Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2010, 3, 1029-1038 JBiSE doi:10.4236/jbise.2010.310134 Published Online October 2010 (http://www.SciRP.org/journal/jbise/). Published Online October 20 10 in SciRes. http://www.scirp.org/journal/jbise The mode of action of electrical high frequency stimulation Miriam Kammerer, Jonas M. Hebel, Thomas J. Feuerstein Section of Clinical Neuropharmacology, Department of Neurosurgery, University Hospital, Freiburg, Germany. Email: thomas.feuerstein@uniklinik-freiburg.de Received 19 March 2010; revised 6 April; accepted 28 July 2010. ABSTRACT This article analyses, on the basis of the patho- physiological grounds of various syndromes treated with deep brain stimulation, whether there is a col- lective explanation of the mode of action of the ap- plied regional stimulations with high frequencies (HFS). This proposed hypothesis assumes that HFS selectively releases GABA. The selective GABA re- lease can explain the efficacy and the side effects of HFS in the various target regions according to the maxim of the philosopher William of Ockham that the simplest explanation is probably the correct explanation. Keywords: HFS, DBS, Parkinson’s disease, Essential tremor, Huntington’s disease, Depression 1. INTRODUCTION Deep brain stimulation (DBS) mostly reflects high fre- quency stimulation (HFS, > 100 Hz); low frequency stimulation (LFS, < 30 Hz) is rarely linked to the term DBS. Parkinsonian tremor was the first syndrome which was beneficially treated with HFS (130 Hz) in the ven- tral intermediate thalamic nucleus, in the year 1987 [1]. Today, i.e. 22 years after this first application of HFS, its mechanism of action is still unclear [2]. DBS is applied in a multitude of clinical conditions, e.g. Parkinson’s disease, Chorea Huntington, dystonia, depression, Gilles de la Tourette syndrome, and obsessive compulsive dis- order. For the treatment of each disorder a unique target brain area needs to be stimulated. Therefore, many brain target regions exist, e.g . the subthalamic nucleus (STN), the globus pallidus medialis (GPmed), and the ventral intermediate thalamic nucleus (VIM). Hitherto, the following assumptions about the mode of action of HFS and their contradictions are discussed: HFS is thought to inactivate the stimulated structures (see [3]). However, the decreased activity of thalamic neurons upon GPmed-HFS [4] (evaluation in awake monkeys) rather goes in the opposite direction. The axon terminals from GABAergic GPmed neurons impinge on glutamatergic thalamic neurons. Thus, their decreased activity must be explained by a GABAA receptor- medi- ated inhibition due to increased GABA release. The axon terminals of GPmed neurons release GABA upon activa- tion, not upon inhibition, by HFS. Thus, HFS may acti- vate the neurons of the stimulated structure GPmed. HFS activates the stimulated structures [5]. This is at variance with the increased activity of STN neurons in Parkinson patients [6]. In addition, HFS has been re- ported to reduce the STN firing rate [7]. An even higher activity due to HFS of subthalamic glutamatergic neu- rons seems counterproductive pathophysiologically: Even more drive of the basal ganglia output nuclei leads to even stronger retardation of thalamic neurons, being less active in the hypokinetic parkinsonian state anyway (see Figure 1). Thus, HFS may inactivate the neurons of the stimulated structure STN to alleviate hypokinesia. 2. HFS: EXCITATION OR INHIBITION OF NERVE TERMINALS OR AXONS? Stimulation of a brain region is normally expected to re- sult in excitatory symptoms (e.g. muscle twitchings, flashes of light [8]). But, as Benabid et al. [2] have shown, clinical benefits from HFS-DBS often resemble those of earlier therapeutic lesions in the target brain areas. This observation leads to the assumption that HFS —corre- sponding to a functional removal of active neurons or their effects—may be similar to an inhibition of these neurons. As such a great variety of brain target regions, involved neurons and treated disorders exists, it seems quite impossible to find a single common denominator for a possible mode of action. Nevertheless, one may ask: Are there any mechanistics hints to solve the question of the HFS mechanism of action? STN, GPmed and VIM are the most often targeted DBS regions in advanced Parkinson’s disease [9]. Frequency is the most important parameter accounting for the thera- peutic effects: Only HFS is efficacious, not low fre- quency stimulation (LFS, ~20 Hz) [8]. Stimulation at 5-10 Hz even worsens Parkinsonism and no significant 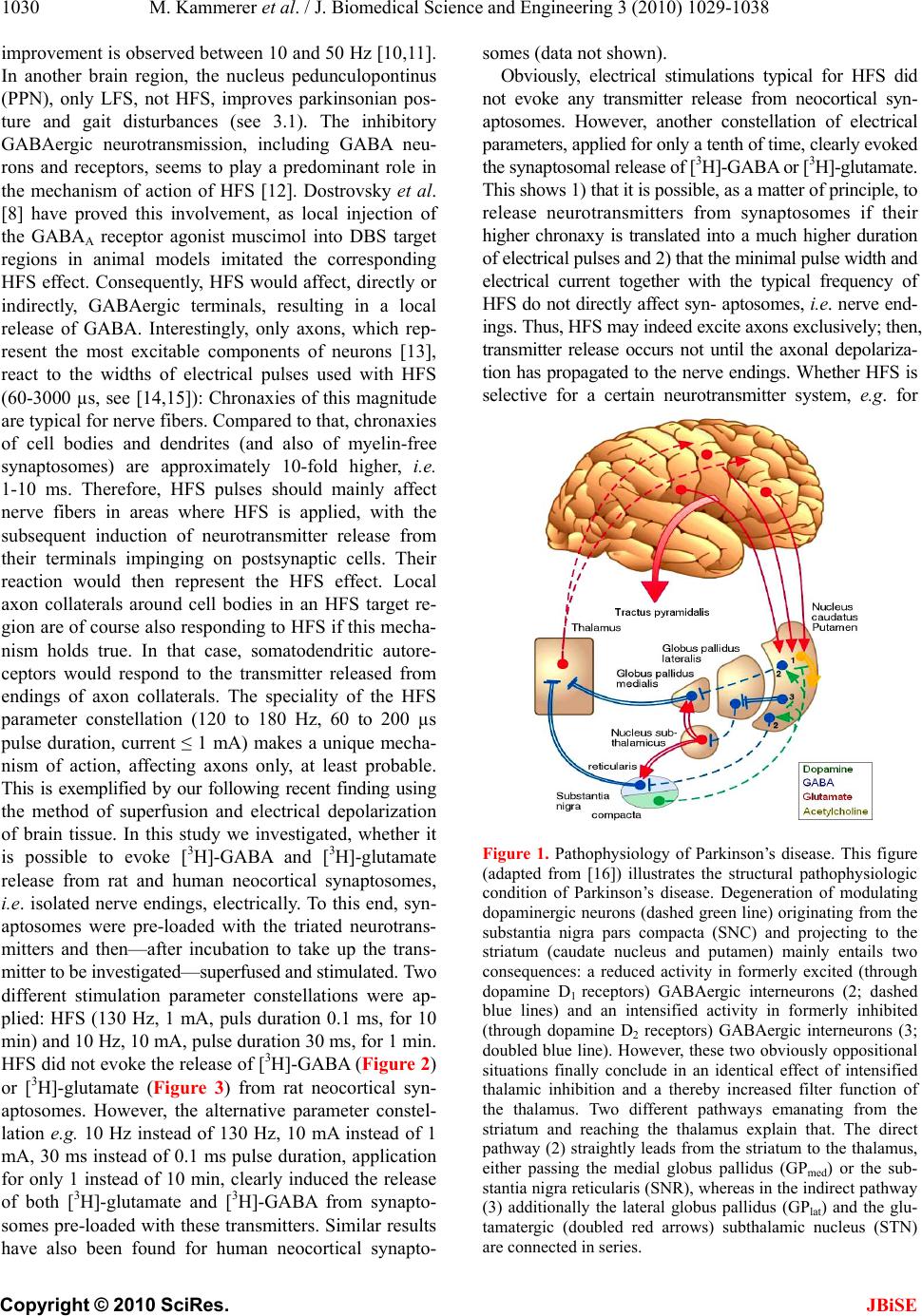 M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1030 improvement is observed between 10 and 50 Hz [10,11]. In another brain region, the nucleus pedunculopontinus (PPN), only LFS, not HFS, improves parkinsonian pos- ture and gait disturbances (see 3.1). The inhibitory GABAergic neurotransmission, including GABA neu- rons and receptors, seems to play a predominant role in the mechanism of action of HFS [12]. Dostrovsky et al. [8] have proved this involvement, as local injection of the GABAA receptor agonist muscimol into DBS target regions in animal models imitated the corresponding HFS effect. Consequently, HFS would affect, directly or indirectly, GABAergic terminals, resulting in a local release of GABA. Interestingly, only axons, which rep- resent the most excitable components of neurons [13], react to the widths of electrical pulses used with HFS (60-3000 µs, see [14,15]): Chronaxies of this magnitude are typical for nerve fibers. Compared to that, chronaxies of cell bodies and dendrites (and also of myelin-free synaptosomes) are approximately 10-fold higher, i.e. 1-10 ms. Therefore, HFS pulses should mainly affect nerve fibers in areas where HFS is applied, with the subsequent induction of neurotransmitter release from their terminals impinging on postsynaptic cells. Their reaction would then represent the HFS effect. Local axon collaterals around cell bodies in an HFS target re- gion are of course also responding to HFS if this mecha- nism holds true. In that case, somatodendritic autore- ceptors would respond to the transmitter released from endings of axon collaterals. The speciality of the HFS parameter constellation (120 to 180 Hz, 60 to 200 µs pulse duration, current ≤ 1 mA) makes a unique mecha- nism of action, affecting axons only, at least probable. This is exemplified by our following recent finding using the method of superfusion and electrical depolarization of brain tissue. In this study we investigated, whether it is possible to evoke [3H]-GABA and [3H]-glutamate release from rat and human neocortical synaptosomes, i.e. isolated nerve endings, electrically. To this end, syn- aptosomes were pre-loaded with the triated neurotrans- mitters and then—after incubation to take up the trans- mitter to be investigated—superfused and stimulated. Two different stimulation parameter constellations were ap- plied: HFS (130 Hz, 1 mA, puls duration 0.1 ms, for 10 min) and 10 Hz, 10 mA, pulse duration 30 ms, for 1 min. HFS did not evoke the release of [3H]-GABA (Figure 2) or [3H]-glutamate (Figure 3) from rat neocortical syn- aptosomes. However, the alternative parameter constel- lation e.g. 10 Hz instead of 130 Hz, 10 mA instead of 1 mA, 30 ms instead of 0.1 ms pulse duration, application for only 1 instead of 10 min, clearly induced the release of both [3H]-glutamate and [3H]-GABA from synapto- somes pre-loaded with these transmitters. Similar results have also been found for human neocortical synapto- somes (data not shown). Obviously, electrical stimulations typical for HFS did not evoke any transmitter release from neocortical syn- aptosomes. However, another constellation of electrical parameters, applied for only a tenth of time, clearly evoked the synaptosomal release of [3H]-GABA or [3H]-glutamate. This shows 1) that it is possible, as a matter of principle, to release neurotransmitters from synaptosomes if their higher chronaxy is translated into a much higher duration of electrical pulses and 2) that the minimal pulse width and electrical current together with the typical frequency of HFS do not directly affect syn- aptosomes, i.e. nerve end- ings. Thus, HFS may indeed excite axons exclusively; then, transmitter release occurs not until the axonal depolariza- tion has propagated to the nerve endings. Whether HFS is selective for a certain neurotransmitter system, e.g. for Figure 1. Pathophysiology of Parkinson’s disease. This figure (adapted from [16]) illustrates the structural pathophysiologic condition of Parkinson’s disease. Degeneration of modulating dopaminergic neurons (dashed green line) originating from the substantia nigra pars compacta (SNC) and projecting to the striatum (caudate nucleus and putamen) mainly entails two consequences: a reduced activity in formerly excited (through dopamine D1 receptors) GABAergic interneurons (2; dashed blue lines) and an intensified activity in formerly inhibited (through dopamine D2 receptors) GABAergic interneurons (3; doubled blue line). However, these two obviously oppositional situations finally conclude in an identical effect of intensified thalamic inhibition and a thereby increased filter function of the thalamus. Two different pathways emanating from the striatum and reaching the thalamus explain that. The direct pathway (2) straightly leads from the striatum to the thalamus, either passing the medial globus pallidus (GPmed) or the sub- stantia nigra reticularis (SNR), whereas in the indirect pathway (3) additionally the lateral globus pallidus (GPlat) and the glu- tamatergic (doubled red arrows) subthalamic nucleus (STN) are connected in series. 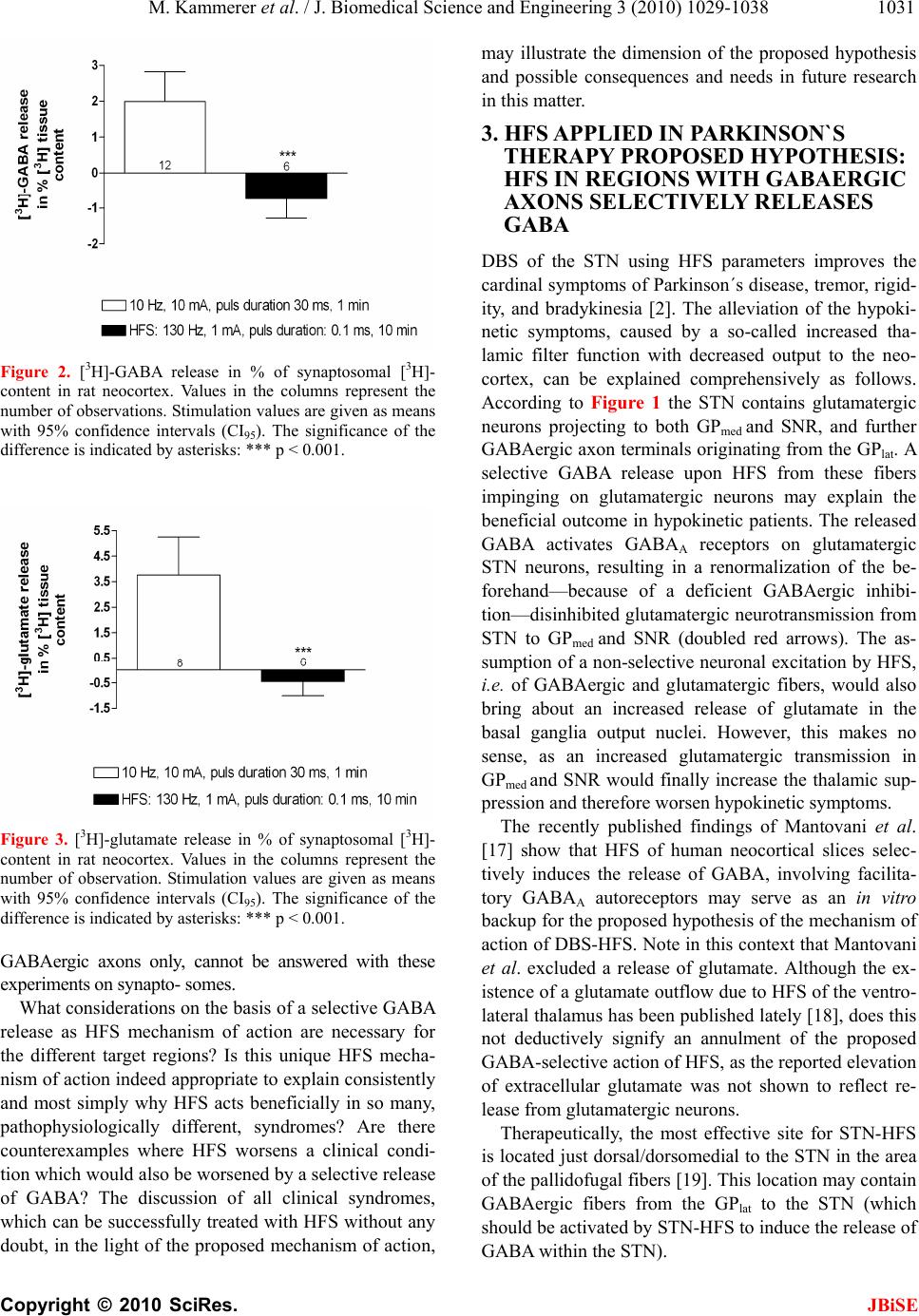 M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1031 Figure 2. [ 3H]-GABA release in % of synaptosomal [3H]- content in rat neocortex. Values in the columns represent the number of observations. Stimulation values are given as means with 95% confidence intervals (CI95). The significance of the difference is indicated by asterisks: *** p < 0.001. Figure 3. [3H]-glutamate release in % of synaptosomal [3H]- content in rat neocortex. Values in the columns represent the number of observation. Stimulation values are given as means with 95% confidence intervals (CI95). The significance of the difference is indicated by asterisks: *** p < 0.001. GABAergic axons only, cannot be answered with these experiments on synapto- somes. What considerations on the basis of a selective GABA release as HFS mechanism of action are necessary for the different target regions? Is this unique HFS mecha- nism of action indeed appropriate to explain consistently and most simply why HFS acts beneficially in so many, pathophysiologically different, syndromes? Are there counterexamples where HFS worsens a clinical condi- tion which would also be worsened by a selective release of GABA? The discussion of all clinical syndromes, which can be successfully treated with HFS without any doubt, in the light of the proposed mechanism of action, may illustrate the dimension of the proposed hypothesis and possible consequences and needs in future research in this matter. 3. HFS APPLIED IN PARKINSON`S THERAPY PROPOSED HYPOTHESIS: HFS IN REGIONS WITH GABAERGIC AXONS SELECTIVELY RELEASES GABA DBS of the STN using HFS parameters improves the cardinal symptoms of Parkinson´s disease, tremor, rigid- ity, and bradykinesia [2]. The alleviation of the hypoki- netic symptoms, caused by a so-called increased tha- lamic filter function with decreased output to the neo- cortex, can be explained comprehensively as follows. According to Figure 1 the STN contains glutamatergic neurons projecting to both GPmed and SNR, and further GABAergic axon terminals originating from the GPlat. A selective GABA release upon HFS from these fibers impinging on glutamatergic neurons may explain the beneficial outcome in hypokinetic patients. The released GABA activates GABAA receptors on glutamatergic STN neurons, resulting in a renormalization of the be- forehand—because of a deficient GABAergic inhibi- tion—disinhibited glutamatergic neurotransmission from STN to GPmed and SNR (doubled red arrows). The as- sumption of a non-selective neuronal excitation by HFS, i.e. of GABAergic and glutamatergic fibers, would also bring about an increased release of glutamate in the basal ganglia output nuclei. However, this makes no sense, as an increased glutamatergic transmission in GPmed and SNR would finally increase the thalamic