 Vol.2, No.10, 1138-1147 (2010) Natural Science http://dx.doi.org/10.4236/ns.2010.210141 Copyright © 2010 SciRes. OPEN ACCESS Correlated mutations in the four influenza proteins essential for viral RNA synthesis, host adaptation, and virulence: NP, PA, PB1, and PB2 Wei Hu Department of Computer Science, Houghton College, Houghton, USA; wei.hu@houghton.edu. Received 12 July 2010; revised 20 August 2010; accepted 23 August 2010. ABSTRACT The NP, PA, PB1, and PB2 proteins of influenza viruses together are responsible for the tran- scription and replication of viral RNA, and the latter three proteins comprise the viral poly- merase. Two recent reports indicated that the mutation at site 627 of PB2 plays a key role in host range and increased virulence of influenza viruses, and could be compensated by multiple mutations at other sites of PB2, suggesting the association of this mutation with those at other sites. The objective of this study was to analyze the co-mutated sites within and between these important proteins of influenza. With mutual information, a set of statistically significant co- mutated position pairs (P value = 0) in NP, PA, PB1, and PB2 of avian, human, pandemic 2009 H1N1, and swine influenza were identified, based on which several highly connected net- works of correlated sites in NP, PA, PB1, and PB2 were discovered. These correlation net- works further illustrated the inner functional dependence of the four proteins that are critical for host adaptation and pathogenicity. Mutual information was also applied to quantify the correlation of sites within each individual pro- tein and between proteins. In general, the inter protein correlation of the four proteins was stronger than the intra protein correlation. Fi- nally, the correlation patterns of the four pro- teins of pandemic 2009 H1N1 were found to be closer to those of avian and human than to swine influenza, thus rendering a novel insight into the interaction of the four proteins of the pandemic 2009 H1N1 virus when compared to avian, human, and swine influenza and how the origin of these four proteins might affect the correlation patterns uncovered in this analysis. Keywords: Co-mutation; Entropy; Influenza; Mutation; Mutual Information; Pandemic 2009 H1N1; Polymerase 1. INTRODUCTION There are eight single-stranded RNA gene segments in the influenza A virus, which are present as ribonucleo- protein complexes (vRNPs) with nucleoprotein (NP) and polymerase within the virus particle. The viral poly- merase itself is a heterotrimer composed of two basic subunits PB1 and PB2, and one acidic subunit PA, which catalyzes the transcription of viral RNA (vRNA) to mRNA and the replication of vRNA to complementary RNA (cRNA). The primary function of NP is to assem- ble the RNA gene segments into a helical nucleocapsid to provide structural support in vRNPs. After infection, vRNPs are transported into the nucleus where the tran- scription and replication of the viral genome take place, which means it is the vRNP, rather than the vRNA, that is utilized as the template for transcription and replica- tion. Moreover, NP could also function as a multifunc- tional key adaptor for interactions between virus and host cell [1,2]. The influenza polymerase also plays an important role in host adaptation and pathogenicity, and mutations at sites 627,701, and 714 in PB2, 615 in PA, and 319 in NP could result in enhanced polymerase activity to facilitate cross species transmission and virulence [3]. A focal poultry outbreak in Manipur, India in 2007 was caused by a unique influenza A (H5N1) virus that contained two unique amino acid mutations, K116R and I411M, in the PB2 protein [4]. Additionally, several