 µM 1400W (14W), were treated with Gh at 0.5 µg/ml and incubated for 30 min in the presence of 100 ng/ml of LPS. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. ***P < 0.05 com- pared with that of Gh LPS. phorylation revealed that the LPS-induced up-regulation in IKK-β activity was associated with a marked increase in IKK-β phosphorylation and the suppression in cNOS protein phosphorylation (Figure 6). Moreover, while preincubation with ghrelin lead to the increase in cNOS phosphorylation that was susceptible to suppression by Akt inhibitor, SH-5, ghrelin produced no apparent effect of the extent of the LPS-induced IKK-β phosphorylation (Figure 6). Thus, the countering effect of ghrelin on H. pylori LPS-induced up-regulation in gastric mucosal IKK-β activation shows an apparent dependence on NO generated by the cNOS system. To ascertain further the role of NO generated by cNOS system in the modulatory influence of ghrelin on H. py- lori LPS-induced up-regulation in gastric mucosal IKK-β activation, we assessed the effect of nitrosothiols reduc- ing agent, ascorbate. As show in Figure 7, preincubation with ascorbate lead to a significant relieve in the inhibi- tory effect of ghrelin on the LPS-induced IKK-β activity. However, ascorbate produced no discernible effect on the extent of the LPS-induced IKK-β activation. More- over, we also examined the dependence of IKK-β S-ni- trosylation on the ghrelin-induced cNOS activation through Akt-mediated phosphorylation by the biotin switch method [29,30]. The gastric mucosal cells were incubated with H. pylori LPS or ghrelin + LPS in the absence and presence of Akt inhibitor, SH-5, and the lysates following the biotin switch procedure were ex- amined for IKK-β protein S-nitrosylation (Figure 8). Western blot analysis revealed that ghrelin countering effect on the LPS-induced up-regulation in IKK-β active- Copyright © 2011 SciRes. OJCB  B. L. SLOMIANY ET AL. 6 Figure 6. Effect of H. pylori LPS and ghrelin (Gh) on cNOS and IKK-β phosphorylation in gastric mucosal cells ex- posed to Akt inhibitor, SH-5 (SH). The cells were preincu- bated for 30 min with Gh at 0.5 µg/ml, or SH (20 µM) + Gh, and incubated for 1 h in the presence of 100 ng/ml of LPS. Cell lysates were resolved on SDS-PAGE, transferred to nitrocellulose and probed with phosphorylation specific cNOS (pcNOS) and pIKKβ antibody, and after stripping reprobed with anti-cNOS and anti-IKKβ antibody. The immunoblots shown are representative of three experi- ments. Figure 7. Effect of ascorbate on the ghrelin (Gh)-induced changes in the expression of IKK-β activity in gastric mu- cosal cells exposed to H. pylori LPS. The cells, preincubated with 300 µM ascorbate (As), were treated with Gh at 0.5 µg/ml and incubated for 30 min in the presence of 100 ng/ml LPS. Values represent the means ± SD of five ex- periments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. ***P < 0.05 com- pared with that of Gh LPS. ity was manifested in a marked increase in the enzyme protein S-nitrosylation, and that this effect of ghrelin on IKK-β S-nitrosylation was subject to suppression by Akt inhibitor, SH-5. These data suggest that ghrelin counter- ing effect of the detrimental consequences of up-regula- tion in IKK-β activity by H. pylori LPS is mediated through cNOS-dependent IKK-β protein S-nitrosylation. 