sup- pression and therefore worsen hypokinetic symptoms. The recently published findings of Mantovani et al. [17] show that HFS of human neocortical slices selec- tively induces the release of GABA, involving facilita- tory GABAA autoreceptors may serve as an in vitro backup for the proposed hypothesis of the mechanism of action of DBS-HFS. Note in this context that Mantovani et al. excluded a release of glutamate. Although the ex- istence of a glutamate outflow due to HFS of the ventro- lateral thalamus has been published lately [18], does this not deductively signify an annulment of the proposed GABA-selective action of HFS, as the reported elevation of extracellular glutamate was not shown to reflect re- lease from glutamatergic neurons. Therapeutically, the most effective site for STN-HFS is located just dorsal/dorsomedial to the STN in the area of the pallidofugal fibers [19]. This location may contain GABAergic fibers from the GPlat to the STN (which should be activated by STN-HFS to induce the release of GABA within the STN).  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1032 It is possible to reduce the levodopa dosage of park- insonian patients treated with STN-HFS by more than 50% and to accomplish an alleviation of levodopa-in- duced hyperkinesias with this reduction [20]. Without lowering the administered levodopa dose, STN-HFS even worsens the dyskinesias [20-22], which can be as- cribed to a decreased filter function of the thalamus, as both levodopa and STN-HFS may diminish the eventual GABAergic neurotransmission to the thalamus. Levodopa should renormalize the patho-physiological condition in the striatum (see Figure 1; dashed green pathways). Concomitantly, and in accordance with our hypothesis, STN-HFS may throttle the glutamatergic output of the STN. Ultimately, an additionally reduced activation of the output nuclei, GPmed and SNR, results. 3.1. LFS Treatment of Postural Imbalance and Gait Disturbance Postural instability and gait disturbance are two very handicapping symptoms in Parkinson’s disease, but can be markedly ameliorated by applying LFS to the pedun- culopontine nucleus (PPN) [23,24]. In contrast to this, DBS of the cholinergic and glutamatergic PPN with a frequency of 100 Hz (approaching HFS) induced Park- inson-like akinesia and postural imbalance in non-human primates [25]. The described impairment may be attrib- uted to a GABAergic inhibition of excitatory PPN neu- rons. This assumption is reinforced by the fact that in a monkey model of Parkinson’s disease PPN lesioning also induced akinesia and postural instability [24]. This lesioning presumably means a withdrawal of PPN pro- jection fibers; this would correspond with a HFS-in- duced local GABAergic inhibition of cholinergic and glutamatergic PPN neurons. Further, both local applica- tion of bicucullin—a GABAA receptor antagonist—into the PPN and stimulation of the PPN with low frequency (~20 Hz) annihilated the previous HFS-induced symp- toms [24]. Summing up, the effects of HFS and LFS seem to be antithetic. LFS may coincide with the ex- pected effects of electrical stimulation of brain struc- tures, i.e. in case of the PPN an augmentation of excita- tory neuron activity, whereas HFS would abolish these effects, most likely through selective GABA release and activation of GABAA receptors on cholinergic and glu- tamatergic PPN neurons. Altoghether, this is an example for an in vivo correlation between HFS and GABAA re- ceptor agonism (see [17] for an in vitro correlate). Note that PPN-LFS primarily leads to a melioration of the parkinsonian symptoms gait disturbance and postural instability, but rarely of other typical parkinsonian symp- toms like rigidity or bradykinesia [23,26]. The last-men- tioned authors recommended a combination of bilateral STN-HFS and PPN-LFS in appropriate Parkinson pa- tients. 3.2. Diversity of Effects of HFS in the GPmed The GABAergic GPmed projection neurons send their axons to the ventral tier thalamic nuclei and to the PPN; some of these GPmed “motor” neurons additionally pro- ject to the CM/Pf thalamic complex [27]. The axons of other GPmed neurons, termed “limbic” neurons by Parent and Parent (2002), arborize principally within the lateral habenular nucleus (LHb) with some collaterals to the anterior thalamic nuclei. Afferents to the GPmed include the GABAergic direct striato-pallidal monosynaptic path- way and the glutamatergic subthalamo-pallidal part of the indirect polysynaptic pathway. In addition, a major- ity of GABAergic GPlat efferents have been shown to project through the GPmed en route to the STN [27,28]. According to the literature, HFS of various GPmed tar- gets may induce different and even contrary clinical ef- fects. Unintentional co-stimulation of GPlat areas may play a role here. GPmed-HFS is reported to alleviate hypokinesias as well as hyperkinesias [29,30]. GPmed-HFS improves ab- normal involuntary movements, though without the pos- sibility to reduce the dosage of levodopa essentially, in contrast to the case of STN-HFS [31]. When the GPlat is stimulated instead of the GPmed, HFS usually does not improve abnormal involuntary movements [32]. When, however, GPlat-HFS affects axons of striatal neurons of the indirect pathway to the GPlat, HFS may increase a too low GABAergic impulse flow in the hyperkinetic state to improve hyperkinesias (see below). Bejjani et al. [33] and Krack et al. [34] reported that GPmed-HFS within the most ventral contacts, lying at the ventral margin of, or just below, the GPmed, led to a pronounced improvement in rigidity and a complete arrest of levodopa-induced abnormal involuntary movements. The anti-akinetic effect of levodopa, however, was blocked and the patients became severely akinetic. Stimulation of the most dorsal contacts, lying at the dorsal border of the GPmed or inside the GPlat, usually led to moderate improvement of off-drug akinesia and in- duced dyskinesias in some patients. Tronnier et al. [35] reported a reduction of dyskinesias, but a worsening of hypokinetic parkinsonian symptoms upon GPmed-HFS, in contrast to other studies (see [29]). Thus, multiple sites, possibly not confined to the GPmed, but involving also the GPlat, seem to be responsible for partly contrasting clinical effects [33]. Obviously, the GPmed does not represent a uniform HFS object. Two GPmed-HFS target regions have been distinguished by Bejjani et al. [33] and by Krack et al. [34]. There may be even more HFS targets within, and in the close vicinity of, the GPmed: 1) HFS may affect thalamopetal axons of GPmed neu-  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1033 rons and thereby improve a hyperkinetic syndrome. 