other mutations in PA, PB1, and PB2 were also shown to influence the po- lymerase activity [5-12]. Furthermore, the interaction of NP and PB2 with Importin α1 was found to be a deter- minant of host range as well [13]. The well-known mutation E627K in PB2 is a deter- minant marker for host shifts between avian and human  W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 113 1139 viruses and increased virulence. However, accumulating evidence demonstrated that the mutation at position 627 in PB2 could be compensated by multiple mutations at other sites of PB2 [14,15], implying that mutations in proteins tend to co-occur at different sites to compensate each other in order to maintain the structural and func- tional constraints. To extend our knowledge of the co- mutations in the proteins of influenza, this study em- ployed entropy and mutual information to analyze co- varying sites in NP, PA, PB1, and PB2 and to uncover a set of statistical significant co-mutated sites to reveal and quantify the interactions of these proteins that play a key role in the life cycle of the influenza viruses. Information theory including entropy and mutual in- formation (MI) enjoyed wide applications in sequence analysis. Mutual information was employed to identify groups of covariant mutations in the sequences of HIV-1 protease and to distinguish the correlated amino acid polymorphisms resulting from neutral mutations and those induced by multi-drug resistance [16]. With en- tropy, a simple informational index was proposed in [17] to reveal the patterns of synonymous codon usage bias. Further, mutual information was utilized in the construc- tion of site transition network based on 4064 HA1 of A/H3N1 sequences from 1968 to 2008, which was able to model the evolutionary path of the influenza virus and to predict seven possible HA mutations for the next an- tigenic drift in the 2009-2010 season [18]. Recently, en- tropy and mutual information were also applied to in- dentify critical positions and co-mutated positions on HA for predicting the antigenic variants [19]. In another report, sequence data of 1032 complete genomes of in- fluenza A virus (H3N2) during 1968-2006 were used to construct networks of genomic co-occurrence to describe H3N2 virus evolutionary patterns and dynamics. It sug- gested that amino acid substitutions corresponding to nucleotide co-changes cluster preferentially in known antigenic regions of HA [20]. To investigate the co-mutations in the proteins of in- fluenza, three separate tasks were performed in this study. The first task was to uncover the variation and co-variation patterns of proteins NP, PA, PB1, and PB2 by the entropy and mutual information computed from their concatenated amino acid sequences. The distribu- tions of entropy and MI obtained reflected the unique sequence characteristics of each protein of avian, human, pandemic 2009 H1N1, and swine influenza viruses, based on which a comparative analysis could be con- ducted to reveal the variation signature of each influenza species. The second task was to zoom in onto each posi- tion pairs in the four proteins to identify a set of statisti- cally significant co-mutated residue pairs (P value = 0), from which several networks of highly correlated sites could be inferred. These correlated pairs and networks of correlated sites presented, at a different scale, finer in- formation about the co-variation of these four proteins than that from task one. In a sense, the correlation in- formation obtained from task two was pair dependent, i.e., it was about pairs. The third task was to mine the association of these four proteins with a pair independent approach, where the locations of pairs with positive MI values were counted according to each protein or to each functional domain in a protein as described in [21]. The strength of association was measured by the averaged counts of correlated pairs located within each protein or between proteins. 