4. Discussion Infection with gastric pathogen, H. pylori, initiates the course of mucosal inflammatory events that increases the risk of chronic gastritis, peptic ulcer disease, and gastric Figure 8. Effect of H. pylori LPS and ghrelin (Gh) on IKK-β S-nitrosylation in gastric mucosal cells exposed to Akt in- hibitor, SH-5 (SH). The cells, were preincubated for 30 min with Gh at 0.5 µg/ml, or SH at (20 µM) + Gh, and incubated for 1 h in the presence of 100 ng/ml LPS. A portion of the cell lysates was processed by biotin switch procedure for protein S-nitrosylation and, along with the reminder of the lysates, resolved on SDS-PAGE, transferred to nitrocellulose probed with anti-IKKbantibody. The immunoblots shown are representative of three experiments. adenocarcinoma [1-3,12]. Indeed, colonization of gastric mucosa by H. pylori in humans or stimulation of gastric mucosal cells with H. pylori LPS is known to induce up-regulation in proinflammatory cytokine release, en- hances the epithelial cell apoptosis, and causes the exces- sive NO generation due to disturbances in NOS isozyme system [4-7,9]. These events define the extent of gastric mucosal inflammatory involvement, and are carefully regulated at the early response stage by a family of tran- scriptional factors, known as NF-κB, which transduce the inflammatory stimulus into the activation of a variety genes involved in ant-microbial responses, including those accountable for NO generation [6, 10-12, 31,32]. As gas- tric mucosal inflammatory responses to H. pylori as well as its LPS are reflected in continual induction of iNOS and the suppression of cNOS activation, and a peptide hormone, ghrelin, is recognized as an important regulator of NOS system [4,5,7,9,23-26], the objective of this stu- dy was to ascertain the influence of ghrelin on the proc- esses associated with NF-κB activation by H. pylori LPS. The data obtained with rat gastric mucosal cells re- vealed that H. pylori LPS-induced enhancement in the expression of iNOS and the impairment in cNOS activity was associated with up-regulation in IKK-β activation through phosphorylation, rise in IκB-α degradation, and the increase in NF-κB nuclear translocation. These find- ings are thus in keeping with the preponderant literature evidence as to the involvement of bacterial LPS in the classical signaling pathway of NF-κB activation [14-17]. In this pathway, NF-κB activity is regulated by cytosolic sequestration through interaction with inhibitory IκB proteins. Activation of NF-κB by proinflammatory stim- uli, such as LPS or TNF-α, involves phosphorylation of the inhibitory IκB-α at two conserved N-terminal serine residues (Ser32 and Ser36) by IκB-kinase (IKK) complex, a process that signals IκB-α for degradation by the ubiq- uitin-proteasomal pathway [15-18]. Then, the released Copyright © 2011 SciRes. OJCB  B. L. SLOMIANY ET AL. Copyright © 2011 SciRes. OJCB 7 activated NF-κB, usually consisting of p50 and p65 sub- units, translocates to the nucleus, and activates the tran- scription of various proinflammatory target genes, in- cluding those coding for cytokines, apoptotic factors, and iNOS [10-12,31,32]. Indeed, we have found earlier that the induction in gastric mucosal iNOS enzyme protein expression in response to H. pylori LPS was susceptible to suppression by the inhibitor of NF-κB activation, PM-18 as well as peptide hormone, ghrelin [9,33]. Fur- thermore, as shown herein, ghrelin exerted the inhibitory effect on H. pylori LPS-induced rise in gastric mucosal IkB-β degradation and NF-κB nuclear translocation. Hence, to reveal further insights into the mechanism by which ghrelin exerts modulatory control over gastric mucosal inflammatory disturbances caused by H. pylori infection, we examined the influence of ghrelin on H. pylori LPS-induced changes in the activity of IKK-β a