2) Alternatively, HFS can reach the pallid opetal fibers of striatal neurons of the direct pathway to increase their too low GABAergic impulse flow in the hypokinetic state; in this case, it alleviates hypokinesia. 3) At a still other site within the GPmed, HFS may stimulate the en route fibers from GPlat to STN running within the GPmed. Then, GPmed-HFS mirrors STN-HFS and also improves hypokinesia. 4) Further, GPmed-HFS may (also) affect nearby palli- dopetal fibers of striatal neurons of the indirect pathway to the GPlat. In this last case, HFS increases the too low GABAergic impulse flow in the hyperkinetic state and improves hyperkinesia. The pathophysiological assumptions behind (A)—(D) are the following: a) HFS of th alamopetal axons Abnormal involuntary movements, i.e. hyperkinesias, of the original hypokinetic Parkinson syndrome corre- spond with a reduced filter function of the thalamus, i.e. an insufficient GABAergic inhibition of thalamic neu- rons. Accordingly, a reduced neuronal activity in GPmed during levodopa-induced dyskinesia has been shown in parkinsonian monkeys [36]. Thus, GPmed-HFS dimin- ishes the abnormal involuntary movements in advanced Parkinson’s disease if the HFS-mediated selective GABA release is paralleled by a less diminished, i.e. normalized, GABAergic projection to the thalamus. It was shown in human neocortex slices that HFS in- duces action potentials in GABAergic fibers and subse- quent terminal release of GABA with subsequent activa- tion of facilitatory GABAA autoreceptors. GABAA re- ceptor blockade, changing the plasmalemmal chloride gradient of GABAA receptor channels and tetrodotoxin (which abolishes action potentials) antagonized this HFS-evoked GABA release [17,37]. Thus, orthodromic action potentials may be induced in thalamopetal GABAergic axons by HFS within the GPmed with sub- sequent release of GABA from their thalamic terminals; even more GABA release is due to activation by released GABA of facilitatory GABAA autoreceptors on these terminals. In addition, one can suppose antidromic ac- tion potentials due to HFS. These antidromic action po- tentials excite the soma of the GPmed neuron or travel backwards to reach recurrent axon collaterals with sub- sequent release of GABA in the somatodendritic region of the GABAergic cell. GABA may increase the firing rate of the GABAergic neuron through facilitatory somatodendritic GABAA autoreceptors. These GABAA autoreceptors have been demonstrated in human neocor- tical slices [17]. Whether these somatodendritic autore- ceptors are facilitatory, like those on GABAergic termi- nals, or inhibitory, as usual for GABAA receptors, de- pends on the local somatodendritic chloride gradient. Using the pharmacological tool furosemide to change the plasmalemmal chloride gradient, Mantovani et al. [17] did not differentiate between the involvement of somatodendritic and/or terminal autoreceptors in the mode of action of HFS. In any case, the overall effect of altering the chloride gradient was a decrease of HFS- induced GABA release. The terminal GABAA autore- ceptors were clearly facilitatory (as shown on isolated nerve endings, see [17]); it may well be that the facilita- tory terminal autoreceptors have overridden inhibitory somatodendritic autoreceptors of minor importance for the overall HFS-induced GABA release. Possibly, also both terminal and somatodendritic GABAA autorecep- tors are facilitatory and cooperate to realize the HFS- induced GABA release. Regardless of the somatoden- dritic autoreceptor being inhibitory or excitatory, the facilitatory feature of the terminal GABAA autoreceptors enabled HFS to induce an increased release of GABA. In the case of GPmed-HFS the increase in GABA release from terminals in the thalamus may either be the positive net effect of facilitatory terminal and inhibitory somato- dendritic GABAA autoreceptors or the sum of the effects of facilitatory terminal and facilitatory somatodendritic receptors. Boraud et al. [38] found that GPmed-HFS re- duced the firing frequency of GPmed neurons in the N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)- treated parkinsonian monkey; this would correspond to the combination of facilitatory terminal and inhibitory somatodendritic GABAA autoreceptors. b) HFS of pallidopetal fibers HFS in the dorsal GPmed and/or inside the GPlat may activate the GABAergic fibers from striatum through GPlat to GPmed of the direct pathway (see Figure 1; dashed blue projection from the striatum to the GPmed). Then, the striato-pallidal GABAergic transmission of the direct pathway is strengthened, GABAA receptors on GPmed projection neurons are activated, i.e. the pal- lido-thalamic neurotransmission is diminished, the filter function of the thalamus decreases, and hypokinesia im- proves. In the end, activating these striato-pallidal fibers of the direct pathway should correspond to STN-HFS. c) HFS of en route fibers from GPlat to STN GPmed- or GPlat-HFS matches STN-HFS when axons from GPlat neurons with terminals in the STN are stimu- lated (see Figure 1; dashed blue projection within the GPlat to the STN). Then, GABAergic axon terminals within the STN will release more GABA, the glutama- tergic subthalamo-pallidal and -nigral neurotransmis- sions decrease, the firing rate of the basal ganglia output nuclei is less activated, and hypokinetic parkinsonian symptoms improve as the filter function of the thalamus decreases.  