2. MATERIALS AND METHODS 2.1. Sequence Data The protein sequences of influenza A virus employed in this study were downloaded from the Influenza Virus Resource of the National Center for Biotechnology In- formation (NCBI). All the NP, PA, PB1, and PB2 protein sequences from the same isolates were concatenated into single sequences, and there were 1520 such concatenated sequences of avian viruses, 1928 of human viruses, 164 of pandemic 2009 H1N1, and 232 of swine viruses. These concatenated sequences, rather than the individual protein sequences, were used in our analysis. All the se- quences utilized in the study were aligned with MAFFT [22]. 2.2. Entropy and Mutual Information In information theory [23,24], entropy is a measure of the uncertainty associated with a random variable. Let x be a discrete random variable that has a set of possible values 123 {,, ,} n aaa awith probabilities123 ,,, n pp ppwhere the entropy H of is ii i xplogp The mutual information of two random variables is a quantity that measures the mutual dependence of the two variables or the average amount of information that conveys about y, which can defined as x,y HxHyHx,y where H(x) is the entropy of x, and H(x,y) is the joint entropy of x and y. I (x, y) = 0 if and only if x and y are independent random variables. In the current study, each of the N columns in a multi- ple sequence alignment of a set of influenza protein se- quences of length N is considered as a discrete random variable i (1 ≤ i ≤ N) that takes on one of the 20 (n = 20) amino acid types with some probability. H(i ) has its minimum value 0 if all the amino acids at position i are the same, and achieves its maximum if all the 20  W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 1140 amino acid types appear with equal probability at posi- tion i, which can be verified by the Lagrange multiplier technique. A position of high entropy means that the amino acids are often varied at this position. While H (i ) measures the genetic diversity at position i in our current study, I (i , y) measures the correlation be- tween amino acid substitutions at positions i and j. 2.3. Mutual Information Evaluation In order to assess the significance of the mutual in- formation value of two positions in a multiple sequence alignment, it is necessary to show that this value is sig- nificantly higher than that based on random sequences. For each pair of positions in a multiple protein sequence alignment, we randomly permuted the amino acids from different sequences at the two positions and calculated the mutual information of these random sequences. This procedure was repeated 1000 times. The P value was calculated as the percentage of the mutual information values of the permuted sequences that were higher than those of the original sequences. 3. RESULTS 3.1. Entropy and Mutual Information of NP, PA, PB1, and PB2 To gain a global view of the sequence variation and co-variation of these four proteins, the entropy and mu- tual information of their concatenated sequences were calculated (Figure 1). The entropy distributions revealed that the swine influenza had the highest overall sequence variation and the pandemic 2009 had the least variation, with avian and human influenza being in the middle. Within each individual influenza species, it appeared that PA had the highest entropy average among the four proteins, with the exception of