key enzyme of NF-κB activation pathway that controls the extent of IκB-α phosphorylation and its proteasomal degradation [12,14,17]. We found that countering effect of ghrelin on the LPS-induced up-regulation in gastric mucosal cell IKK-β activity was subject to suppression by Src inhibitor, PP2 as well as Akt inhibitor, SH-5. A significant decrease in the effectiveness of ghrelin to counter the LPS-induced IKK-β activation was also ob- served in the presence of cNOS inhibitor, L-NAME, while the inhibitor of iNOS, 1400 W, had no effect. Fur- thermore, in consonance with the documented involve- ment of Akt in rapid cNOS activation through phos- phorylation at Ser1179 [9,24,33], ghrelin-induced up- regulation in cNOS activity was reflected in the increase of enzyme phosphorylation that was susceptible to sup- Figure 9. Schematic diagram illustrating modulatory mechanism of ghrelin action in countering the gastric mucosal proin- flammatory events triggered by H. pylori LPS-induced NF-κB activation. Binding of the LPS to Toll-like receptor 4 (TLR4)/ MD2 leads to IKK complex activation by phosphorylation at its IKK-βcatalytic subunit, thus triggering up-regulation in IkB-α phosphorylation and its proteasomal degradation. This causes the increase in NF-κB translocation to the nucleus to promote iNOS gene transcription. Ghrelin, through binding to growth hormone secretagogue receptor (GHSR), inhibits the LPS-in- duced IKK activation via up-regulation in Src/Akt-dependent cNOS activation that results in cNOS-mediated IKK-β S-nitro- sylation, thus leading to suppression of IκB-α phosphorylation and degradation, inhibition of NF-κB nuclear translocation, and the repression of iNOS gene transcription.  B. L. SLOMIANY ET AL. Copyright © 2011 SciRes. OJCB 8 pression by Akt inhibitor, SH-5. However, the hormone produced no apparent effect on the extent of the LPS- induced phosphorylation of IKK-β. From this, we con- cluded that the countering effect of ghrelin on H. pylori LPS-induced up-regulation in gastric mucosal IKK-β activation and iNOS induction occurs with the involve- ment of Akt-mediated cNOS activation. Indeed, signal- ing through Src/Akt pathway is known to occupy a cen- tral stage in the receptor (GHS-R)-mediated responses to ghrelin stimulation [26,34], and the recent reports sug- gest that the activity of IKK complex may also be subject to inhibition through S-nitrosylation of a specific cys- teine residue within the activation loop of IKK-β by the endogenous NO donors [21,22,31]. Therefore, to address further the role of cNOS in the regulation of IKK-β activation by ghrelin, we assessed the effect of nitrosothiol reducing agent, ascorbate. We found that while ascorbate produced no discernible effect on the extent of the LPS-induced IKK-β activation, the agent elicited a marked relieve in the inhibitory effect of ghrelin on the LPS-induced IKK-β activity. Hence, tak- ing into consideration known susceptibility of S-nitro- sylated proteins to reduction by ascorbic acid [26,29,30, 33], we surmised that the countering effect of ghrelin on H. pylori LPS-induced IKK-β activation are intimately linked to the events of IKK-β protein S-nitrosylation by NO generated by the cNOS system. Indeed, examination of IKK-β S-nitrosylation by the biotin switch method, revealed that ghrelin countering effect on the LPS-in- duced up-regulation in IKK-β activity was manifested by a marked increase in the kinase protein S-nitrosylation, and subject to suppression by Akt inhibitor, SH-5. These findings, together with the results of IKK-β activity as- says, suggest