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1034 d) HFS of pallidopetal fibers of striatal neurons of the indire ct pathway GPlat-HFS may, either intentionally or not in the course of GPmed-HFS, target the GABAergic striato- pallidal fibers of the indirect pathway (see Figure 1; doubled blue projection (3) from striatum to GPlat). In the hypokinetic parkinsonian condition these over- active striato-pallidal fibers strongly decelerate the pal- lido-subthalamic neurons. Then, the axons of these stri- ato-pallidal neurons either may react to HFS with even more increased GABA release from their terminals or the increased GABA release is already maximal without a further deceleration of the pallido-subthalamic neurons. Again, also antidromic action potentials due to HFS must be supposed; they excite the soma of the striatal GABAergic neuron or reach recurrent axon collaterals which subsequently release GABA in the somatoden- dritic region in the striatum. In hyperkinesia, the inhibition by the GABAergic output nuclei GPmed and SNR of the thalamus is medi- ated through the too strong dopamine D1 receptor-driven GABAergic transmission in the monosynaptic direct striato-pallidal and striato-nigral pathway. The resulting reduction of the filter function of the thalamus is intensi- fied by the D2 receptor-initiated decrease in the activity of the polysynaptic indirect pathway to the output nuclei with an increased GABA release in the STN and, subse- quently, a reduced subthalamic drive of GPmed and SNR. The proposed hypothesis predicts an increased release of GABA upon HFS: Indeed, GPmed-HFS enhanced the concentration of GABA in the ventricular cerebrospinal fluid during stimulation. In addition, the GABA level correlated with the degree of HFS-induced clinical ef- fects against tremor, rigidity, and drug-induced dyskine- sia [39]. 3.3. HFS in the Treatment of Parkinsonian Tremor Parkinsonian tremor can be treated by HFS in the ventral intermediate thalamic nucleus (VIM, see 4.) and by STN-HFS. An even better anti-tremor efficacy in Park- inson patients seems to result from HFS in the centrum medianum and parafascicularis thalamic nucleus (CM/Pf) [40]. The thalamic neurons of the CM/Pf are retarded by both GABAergic afferents from the GPmed and axon col- laterals of GABAergic interneurons and activated by glutamatergic afferents, e.g. from the cerebellum. Ac- cording to these circumstances, a selective GABA re- lease due to HFS in the CM/PF either inhibits a tremor- transmitting cerebellar projection and/or local glutama- tergic tremor cells. Dyskinesias can also be improved using CM/Pf-HFS, as shown by Krauss et al. [41], reminding of earlier antidyskinetic outcomes of medial thalamotomies [42]. Also in this case, a local inhibition of glutamatergic neurons due to a selective GABA release may explain the HFS mode of action. 4. HFS IN THE TREATMENT OF ESSENTIAL TREMOR Excitatory afferences from the deep cerebellar nuclei project to the VIM, which is their thalamic relay. Park- insonian tremor [1] as well as essential tremor [43] is improved due to the application of HFS to the VIM. Es- sential tremor can also be improved by local injection of the GABAA receptor agonist muscimol into the VIM, as shown by Pahapill et al. [43]. VIM-HFS as well as the application of muscimol results in improvement of tremor; this leads us to presume that the selective GABA release is the most likely HFS mode of action also in this target area. In the VIM, a selective GABA release from axon terminals of thalamic reticular neurons and VIM interneurons inhibits the thalamic relay cells which are driven by cerebellar afferents (for anatomical connec- tions see [44]). Consequently, released GABA seems to activate inhibitory GABAA receptors on glutamatergic cerebellar afferents and on glutamatergic thalamic relay neurons. 5. HFS IN THE TREATMENT OF DYSTONIA AND HUNTINGT ON’S DISEASE A similar reasoning as for the therapy of hyperkinesias in Parkinson’s disease, i.e. a strengthening of the GABAergic pallido-thalamic projection, explains hypo- thetically the efficacy of GPmed-HFS on other hyperkine- sias, e.g. on dystonia and Huntington´s disease. One, or even the most important, pathophysiological basis of these hyperkinesias is also a reduced filter function of the thalamus, to be reversed therapeutically. Besides using HFS to treat chorea of a Huntington patient, Moro et al. [45] also applied 40 Hz. 130 Hz improved cho- reatic symptoms more than 40 Hz; the concomitant bra- dykinesia, however, was rarely affected. The bradykine- sia ameliorated with 40 Hz, admittedly at the expense of the chorea reduction. A corresponding clinical difference between HFS and 40 Hz-stimulation was also observed by Fasano et al. [46]. Thus, 40 Hz induce other, possibly opposed, effects as the HFS-typical 130 Hz. 6. HFS IN DEPRESSION 6.1. HFS in the Treatment of Major Depression In depression the subgenual gyrus cinguli (Brodman area 25) is metabolically overactive. This overactive metabo- lism is being decreased due to antidepressant medication [47]. HFS of the white matter of the subgenual gyrus  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1035 cinguli was applied to reduce this elevated activity and successfully improved treatment-resistant major depres- sion. Therefore white matter of the subgenual gyrus cinguli-HFS may lead to an increased release of GABA from axon terminals in the Brodman area 25. GABA then activates inhibitory GABAA receptors on overactive postsynaptic neurons which calms these neurons. 6.2. Suicidality as Side-effect of STN-HFS A depression-like behaviour is aggravated in the forced swim test due to STN-HFS; the forced swim test is a widely used and validated rodent model of depression. On the level of neuronal activities, the firing rate of 5-HT neurons in the dorsal raphe nucleus (NDR) of rats is inhibited following STN-HFS [48]. Muscimol, being infused into the STN, imitated the effects of STN-HFS on the firing rate of 5-HT- neurons. Voon et al. [49] have recently confirmed that suicide is one of the most im- portant risks for mortality following STN-HFS in ad- vanced Parkinson’s disease. This serious adverse effect’s pathophysiology may allow us to draw conclusions about the mode of action of HFS. Anatomically, the following connections between STN and the 5-HT neurons of the NDR exist (see [50,51]; : excitation, ┤: inhibition, ncl. habenulae lat- eralis: LHb, GABA interneurons of NDR: NDRGABA, 5-HT neurons of NRD: NDR5-HT): The physiological condition is reflected by the fol- lowing chain of neuronal impacts: STN GPmed ┤ LHb NDRGABA ┤ NDR5-HT. The condition in patients suffering from Parkinson’s disease, however