pandemic 2009 H1N1 (Table 1). Mutual information measures the correlation of the amino acids at two sites in a multiple sequence alignment. Therefore, to offer the information of how each site co-mutated with all other sites within each in- dividual protein and between proteins, for each site, all the MI values associated with this site were summed (Figure 1). These MI values represented the association between one site and all other sites in the four proteins. The pat- terns of MI distributions were quite different from those of entropy, where the ranking of the overall average MI values was swine (5.9533), human (3.6590), avian (0. 8298), and pandemic 2009 H1N1 (0.0165), suggesting that variation and co-variation were two distinct meas- urements of protein sequence changes. The most co- varying protein in each influenza species was PA in avian, PB1 in human, PA in pandemic 2009 H1N1, and PA in swine (Table 1). 3.2. Highly Correlated Sites in NP, PA, PB1, and PB2 In order to provide more detailed information about the highly correlated sites, top 50 MI sites in the con- catenated sequences from the four proteins of each in- fluenza species were selected from the sites in Figure 1. Among the top 50 MI sites (Figure 2), there were sev- eral sites that were shared between two different influ- enza species indicating their significant correlation. These top 50 MI sites represented their correlation in a pair independent manner. Next, top 30 MI co-mutated resi- due pairs of highest MI values (P value = 0) from each influenza species were identified (Table 2), and a collec- tion of highly connected networks of co-varying sites in the four proteins were established based on these 30 sta- tistically significant pairs. There were two avian, one human, two pandemic 2009 H1N1, and one swine cor- relation networks (Figure 3). All these networks from various influenza species exhibited their preferred con- nectivity among the four proteins. In the two avian net- works, one had only sites from PA and PB1, and the other contained only those from NP, PB1, and PB2. The human network had PA, PB1, and PB2 sites, where the most connected sites were PA_32, PB1_61, and PB1_63. In the two pandemic 2009 networks, the first had sites selected from all four proteins, while the second had only sites from PA, PB1, and PB2. The swine network had sites from all four proteins, where the most con- nected sites were PA_580 and PB2_661. These top 30 MI residue pairs and networks of associated sites pre- sented their correlation in a pair dependent manner. PB2_627 is a key site for host switches and virulence, which is also the most extensively studied site. Never- theless, only avian viruses had it as one of their top 50 MI sites (Figure 2). To find those sites that related to PB2_627, a set of sites that had high MI values with Table 1. Averaged entropy and MI of the four proteins. Entropy NP PA PB1 PB2 Overall Average Avian 0.04070.04990.0351 0.0426 0.0420 Human 0.04760.05100.0448 0.0471 0.0476 Pandemic 2009 0.00400.00460.0039 0.0050 0.0044 Swine 0.08840.10560.0751 0.0911 0.0900 MI NP PA PB1 PB2 Overall Average Avian 0.75600.93870.7888 0.8358 0.8298 Human 3.38193.81604.1865 3.2518 3.6590 Pandemic 2009 0.01370.01890.0157 0.0177 0.0165 Swine 4.73587.57615.4241 6.0774 5.9533 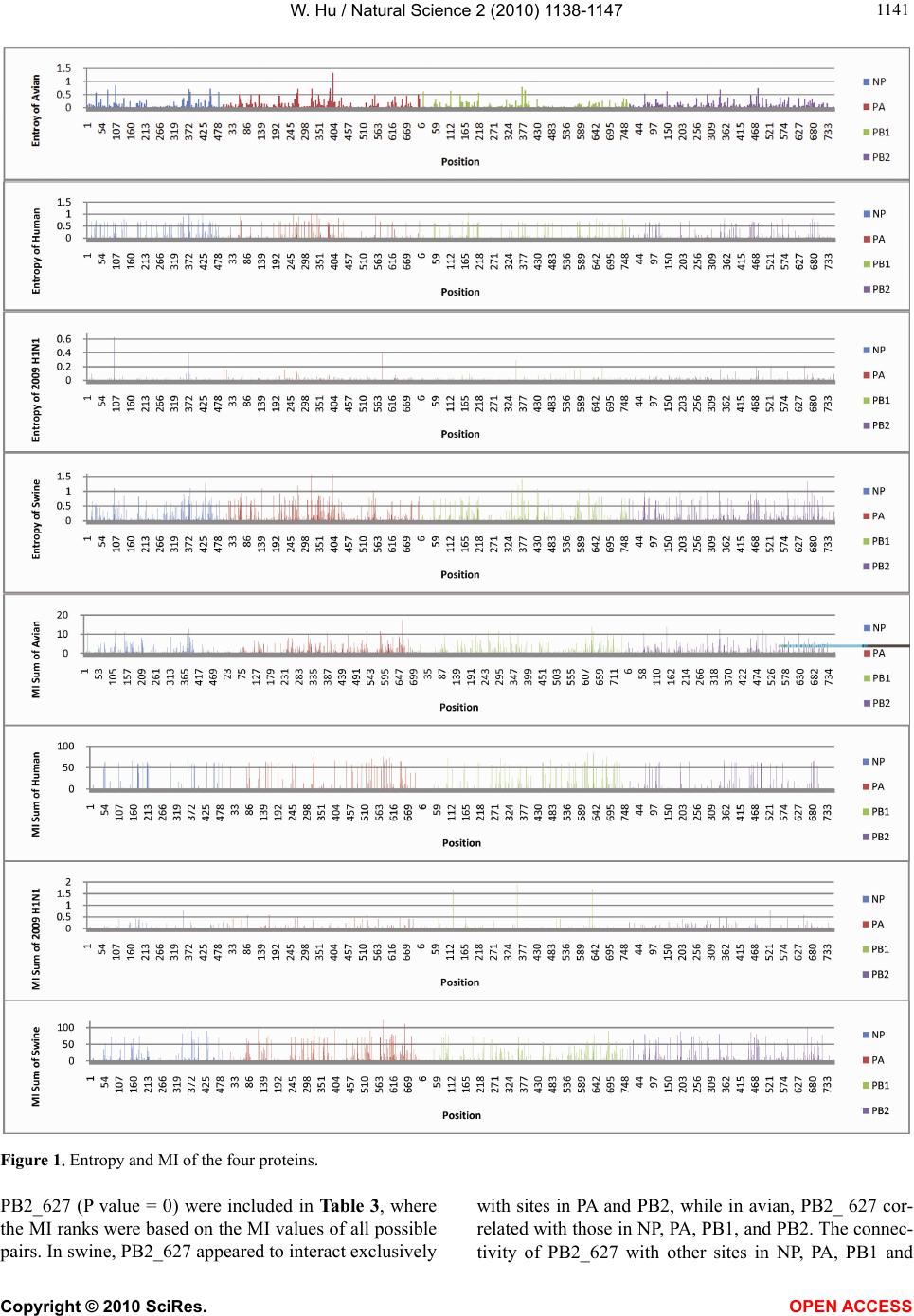 W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 114 1141 Figure 1.Entropy and MI of the four proteins. PB2_627 (P value = 0) were included in Table 3, where the MI ranks were based on the MI values of all possible pairs. In swine, PB2_627 appeared to interact exclusively with sites in PA and PB2, while in avian, PB2_ 627 cor- related with those in NP, PA, PB1, and PB2. The connec- tivity of PB2_627 with other sites in NP, PA, PB1 and 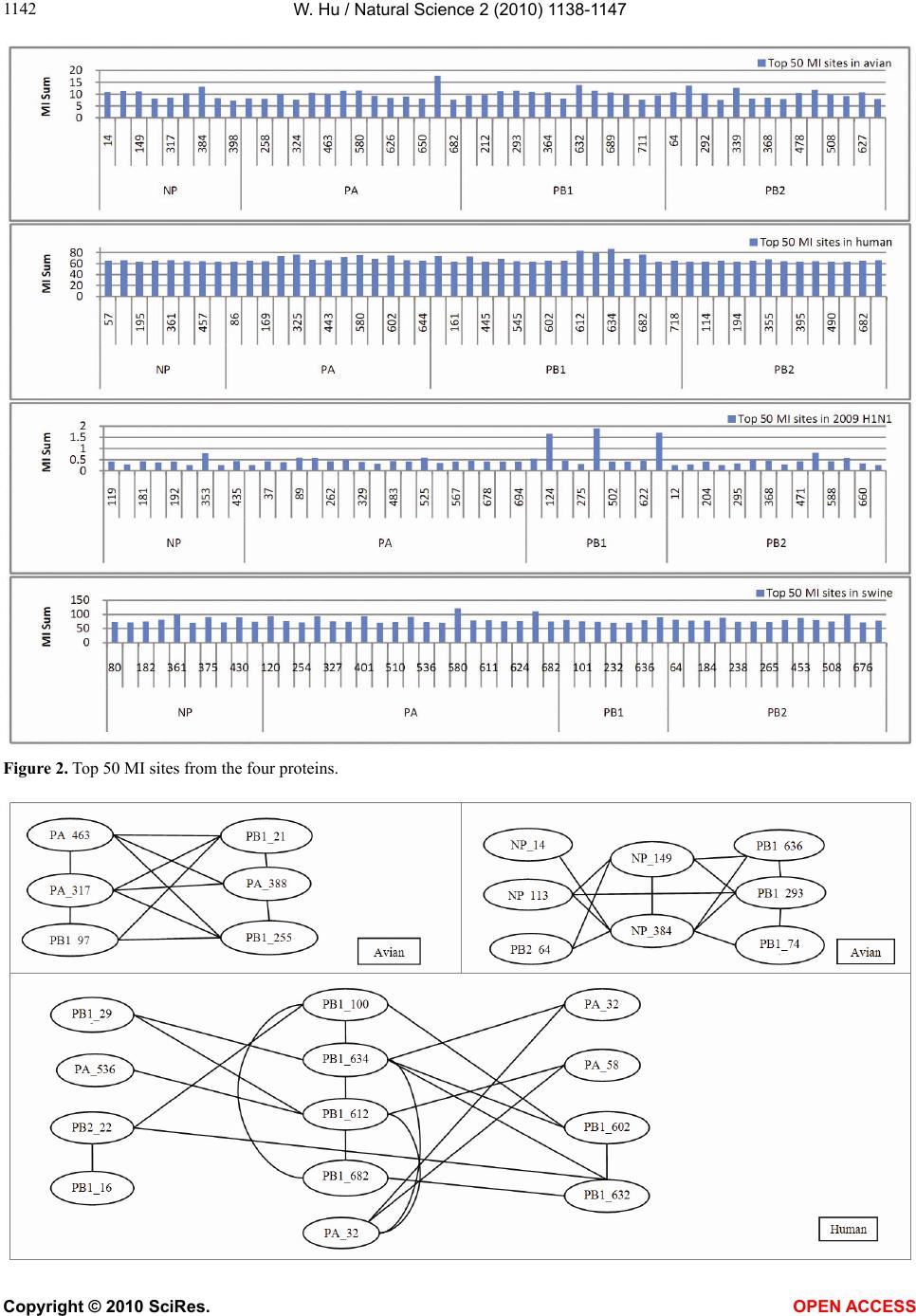 W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 1142 Figure 2. Top 50 MI sites from the four proteins. 