that proinflammatory events elicited in gastric mucosa by H. pylori and manifested by the induc- tion in iNOS expression are the result of the LPS-in- duced up-regulation in IKK-β activation through phos- phorylation. Further, it is also evident that ghrelin exerts the inhibitory effect on the LPS-induced activation IKK-β via cNOS-mediated IKK-β S-nitrosylation, thus leading to suppression in IκB-α phosphorylation and degradation, inhibition in of NF-κB nuclear translocation, and the repression of iNOS gene transcription. Conse- quently, the role of ghrelin in modulation of the extent of gastric mucosal inflammatory involvement appears to be intimately linked to signaling events associated with Src/Akt-mediated cNOS activation through phosphoryla- tion. Taken together, our results suggest that the initial phase gastric mucosal proinflammatory events triggered by H. pylori involves the disturbances in Src/Akt kinase-de- pendent cNOS phosphorylation that leads to abrogation of cNOS-mediated control over IKKβ activity through S-nitrosylation and the increase in proteasomal degrada- tion of IκB-α, thus allowing NF-κB translocation to the nucleus to promote iNOS gene transcription. We also re- port that ghrelin counters these untoward consequences of H. pylori LPS via up-regulation in Src/Akt-dependent cNOS activation through phosphorylation that results in up-regulation in cNOS-mediated IKK-β S-nitrosylation which interferes with IκB-α phosphorylation and protea- somal degradation, thereby preventing the nuclear trans- location of NF-κB and the activation of iNOS gene tran- scription. A schematic diagram illustrating the proposed mechanism of anti-H. pylori action of ghrelin is depicted in Figure 9. 5. References [1] M. Stolte and S. Edit, “Helicobacter Pylori and Evolution of Gastritis,” Scandinavian Journal of Gastro-enterology, Vol. 31, No. 214, 1996, pp. 13-16. doi:10.3109/00365529609094508 [2] J. Piotrowski, E. Piotrowski, D. Skrodzka, A. Slomiany and B. L. Slomiany, “Induction of Acute Gastritis and Epithelial Cell Apoptosis by Helicobacter pylori lipopoly- saccharide,” Scandinavian Journal of Gastroenterology, Vol. 32, 1997, pp. 203-211. doi:10.3109/00365529709000195 [3] W. A. de Boer, “Helicobacter Pylori Infection: Focus on a ‘Search-And-Treat’ Strategy for Ulcer Disease,” Scan- dinavian Journal of Gastroenterology, Vol. 35, No. 232, 2000, pp. 4-9. [4] F. Fu, K. S. Ramanujan, A. Wong, et al., “Increased Ex- pression and Cellular Localization of Inducible Nitric O- xide Synthase and Cyclooxygenase-2 in Helicobacter pylori Gastritis,” Gastroenterology, Vol. 116, No. 6, 1999 pp. 1319-1329. doi:10.1016/S0016-5085(99)70496-8 [5] B. L. Slomiany, J. Piotrowski and A. Slomiany, “Gastric Mucosal Inflammatory Responses to Helicobacter pylori lipopolysaccharide: Down-Regulation of Nitric Oxide Synthase-2 and Caspase-3 by Sulglycotide,” Biochemi- caland Biophysical Research Communications, Vol. 261, No. 1, 1999, pp. 15-20. doi:10.1006/bbrc.1999.1003 [6] R. A. Gupta, D. B. Polk, U. Krishna, et al., “Activation of Peroxisome Proliferator-Activated Receptor g Suppresses Nuclear Factor kB-Mediated Apoptosis Induced by Heli- cobacter pylori in Gastric Epithelial Cells,” Journal of Biological Chemistry, Vol. 276, No. 33, 2001, pp. 31059- 31066. doi:10.1074/jbc.M104141200 [7] G. Reider, J. A. Hofmann, R. A. Hatz, M. Stolte and G. A. Enders, “Up-Regulation of Inducible Nitric Oxide Syn- thase in Helicobacter pylori-Associated Gastritis may Represent an Increased Risk Factor to Develop Gastric Carcinoma of the Intestinal Type,” International Journal of