is different. In the hypokinetic state the STN is disinhibited, which changes the above-mentioned chain. ( means increased, () decreased, excitation; ╢ means increased, (┤) decreased, inhibition): STN GPmed ╢ LHb () NDRGABA (┤) NDR5-HT. A reduced inhibition of NDR 5-HT neurons is the re- sult of the disinhibited STN. The condition after STN-HFS, however, may vary as follows: STN-HFS () GPmed (┤) LHb NDRGABA ╢ NRD5-HT. Obviously, STN-HFS increases the inhibition of 5-HT neurons of the NDR which results in a lowering of the serotonergic neurotransmission to cortical areas. Defi- ciencies in the monoamine neurotransmission is the cur- rent hypothesis underlying major depression. Conse- quently, this decrease may explain the increased sui- cidality of Parkinson patients after STN-HFS. Not only 5-HT neurons in the NDR, but also nora- drenergic neurons in the locus coeruleus (LC), which project to the cortical areas, are influenced by STN-HFS. Pathophysiological condition in the hypokinetic Park- inson syndrome: STN GPmed ╢ LHb () LCGABA (┤) LCNA. Condition in Parkinson patients after STN-HFS: STN-HFS () GPmed (┤) LHb LCGABA ╢ LCNA. An increased inhibition due to a lowered noradrener- gic neurotransmission also promotes the occurrence of depression [52]. Note that the coincidence of a decrease of the sero- tonergic as well as the noradrenergic neurotransmission may lead to a substantially increased risk of the occur- rence of depression. GPmed-HFS is also used in the treatment of Parkin- son’s disease. Does suicidality also occur in GPmed-HFS as an adverse effect? Rodriguez-Oroz et al. [53] have compared the clinical occurence of depression in Park- inson patients treated with these two different HFS methods, i.e. STN-HFS and GPmed-HFS. A lower rate of depressions after GPmed-HFS compared to STN-HFS was found. This could be the result of a decreased inhi- bition of NDR5-HT after GPmed-HFS which ends in an undiminished serotonergic neurotransmission to cortical areas. Therefore, depression is not likely to occur after GPmed-HFS. Condition of Parkinson patients after GPmed-HFS: GPmed-HFS ╢ LHb () NDRGABA (┤) NRD5-HT. 7. HFS IN THE GILLES DE LA TOURETTE SYNDROME According to Servello et al. [54] it is possible to suc- cessfully treat patients suffering from Tourette syndrome by applying HFS to the centrum medianum/ parafas- cicular nucleus (CM/Pf) of the thalamus and to the ven- tral oral anterior thalamic nucleus (Voa). Although the pathophysiological knowledge about the exact network of neurotransmitters acting in the Gilles de la Tourette syndrome is limited, one may assume a GABAergic in- hibition of glutamatergic (CM/Pf and Voa) and choliner- gic (CM/Pf) neurons through GABAA receptors. The GABAergic afferents in this case come from the GPmed. While applying HFS to the CM/Pf and Voa, a terminal release of GABA is induced in afferent fibers to these nuclei and, by this, neurons in the HFS target structures are inhibited. Thus, the above stated assumption leads to the proposal that CM/Pf- and Voa-efferents, projecting to both striatum and neocortex, there may trigger the Gilles de la Tourette syndrome. 8. HFS IN OBSESSIVE COMPULSIVE DISORDER Corresponding to a recent publication by Greenberg et al. [55] DBS of the ventral internal capsule (VC) and the ventral striatum (VS) with HFS parameters meliorates the symptoms of patients with Obsessive Compulsive Disorder. Also here the particular pathophysiology is  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1036 unclear, but, in compliance with our hypothesis of the mode of functioning of HFS, GABA release in the VS as well as from VC fiber endings would take place. Now, on the one hand, axon collaterals from GABAergic inter or projection neurons of the VS could be excited by HFS and, on the other hand, postsynaptic neurons in target areas of VC fibers could be influenced in an inhibitory manner by the released GABA. 9. HFS IN EPILEPSIES Various human epilepsies have also been experimentally treated with DBS and by subdural neocortical stimula- tion (target regions: hippocampus, cerebellum, thalamus, STN, neocortex; see [56]). Comparing the efficacies, a higher rate of electrical stimulation approaches in animal models of epilepsies than of the corresponding clinical applications have displayed anticonvulsant properties (e.g. STN-HFS against absence-like seizures, cortex piriformis-LFS in kindled animals, hippocampus-HFS and -LFS). Taken together, electrical stimulation meth- ods in the treatment of epilepsies seem to be more re- mote from a common clinical application than the other clinical syndromes mentioned above. Mechanistically, however, just epilepsies could offer interesting aspects for the implementation of HFS inducing selective GABA release, as regards the pathophysiological role of the opponents GABA and glutamate in these disorders. 10. WHAT IS THE OVERALL IMPACT OF THE HYPOTHESIS OF A SELECTIVE GABA RELEASE BY HFS? Various treatment locations and options for HFS against different neurological and psychiatric syndromes are discussed above; there are detailed pathophysiological conceptions for most of these syndromes and, addition- ally for the suicidality following STN-HFS [49]. The hypothesis of a selective GABA release due to HFS is in line with these conceptions (see underlined pathophysi- ological basis in the following) by explaining the effica- cies and side effects of HFS according to the stimulated regions. (A) Augmented filter function of the thalamus (see 3.): GABA, released in the STN from axon termi- nals of neurons from the GPlat, reduces the disinhibition of the glutamatergic neurotransmission from STN to GPmed and to SNR and thus (re-) normalizes their tha- lamopetal projections. (B) Reduced filter function of the thalamus (see 3.2, 5.): GPmed-HFS increases the GABA- ergic transmission of projection neurons to the thalamus. (C) In contrast to STN-HFS, GPmed-HFS is not depres- siogenic, according to the pathophysiological conception of the transmission from GPmed to LHb to both NDR and LC. (D) In opposition to PPN-LFS (3.1), PPN-HFS im- pairs gait disturbance and postural instability of the Park- inson syndrome, as HFS induces a GABAergic inhibi- tion of excitatory PPN neurons. (E) CM/Pf-HFS and VIM-HFS are