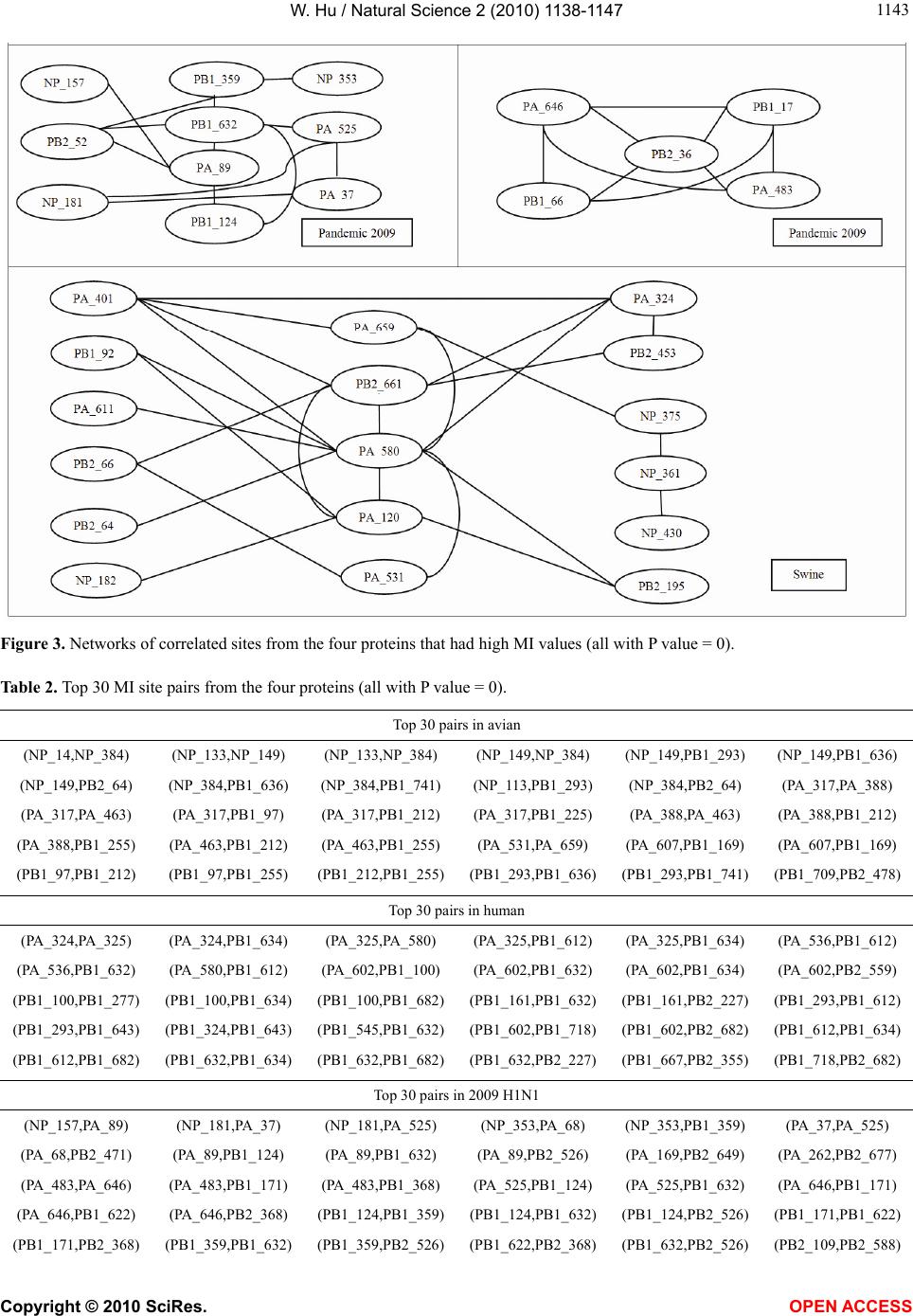 W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 114 1143 Figure 3. Networks of correlated sites from the four proteins that had high MI values (all with P value = 0). Table 2. Top 30 MI site pairs from the four proteins (all with P value = 0). Top 30 pairs in avian (NP_14,NP_384) (NP_133,NP_149) (NP_133,NP_384) (NP_149,NP_384) (NP_149,PB1_293) (NP_149,PB1_636) (NP_149,PB2_64) (NP_384,PB1_636) (NP_384,PB1_741) (NP_113,PB1_293) (NP_384,PB2_64) (PA_317,PA_388) (PA_317,PA_463) (PA_317,PB1_97) (PA_317,PB1_212) (PA_317,PB1_225) (PA_388,PA_463) (PA_388,PB1_212) (PA_388,PB1_255) (PA_463,PB1_212) (PA_463,PB1_255) (PA_531,PA_659) (PA_607,PB1_169) (PA_607,PB1_169) (PB1_97,PB1_212) (PB1_97,PB1_255) (PB1_212,PB1_255)(PB1_293,PB1_636)(PB1_293,PB1_741) (PB1_709,PB2_478) Top 30 pairs in human (PA_324,PA_325) (PA_324,PB1_634) (PA_325,PA_580) (PA_325,PB1_612) (PA_325,PB1_634) (PA_536,PB1_612) (PA_536,PB1_632) (PA_580,PB1_612) (PA_602,PB1_100) (PA_602,PB1_632) (PA_602,PB1_634) (PA_602,PB2_559) (PB1_100,PB1_277) (PB1_100,PB1_634) (PB1_100,PB1_682)(PB1_161,PB1_632) (PB1_161,PB2_227) (PB1_293,PB1_612) (PB1_293,PB1_643) (PB1_324,PB1_643) (PB1_545,PB1_632)(PB1_602,PB1_718) (PB1_602,PB2_682) (PB1_612,PB1_634) (PB1_612,PB1_682) (PB1_632,PB1_634) (PB1_632,PB1_682)(PB1_632,PB2_227) (PB1_667,PB2_355) (PB1_718,PB2_682) Top 30 pairs in 2009 H1N1 (NP_157,PA_89) (NP_181,PA_37) (NP_181,PA_525) (NP_353,PA_68) (NP_353,PB1_359) (PA_37,PA_525) (PA_68,PB2_471) (PA_89,PB1_124) (PA_89,PB1_632) (PA_89,PB2_526) (PA_169,PB2_649) (PA_262,PB2_677) (PA_483,PA_646) (PA_483,PB1_171) (PA_483,PB1_368) (PA_525,PB1_124) (PA_525,PB1_632) (PA_646,PB1_171) (PA_646,PB1_622) (PA_646,PB2_368) (PB1_124,PB1_359)(PB1_124,PB1_632)(PB1_124,PB2_526) (PB1_171,PB1_622) (PB1_171,PB2_368) (PB1_359,PB1_632) (PB1_359,PB2_526)(PB1_622,PB2_368) (PB1_632,PB2_526) (PB2_109,PB2_588) 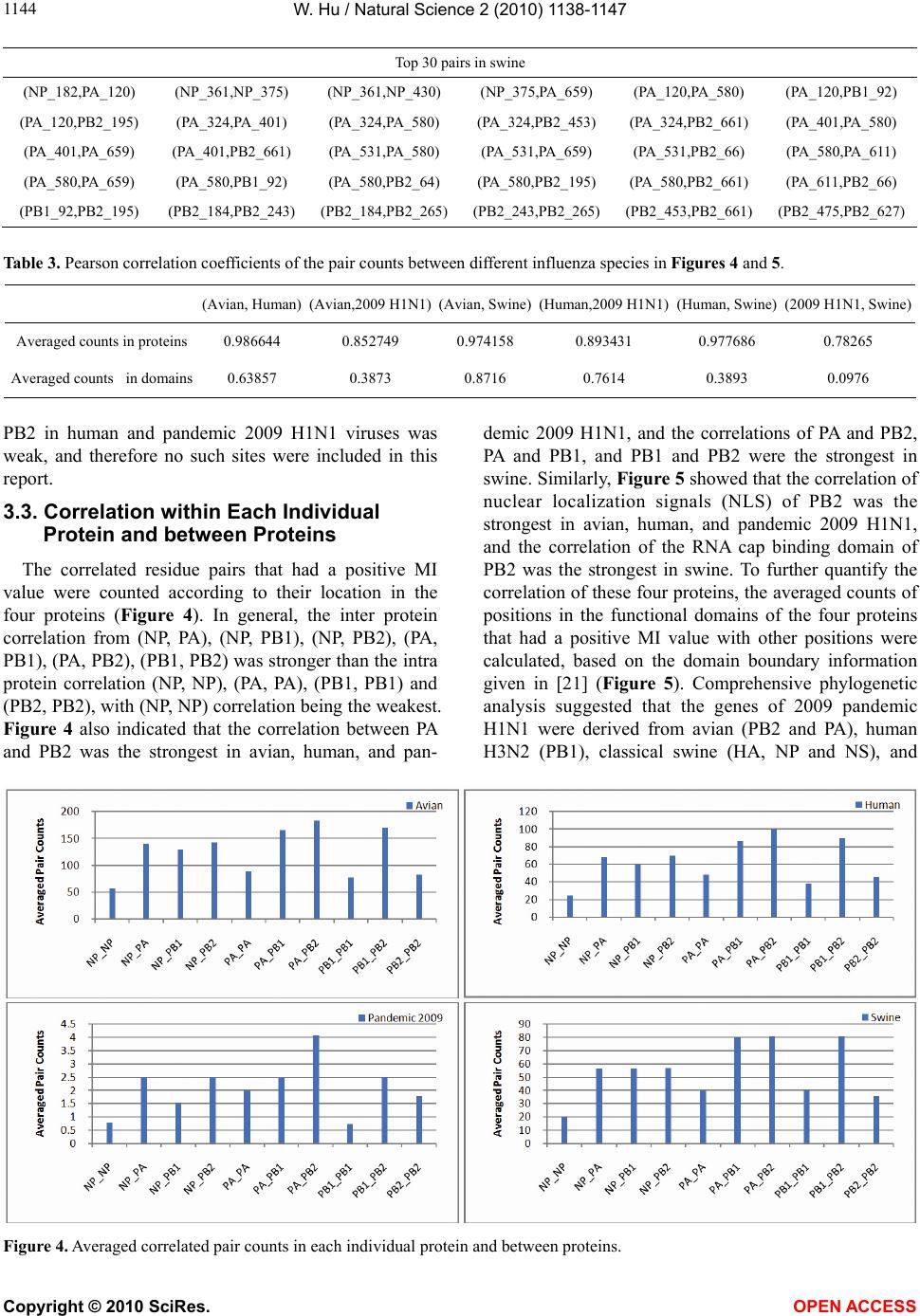 W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 1144 Top 30 pairs in swine (NP_182,PA_120) (NP_361,NP_375) (NP_361,NP_430) (NP_375,PA_659) (PA_120,PA_580) (PA_120,PB1_92) (PA_120,PB2_195) (PA_324,PA_401) (PA_324,PA_580) (PA_324,PB2_453) (PA_324,PB2_661) (PA_401,PA_580) (PA_401,PA_659) (PA_401,PB2_661) (PA_531,PA_580) (PA_531,PA_659) (PA_531,PB2_66) (PA_580,PA_611) (PA_580,PA_659) (PA_580,PB1_92) (PA_580,PB2_64) (PA_580,PB2_195) (PA_580,PB2_661) (PA_611,PB2_66) (PB1_92,PB2_195) (PB2_184,PB2_243) (PB2_184,PB2_265)(PB2_243,PB2_265)(PB2_453,PB2_661) (PB2_475,PB2_627) Table 3. Pearson correlation coefficients of the pair counts between different influenza species in Figures 4 and 5. (Avian, Human) (Avian,2009 H1N1)(Avian, Swine)(Human,2009 H1N1)(Human, Swine) (2009 H1N1, Swine) Averaged counts in proteins 0.986644 0.852749 0.974158 0.893431 0.977686 0.78265 Averaged counts in domains 0.63857 0.3873 0.8716 0.7614 0.3893 0.0976 PB2 in human and pandemic 2009 H1N1 viruses was weak, and therefore no such sites were included in this report. 