Medical Microbiology, Vol. 293, No. 6, 2003, pp. 403- 412. doi:10.1078/1438-4221-00280 [8] S. Cuzzocrea and D. Salvemini, “Molecular Mechanisms Involved in the Reciprocal Regulation of Cyclooxy-  B. L. SLOMIANY ET AL.9 Genase and Nitric Oxide Synthase Enzymes,” Kidney In- ternational, Vol. 71, No. 4, 2007, pp. 290-297. doi:10.1038/sj.ki.5002058 [9] B. L. Slomiany and A. Slomiany, “Helicobacter pylori Induces Disturbances in Gastric Mucosal Akt Activation through Inducible Nitric Oxide Syn-Thase-Dependent S- Nitrosylation: Effect of Ghrelin,” ISRN Gastroenterology, Article No. 308727, 2010. doi:10.5402/2011 [10] S. Brandt, T. Kwok, R. Harting, W. Konig and S. Backert, “NF-κB Activation and Potentiation of Proinflammatory Responses by the Helicobacter pylori CagA Protein,” Proceedings of the National Academy of Sciences of the USA, Vol. 102, No. 26, 2005, pp. 9300-9305. doi:10.1073/pnas.0409873102 [11] R. L. Ferrero, P. Ave, D. Nadiaye, et al., “NF-kB Activa- tion During Acute Helicobacter pylori Infection in Mice,” Infection and Immunity, Vol. 76, No. 2, 2008, pp. 551- 561. doi:10.1128/IAI.01107-07 [12] S. Backert and M. Neumann, “What a Disorder: Proin- Flammatory Signaling Pathways Induced by Helicobacter pylori,” Trends in Microbiology, Vol. 18, No. 11, 2010, pp. 479-486. doi:10.1016/j.tim.2010.08.003 [13] K. W. Kang, S. Y. Choi, M. K. Cho, C. C. Lee and S. G. Kim, “Thrombin Induces Nitric-Oxide Synthase via Ga12/13- Coupled Protein Kinase C-Dependent I-kBa Phos-pho- rylation and JNK-Mediated I-kBa Degradation,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 17368- 17378. doi:10.1074/jbc.M300471200 [14] K. Singh, R. Chaturvedi, M. Asim, D. P. Barry, N. D. Lewis, M. P. Vitek and K. T. Wilson, “The Apolipopro- tein E-Mimetic Peptide COG112 Inhibits the Inflamma- tory Response to Citrobacter rodentium in Colonic Epi- thelial Cells by Preventing NF-kB Activation,” Journal of Biological Chemistry, Vol. 283, No. 24, 2008, pp. 16752- 16761. doi:10.1074/jbc.M710530200 [15] H. Tanaka, N. Fujita and T. Tsuruo, “3-Phospho-Inosi- tide-Dependent Protein Kinase-1-Mediated IκB Kinase β (IKKB) Phosphorylation Activates NF-κB Signaling,” Journal of Biological Chemistry, Vol. 280, No. 49, 2005, pp. 40965-40973. doi:10.1074/jbc.M506235200 [16] J. L. Kang, H. W. Lee, H. J. Kim, H. S. Lee, V. Cas- tranova, C. M. Lim and Y. Koh, “Inhibition of Src Tyro- sine Kinase β Suppresses Activation of Nuclear Fac- tor-kB, and Serine and Tyrosine Phosphorylation of IκB-a in Lipopolysaccharide-Stimulated Raw 264.7 Ma- crophages,” Journal of Toxicology and Environmental Health, Part A, Vol. 68, 2005, pp. 1643-1662. doi:10.1080/15287390500192114 [17] S. C. Gupta, S. Prasad, S. Reuter, et al., “Modification of Cysteine 179 of IkBa Kinase by Nimbolide Leads to Down-Regulation of NF-kB-Regulated Cell Survival and Proliferative Proteins and Sensitization of Tumor Cells to Chemotherapeutic Agents, Journal of Biological Chemis- try, Vol. 285, No. 46, 2010, pp. 35406-35417. doi:10.1074/jbc.M110.161984 [18] H. Nakano, M. Shinodo, S. Sakon, S. Nishinaka, M. Mi- hara, H. Yagita and K. Okumura, “Differential Regula- tion of IκB Kinase a and b by Two Upstreamkinases, NF-κB-Inducing Kinase and Mitogen-Activated Protein Kinase/ERK Kinase-1,” Proceedings of the National Aca- demy of Sciences of the USA, Vol. 95, 1998, pp. 3537- 3542. doi:10.1073/pnas.95.7.3537 [19] S. Gosh and M. Karin, “Missing Pieces in the NF-κB Puzzle,” Cell, Vol. 109, 2002, pp. S81-S96. doi:10.1016/S0092-8674(02)00703-1 [20] C. Rieke, A. Papendieck, O. Sokolova and M. Naumann, “Helicobacter pylori-Induced