effective against tremor and dyskinesias (3.3, 4.) by a GABAergic inhibition of glutamatergic thalamic neurons. (F) HFS in the subgenual gyrus cin- guli inhibits through GABAergic axon terminals overac- tive neurons of the Brodmann area 25 in depression. The error probability of a correct explanation of the overall mechanism of action of HFS (selective GABA release) with regard to its clinical effects may be as- sessed as follows: Together, six independent arguments have been listed (A-F). If this independence of the six arguments is accepted, then a single probability of only 39.5% has to be assumed: The validity of the hypothesis “a selective GABA release explains argument (A) or (B) or … or (F)” corresponds to a significant collective ex- planation for the mode of action of HFS. The error probability for this collective explanation is in that case p = 0.049 = (1 – 0.395)6. Thus, we can state that a selec- tive GABA release significantly explains the mode of action of HFS. The proposed hypothesis corresponds to an optimal simplicity in explaining the observed clinical effects since the collective explanation is in any case simpler than another one which assumes different modes of actions of HFS in different target regions. We should add that the individual evidence of the se- lective GABA release following HFS in the various con- ditions should be separately demonstrated in spite of the present consideration of a collective explanation. Disclosure/Conflict-of-Interest Statement: This re- search was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. REFERENCES [1] Benabid, A.L., Pollak, P., Louveau, A., Henry, S. and Rougemont, J.D. (1987) Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nu- cleus for bilateral Parkinson disease. Applied Neurophysi- ology, 50(1-6), 344-346. [2] Benabid, A.L., Chabarde, S., Mitrofanis, J. and Pollak, P. (2009) Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson disease. Lancet Neurology, 8(1), 67-81. [3] Beurrier, C., Bioulac, B., Audin, J. and Hammond, C. (2001) High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. Journal of Neurophysiology, 85(4), 1351-1356. [4] Anderson, M.E., Postupna, N. and Ruffo, M. (2003) Ef- fects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. Journal of Neurophysiology, 89(2), 1150-1160. [5] Montgomery, E.B. and Baker, K.B. (2000) Mechanisms of deep brain stimulation and future technical developments.  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1037 Neurological Research, 22(3), 259-266. [6] Levy, R., Ashby, P., Hutchison, W.D., Lang, A.E., Lozano, A.M. and Dostrovsky, J.O. (2002) Dependence of subtha- lamic nucleus oscillations on movement and dopamine in Parkinson’s disease. Brain, 125, 1196-1209. [7] Meissner, W., Leblois, A., Hansel, D., Bioulac, B., Gross, C.E., Benazzouz, A. and Boraud T. (2005) Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain, 128, 2372-2382. [8] Dostrovsky, J.O. and Lozano, A.M. (2002) Mechanisms of deep brain stimulation. Movement Disorders, 17(3) S63-S68. [9] Garcia, L., Allesandro, G.D., Bioulac, B. and Hammond, C. (2005) High-frequency stimulation in Parkinson’s dis- ease: More or less? Trends in Neuroscience, 28(4), 209-216. [10] Moro, E., Esselink, R.J., Xie, J., Hommel, M., Benabid, A.L. and Pollak, P. (2002) The impact on Parkinson’s disease of electrical parameter settings in STN stimulation. Neurology, 59(5), 706-713. [11] Rizzone, M., Lamotte, M., Bergamasco, B., Tavella, A., Torre, E., Faccani, G., Melcarne, A. and Lopiano, L. (2001) Deep brain stimulation if the subthalamic nucleus in Parkinson’s disease: Effects of variation in stimulation parameters. Journal of Neurology, Neurosurgery, and Psychiatry, 71(2), 215-219. [12] Benabid, A.L., Benazzous, A. and Pollak, P. (2002) Mechanisms of deep brain stimulation. Movement Disor- ders, 17(3), S73-S74. [13] Nowak, L.G. and Bullier, J. (1998) Axons, but not cell bodies, are activated by electrical stimulation in cortical grey matter. I. evidence from chronaxie measurements. Experimental Brain Research, 118(4), 477-488. [14] Holsheimer, J., Demeulemeester, H., Nuttin, B. and Sutter, P.D. (2000) Identification of the target neuronal elements in electrical deep brain stimulation. European Journal of Neuroscience, 12(12), 4573-4577. [15] Holsheimer, J., Dijkstra, E.A., Demeulemeester, H. and Nuttin, B. (2000) Chronaxie calculated from current-duration and voltage-duration data. Journal of Neuroscience Methods, 97(1), 45-50. [16] Feuerstein, T.J. (2009) Antiparkinsonmittel. Pharmako- therapie des morbus Parkinson. In: Aktories, K., Förster- mann, U., Hofmann, F., Starke, K., Eds., Allgemeine und Spezielle Pharmakologie and Toxikologie, Urban & Fischer, München, 299-305. [17] Mantovani, M., Moser, A., Haas, C.A., Zentner, J. and Feuerstein, T.J. (2009) GABAA autoreceptors enhance GABA release from human neocortex: Towards a me- chanism for high-frequency stimulation (HFS) in brain? Naunyn-Schmiedeberg’s Archives of Pharmacology, 380 (1), 45-58. [18] Chang, S., Shon, Y.M., Agnesi, F. and Lee, K.H. (2009) Microthalamotomy effect during deep brain stimulation: Potential involvement of adenosine and glutamate efflux. Conference Proceedings: 31st Annual International Con- ference of the IEEE EMBS, 3294-3297. [19] Plaha, P., Shlomo, Y.B., Patel, N.K. and Gill, S.S. (2006) Stimulation of the caudal zona incerta is superior to stimulation of the subthalamic nucleus in improving con- tralateral Parkinsonism. Brain, 129, 1732-1747. [20] Moro, E., Scerrati, M., Romito, L.M., Roselli, R., Tonali, P. and Albanese, A. (1999) Chronic subthalamic nucleus stimulation reduces medication requirements in Parkin- son’s disease. Neurology , 53(1), 85-91. [21] Krack, P., Pollak, P., Limousin, P., Hoffman, D., Xie, J., Benazzouz, A. and Benabid, A.L. (1998) Subthalamic nu- cleus or internal pallidal stimulation in young onset Park- inson’s disease. Brain, 121, pp. 451-457. [22] Limousin, P., Pollak, P., Hoffmann, D., Benazzouz, A., Perret, J.E. and Benabid, A.L. (1996) Abnormal involun- tary movements induced by subthalamic nucleus stimula- tion in parkinsonian patients. Movement Disorders, 11 (3 ) , 231-235. [23] Plaha, P. and Gill, S.S. (2005) Bilateral deep brain stimu- lation of the pedunculopontine nucleus for Parkinson’s