3.3. Correlation within Each Individual Protein and between Proteins The correlated residue pairs that had a positive MI value were counted according to their location in the four proteins (Figure 4). In general, the inter protein correlation from (NP, PA), (NP, PB1), (NP, PB2), (PA, PB1), (PA, PB2), (PB1, PB2) was stronger than the intra protein correlation (NP, NP), (PA, PA), (PB1, PB1) and (PB2, PB2), with (NP, NP) correlation being the weakest. Figure 4 also indicated that the correlation between PA and PB2 was the strongest in avian, human, and pan- demic 2009 H1N1, and the correlations of PA and PB2, PA and PB1, and PB1 and PB2 were the strongest in swine. Similarly, Figure 5 showed that the correlation of nuclear localization signals (NLS) of PB2 was the strongest in avian, human, and pandemic 2009 H1N1, and the correlation of the RNA cap binding domain of PB2 was the strongest in swine. To further quantify the correlation of these four proteins, the averaged counts of positions in the functional domains of the four proteins that had a positive MI value with other positions were calculated, based on the domain boundary information given in [21] (Figure 5). Comprehensive phylogenetic analysis suggested that the genes of 2009 pandemic H1N1 were derived from avian (PB2 and PA), human H3N2 (PB1), classical swine (HA, NP and NS), and Figure 4. Averaged correlated pair counts in each individual protein and between proteins. 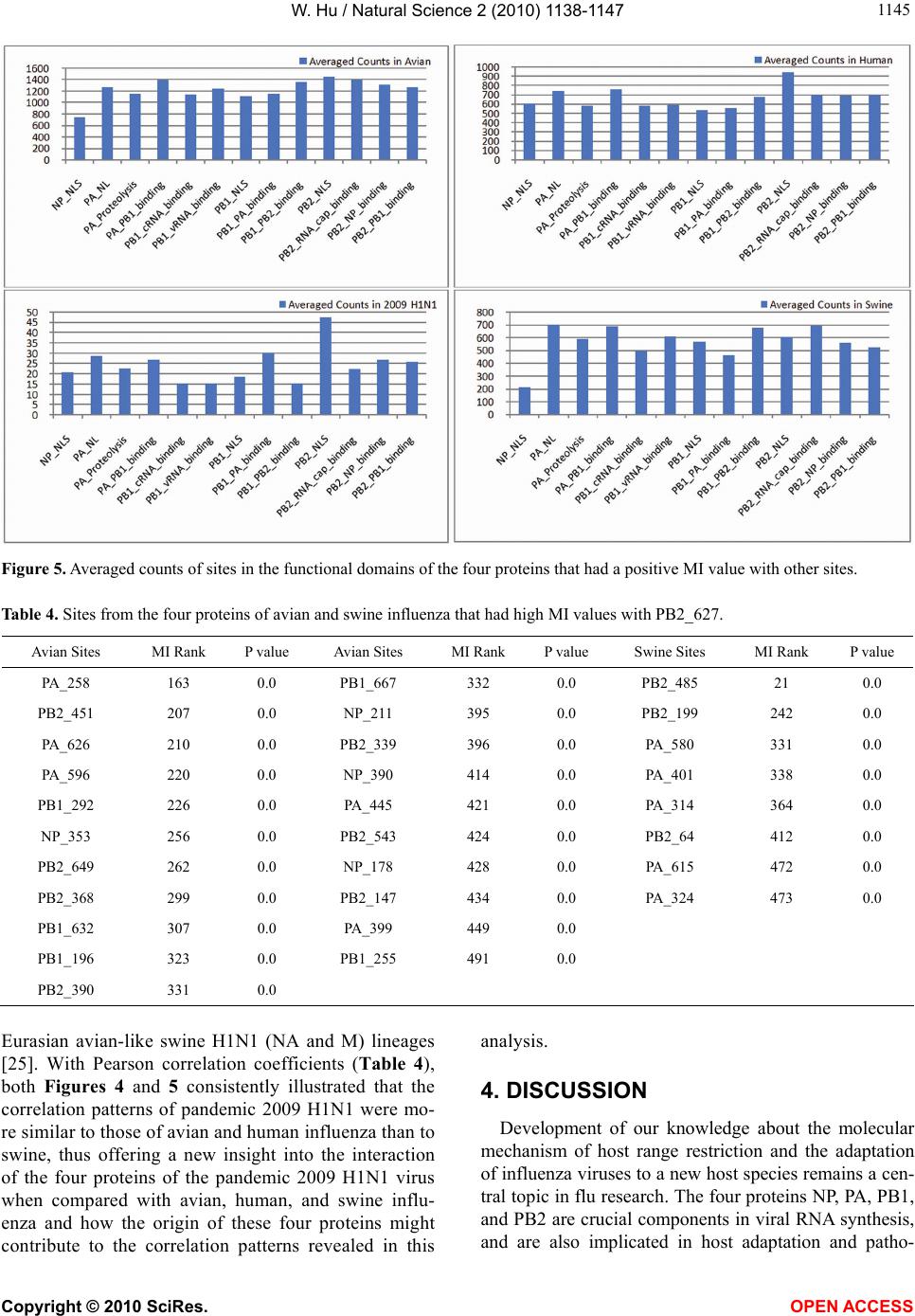 W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 114 1145 Figure 5. Averaged counts of sites in the functional domains of the four proteins that had a positive MI value with other sites. Table 4. Sites from the four proteins of avian and swine influenza that had high MI values with PB2_627. Avian Sites MI Rank P value Avian Sites MI Rank P value Swine Sites MI Rank P value PA_258 163 0.0 PB1_667 332 0.0 PB2_485 21 0.0 PB2_451 207 0.0 NP_211 395 0.0 PB2_199 242 0.0 PA_626 210 0.0 PB2_339 396 0.0 PA_580 331 0.0 PA_596 220 0.0 NP_390 414 0.0 PA_401 338 0.0 PB1_292 226 0.0 PA_445 421 0.0 PA_314 364 0.0 NP_353 256 0.0 PB2_543 424 0.0 PB2_64 412 0.0 PB2_649 262 0.0 NP_178 428 0.0 PA_615 472 0.0 PB2_368 299 0.0 PB2_147 434 0.0 PA_324 473 0.0 PB1_632 307 0.0 PA_399 449 0.0 PB1_196 323 0.0 PB1_255 491 0.0 PB2_390 331 0.0 Eurasian avian-like swine H1N1 (NA and M) lineages [25]. With Pearson correlation coefficients (Table 4), both Figures 4 and 5 consistently illustrated that the correlation patterns of pandemic 2009 H1N1 were mo- re similar to those of avian and human influenza than to swine, thus offering a new insight into the interaction of the four proteins of the pandemic 2009 H1N1 virus when compared with avian, human, and swine influ- enza and how the origin of these four proteins might contribute to the correlation patterns revealed in this analysis. 4. DISCUSSION Development of our knowledge about the molecular mechanism of host range restriction and the adaptation of influenza viruses to a new host species remains a cen- tral topic in flu research. The four proteins NP, PA, PB1, and PB2 are crucial components in viral RNA synthesis, and are also implicated in host adaptation and patho-  W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 1146 genicity. Therefore, clear revelation of the function and action of these four proteins is required. Sequence sur- vey implied that the common host shift markers in the proteins of avian or swine influenza are not present in the pandemic 2009 H1N1 virus. Moreover, introduction of known virulence markers into 2009 H1N1 does not increase its virulence [26,27]. The combination of its pandemic potential and absence of traditional host markers has remained a source for concern and justifies the search for its own host markers outside of the space of classical host markers [28,29]. The PB2 of 2009 H1N1 had a glutamic acid at posi- tion 627, reflecting its avian origin. Typically avian vi- ruses have a glutamic acid (E) at position 627, while human viruses usually have a lysine (K) at this position. Additionally, the presence of a glutamic acid at position 627 in PB2 contributed to the cold sensitivity of poly- merase