Tyrosine Phosphorylation of IKKβ Contributes to NF-κB Activation,” Biological Chemistry, Vol. 392, 2011, pp. 387-393. doi:10.1515/BC.2011.029 [21] N. L. Reynaert, K. Ckless, S. H. Korn, et al., “Nitric Ox- ide Represses Inhibitory κB Kinase through S-Nitrosyla- tion,” Proceedings National Academy of Sciences of the USA, Vol. 101, No. 24, 2004, pp. 8945-8950. doi:10.1073/pnas.0400588101 [22] N. D. Perkins, “Integrating Cell-Signalling Path Ways with NF- and IKK Function,” Nature Reviews Molecular Cell Biology, Vol. 8, No. 1, 2007, pp. 49-62. doi:10.1038/nrm2083 [23] M. Kojima, H. Hosoda, Y. Date, M. Nakazato and K. Kangawa, “Ghrelin is a Growth-Hormone-Releasing A- cylated Peptide from Stomach,” Nature, Vol. 402, No. 6762, 1999, pp. 656-660. doi:10.1038/45230 [24] X. Xu, B. S. Jhun, C. H. Ha and Z. G. Jin, “Molecular Mechanisms of Ghrelin-Mediated Endothelial Nitric- Oxide Synthase Activation. Endocrinology, Vol. 149, No. 8, 2008, pp. 4183-4192. doi:10.1210/en.2008-0255 [25] B. L. Slomiany and A. Slomiany, “Ghrelin Protection against Lipopolysaccharide-Induced Gastric Mucosal Cell Apoptosis Involves Constitutive Nitric Oxide Syn- thase-Mediated Caspase-3 S-Nitrosylation,” Mediators of Inflammation, 2010. doi:1155/2010/280464 [26] B. L. Slomiany and A. Slomiany, “Role of Ghrelin-In- duced cSrc Activation in Modulation of Gastric Mucosal Inflammatory Responses to Helicobacter pylori,” In- flammopharmacology, Vol. 19, No. 4, 2011, pp. 197-204. [27] A. Slomiany and B. L. Slomiany, “Transformations of Phosphatidyl-Inositol Phosphates in the Outer and Inner Nuclear Membrane are Linked to Synthesis and Restitu- tion of Cellular Membranes,” Health, Vol. 3, No. 4, 2011, pp. 187-199. doi:10.4236/health.2011.34035 [28] S. M. Noha, A. G. Atanasov, D. Schuster, et al., “Dis- covery of a Novel IKK-b Inhibitor by Ligand-Based Vir- tual Screening Techniques,” Bioorganic and Medicinal Chemistry Letters, Vol. 21, 2011, pp. 577-583. doi:10.1016/j.bmcl.2010.10.051 [29] S. R. Jaffrey, H. Erdjument-Bromage, D. Ferris, P. Tempst and S. H. Snyder, “Protein S-Nitrosylation: A Physio- logical Signal for Neuronal Nitric Acid,” Nature Cell Bi- ology, Vol. 3, No. 2, 2001, pp. 193-197. doi:10.1038/35055104 [30] M. T. Forrester, M. W. Foster and J. S. Stamler, “As- sessment and Application of the Biotin Switch Technique for Examining Protein S-Nitrosylation under Conditions of Pharmacologically Induced Oxidative Stress,” Journal of Biological Chemistry, Vol. 282, No. 19, 2007, pp. 13977-13983. doi:10.1074/jbc.M609684200 Copyright © 2011 SciRes. OJCB  B. L. SLOMIANY ET AL. Copyright © 2011 SciRes. OJCB 10 [31] H. E. Marshall, D. T. Hess and J. S. Stamler, “S-Nitro- sylation: Physiological Regulation of NF-κB,” Proceed- ings of the National Academy of Sciences of the USA, Vol. 101, 2004, pp. 8841-8842. doi:10.1073/pnas.0403034101 [32] R. Korhonen, A. Lahti, H. Kankaanranta and E. Moilanen, “Nitric Oxide Production and Signaling in Inflamma- tion,” Current Drug Targets: Inflammation and Allergy, Vol. 4, No. 4, 2005, pp. 471-479. doi:10.2174/1568010054526359 [33] B. L. Slomiany and A. Slomiany, “Ghrelin Suppression of Helicobacter pylori-Induced S-Nitrosylation-Depen- dent Gastric Mucosal Akt Inactivation Exerts Modulatory Influence on Gastric Mucin Synthesis,” Inflammophar- macology, Vol. 19, 2011, pp. 89-97. doi:10.1007/s10787-011-0078-4 [34] P. Lodeiro, M. Theodoropoulou, M. Pardo, F. F. Casa-nueva and J. P. Camina, “C-Src Regulates Akt Sig- naling in Response to Ghrelin via β-Arrestin Signaling- Independent and -Dependent Mechanism,” PLoS ONE, Vol. 4, No. 3, 2009, p.e4686.
|