derived from avian viruses in mammalian cells [3]. However, the clinical experience in 2009 demon- strated that this novel virus was able to transmit and rep- licate in humans efficiently. A natural assumption was that some amino acids at other sites in PB2 might be responsible for its efficiency in reproduction and trans- mission. It turned out that two amino acids, serine (S) at site 590 and arginine (R) at site 591, in PB2, termed SR polymorphism, compensate the lack of amino acid lysine at site 627 in PB2 [15]. Although our mutual information analysis illustrated the connectivity was low between PB2_627, PB2_591, PB2_590, and other sites in pandemic 2009 H1N1, this study discovered three sites correlating with PB2_590, which were NP_480 (MI = 0.0219, P value = 0.033, MI rank = 370), PB1_359 (MI = 0.0060, P value = 0.338, MI rank = 611), and PB1_124 (MI = 0.0011, P value = 0.0, MI rank = 1093). With the same approach, associa- tions with other critical sites such as PB2_701 and PB2_271 could also be detected. Host range and virulence of influenza viruses are mul- tigenically determined through interactions between the proteins involved, which could be, in part, elucidated with identification of mutations and co-mutations that might confer increased pathogenicity or transmissibility. The absence of familiar host switch markers in 2009 H1N1 added a new dimension in this effort, and moti- vated the extensive search for other mutations or strate- gies that influenza viruses evolved to develop and adapt. To move this direction, this report revealed and quanti- fied the interactions of NP, PA, PB1, and PB2 of avian, human, pandemic 2009 H1N1, and swine influenza, and identified a collection of statistically significant co- varying sites, not only in each individual protein but also between proteins, for further investigation of their inte- grative biological relevance experimentally. 5. ACKNOWLEDGEMENT We thank Houghton College for its financial support. REFERENCES [1] Neumann, G., Brownlee, G.G., Fodor, E. and Kawaoka, Y. (2004) Orthomyxovirus replication, transcription, and polyadenylation. Current Topics in Microbiology and Im- munology, 283, 121-143. [2] Ng, A.K., Zhang, H., Tan, K., et al. (2008) Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. The FASEB Journal, 22(10), 3638-3647. [3] Jürgen, S. (2008) Influenza A virus polymerase: A deter- minant of host range and pathogenicity. In: Klenk H.D., Matrosovich M.N. and Stech J. Eds., Avian Influenza, Monogr Virol. Basel, Karger, 27, 187-194. [4] Mishra, A.C., Cherian, S.S., Chakrabarti, A.K., et al. (2009) A unique influenza A (H5N1) virus causing a fo- cal poultry outbreak in 2007 in Manipur, India. Journal of Virology, 6(1), 26. [5] Yuan, P.W., Bartlam, M., Lou, Z.Y., et al. (2009) Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site, Nature, 458, 909-913. [6] Fodor, E., Crow, M., Mingay, L.J., et al. (2002) A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. Journal of Virology, 76(18), 8989-9001. [7] Hara, K., Schmidt, F.I., Crow, M. and Brownlee, G.G. (2006) Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease ac- tivity, cap binding, and virion RNA promoter binding. Journal of Virology, 80(16), 7789-7798. [8] Kerry, P.S., Willsher, N. and Fodor, E. (2008) A cluster of conserved basic amino acids near the C-terminus of the PB1 subunit of the influenza virus RNA polymerase is involved in the regulation of viral transcription. Virology, 373(1), 202-210. [9] Dias, A., Bouvier, D., Crépin, T., McCarthy, A.A., et al. (2009) The cap-snatching endonuclease of influenza vi- rus polymerase resides in the PA subunit. Nature, 458 (7240), 914-918. [10] Rolling, T., Koerner, I., Zimmermann, P., Holz, K., et al. (2009) Adaptive mutations resulting in enhanced poly- merase activity contribute to high virulence of influenza A virus in mice. Journal of Virology, 83(13), 6673-6680. [11] Bussey, K.A., Bousse, T.L., Desmet, E.A., Kim, B. and Takimoto, T. (2010) PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. Journal of Virology, 84(9), 4395- 4406. [12] Zhu, H., Wang, J., Wang, P., Song, W., Zheng, Z., Chen, R., Guo, K., Zhang, T., Peiris, J.S., Chen, H. and Guan, Y. (2010) Substitution of lysine at 627 position in PB2 pro- tein does not change virulence of the 2009 pandemic H1N1 virus in mice. Virology, 401(1), 1-5. [13] Gabriel, G., Herwig, A. and Klenk, H.D. (2008) Interac- tion of polymerase subunit PB2 and NP with importin α1  W. Hu / Natural Science 2 (2010) 1138-1147 Copyright © 2010 SciRes. OPEN ACCESS 114 1147 is a determinant of host range of influenza A virus. PLoS Pathog, 4(2), e11. [14] Li, J., Ishaq, M., Prudence, M., et al. (2009) Single muta- tion at the amino acid position 627 of PB2 that leads to increased virulence of an H5N1 avian influenza virus during adaptation in mice can be compensated by multi- ple mutations at other sites of PB2. Virus Research, 144 (1-2), 123-129. [15] Mehle, A. and Doudna, J.A. (2009) Adaptive strategies of the influenza virus polymerase for replication in humans, Proceedings of the National Academy of Sciences, 106 (50), 21312-21316. [16] Liu, Y., Eyal, E. and Bahar, I. (2008) Analysis of corre- lated mutations in HIV-1 protease using spectral cluster- ing. Bioinformatics, 24(10), 1243-1250. [17] Colman, P.M., Hoyne, P.A. and Lawrence, M.C. (1993) Sequence and structure alignment of paramyxovirus he- magglutinin-neuraminidase with influenza virus neura- minidase. Journal of Virology, 67, 2972-2980. [18] Xia, Z., Jin, G.L., Zhu, J. and Zhou, R.H. (2009) Using a mutual information-based site transition network to map the genetic evolution of influenza A / H3N2 virus. Bioin- formatics, 25(18), 2309-2317. [19] Huang, J.-W., King, C.-C. and Yang, J.-M. (2009) Co- evolution positions and rules for antigenic variants of human influenza A / H3N2 viruses. BMC Bioinformatics, 10, S41. [20] Du, X.J., Wang, Z., Wu, A.P., et al. (2008) Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Research, 18, 178-187. [21] Miotto, O., Heiny, A.T., Albrecht, R., García-Sastre, A., et al. (2010) Complete-proteome mapping of human in- fluenza A adaptive mutations: Implications for human transmissibility of zoonotic strains, PLoS One, 5(2), e9025. [22] Katoh, K., Kuma, K., Toh, H. and Miyata, T. (2005) MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Research, 33, 511-518. [23] Cover, T.A. and Thomas, J.A. (1991) Elements of infor- mation theory. John Wiley and Sons, New York. [24] MacKay, D. (2003) Information theory, inference, and learning algorithms. Cambridge University Press. [25] Smith, G.J.D., Vijaykrishna, D., et al. (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature, 459, 1122-1125. [26] Jagger, B.W., Memoli, M.J., Sheng, Z.-M., et al. (2010) The PB2-E627K mutation attenuates viruses containing the 2009 H1N1 influenza pandemic polymerase. mBio, 1 (1), e00067-10. [27] Herfst, S., Chutinimitkul, S., Ye, J.Q., et al. (2010) In- troduction of virulence markers in PB2 of pandemic swine-origin influenza virus does not result in enhanced virulence or transmission. Journal of Virology, 84(8), 3752-3758. [28] Hu, W. (2010) Novel host markers in the 2009 pandemic H1N1 influenza A virus. Journal of Biomedical Science and Engineering, 3(6), 584-601. [29] Hu, W. (2010) Nucleotide host markers in the influenza a viruses. Journal of Biomedical Science and Engineering, 3, 684-699.
|