disease. Neuroreport, 16(17), 1883-1887. [24] Pereira, E.A., Muthusamy, K.A., Pennington, N.D., Joint, C.A. and Aziz, T.Z. (2008) Deep brain stimulation of the pedunculopontine nucleus in Parkinson’s disease. Pre- liminary experience at Oxford. British Journal of Neuro- surgery, 22(1), S41-S44. [25] Nandi, D., Liu, X., Winter, J.L., Aziz, T.Z. and Stein, J.F. (2002) Deep brain stimulation of the pedunculopontine region in the normal non-human primate. Journal of Clinical Neuroscience, 9(2), 170-174. [26] Stefani, A., Lozano, A.M., Peppe, A., Stanzione, P., Galati, S., Tropepi, D., Pierantozzi, M., Brusa, L., Scarnati, E. and Mazzone, P. (2007) Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Park- inson’s disease. Brain, 130, 1596-1607. [27] Parent, M. and Parent, A. (2002) Axonal collateralization in primate basal ganglia and related thalamic nuclei. Thalamus & Related systems, 2(1), 71-86. [28] Sato, F., Lavallée, P., Lévesque, M. and Parent, A. (2000) Single-axon tracing study of neurons of the external seg- ment of the globus pallidus in primate. Journal of Com- parative Neurology, 417(1), 17-31. [29] Volkmann, J. (2004) Deep brain stimulation for the treat- ment of Parkinson’s disease. Journal of Clinical Neuro- physiology, 21(1), 6-17. [30] Siegfried, J. and Lippitz, B. (1994) Bilateral chronic elec- trostimulation of ventroposterolateral pallidum: A new therapeutic approach for alleviating all parkinsonian symp- toms. Neurosurg ery, 35(6), 1126-1129. [31] Perlmutter, J.S. and Mink, J.W. (2006) Deep brain stimu- lation. Annual Review of Neuroscience, 29, 229-257. [32] Peppe, A., Pierantozzi, M., Altibrandi, M.G., Giacomini, P., Stefani, A., Bassi, A., Mazzone, P., Bernardi G. and Stanzione, P. (2001) Bilateral GPi DBS is useful to reduce abnormal involuntary movements in advanced Parkin- son’s disease patients, but its action is related to modality and site of stimulation. European Journal of Neurology, 8 (6), 579-586. [33] Bejjani, B., Damier, P., Arnulf, I., Bonnet, A.M., Vidailhet, M., Dormont, D., Pidoux, B., Cornu, P., Marsault C. and Agid, Y. (1997) Pallidal stimulation for Parkinson’s dis- ease. Two targets? Neurology, 49(6), 1564-9156. [34] Krack, P., Pollak, P., Limousin, P., Hoffman, D., Benaz- zouz A. and. Bas, J.F.L., Koudsie, A. and Benabid, A.L. (1998) Opposite motor effects of pallidal stimulation in Parkinson’s disease. Annals of Neurology, 43(2), 180-192. [35] Tronnier, V.M., Fogel, W., Kronenbuerger, M. and Stein- vorth, S. (1997) Pallidal stimulation: An alternative to pal-  M. Kammerer et al. / J. Biomedical Science and Engineering 3 (2010) 1029-1038 Copyright © 2010 SciRes. JBiSE 1038 lidotomy? Journal of Neurosurgery, 87(5), 700-705. [36] Papa, S.M., Desimone, R., Fiorani, M. and Oldfield, E.H. (1999) Internal global pallidus discharge is nearly sup- pressed during levodopa-induced dyskinesias. Annals of Neurology, 46(5), 732-738. [37] Mantovani, M., Velthoven, V.V., Fuellgraf, H., Feuerstein, T.J. and Moser, A. (2006) Neuronal electrical high fre- quency stimulation enhances GABA outflow from human neocortical slices. Neurochemistry International, 49(4), 347-350. [38] Boraud, T., Bezard, E., Bioulac, B. and Gross, C. (1996) High frequency stimulation of the internal Globus pallidus (GPi) simultaneously improves parkinsonian symptoms and reduces the firing frequency of GPi neurons in the MPTP-treated monkey. Neuroscience Letters, 210(1), 17-20. [39] Ogura, M., Nakao, N., Nakai, E., Uematsu, Y. and Itakura, T. (2004) The mechanism and effect of chronic electrical stimulation of the globus pallidus for treatment of Park- inson disease. Journal of Neurosurgery, 100(6), 997-1001. [40] Peppe, A., Gasbarra, A., Stefani, A., Chiavalon, C., Pier- antozzi, M., Fermi, E., Stanzione, P., Caltagirone, C. and Mazzone, P. (2008) Deep brain stimulation of CM/PF of thalamus could be the new elective target for tremor in advanced Parkinson’s Disease? Parkinsonism & Related Disorders, 14(6), 501-504. [41] Krauss, J.K., Pohle, T., Weigel, R. and Burgunder, J.M. (2002) Deep brain stimulation of the centre median- parafascicular complex in patients with movement disor- ders. Journal of Neurology, Neurosurgery and Psychiatry, 72(4), 546-548. [42] Ohye, C. and Shibazak, T. (2001) Lesioning the thalamus for dyskinesia. Stereotactic and Functional Neurosurgery, 77(1-4), 33-39. [43] Pahapill, P.A., Levy, R., Dostrovsky, J.O., Davis, K.D., Rezai, A.R., Tasker, R.R. and Lozano, A.M. (1999) Tremor arrest with thalamic microinjections of muscimol in patients with essential tremor. Annals of Neurology, 46(2), 249-252. [44] Strafella, A., Ashby, P., Munz, M., Dostrovsky, J.O., Lozano, A.M. and Lang, A.E. (1997) Inhibition of volun- tary activity by thalamic stimulation in humans: Rele- vance for the control of tremor. Movement Disorders, 12(5), 727-737. [45] Moro, E., Lang, A.E., Strafella, A.P., Poon, Y.Y., Arango, P., Dagher, A., Hutchison, W.D. and Lozano, A.M. (2004) Bilateral globus pallidus stimulation for Huntington’s disease. Annals of Neurology, 56(2), 290-294. [46] Fasano, A., Mazzone, P., Piano, C., Quaranta, D., Soleti F. and Bentivoglio, A.R. (2008) GPi-DBS in Huntington’s disease: Results on motor function and cognition in a 72-year-old case. Movement Disorders, 23(9), 1289-1292. [47] Mayberg, H.S., Lozano, A.M., Voon, V., McNeely, H.E., Seminowicz, D., Hamani, C., Schwalb, J.M. and. Ken- nedy, S.H. (2005) Deep brain stimulation for treat- ment-resistant depression. Neuron, 45(5), 651-660. [48] Temel, Y., Boothman, L.J., Blokland, A., Magill, P.J., Steinbusch, H.W., Vandewalle, V.V. and Sharp, T. (2007) Inhibition of 5-HT neuron activity and induction of de- pressive-like behavior by high-frequency stimulation of the subthalamic nucleus. Proceedings of the National Academy of Sciences of the United States of America, 104(43), 17087-17092. [49] Voon, V., Krack, P., Lang, A.E., Lozano, A.M., Dujardin, K., Schüpbach, M., Ambrosia, J.D., Thobois, S., Tamma, F., Herzog, J., Speelman, J.D., Samanta, J., Kubu, C., Rossignol, H., Poon, Y.Y., Cyr, J.A.S., Ardouin, C. and Moro, E. (2008) A multicentre study on suicide outcomes following subthalamic stimulation for Parkinson’s disease. Brain, 131, 2720-2728. [50] Ferraro, G., Montalbano, M.E., Sardo, P. and Grutta, V.L., (1996) Lateral habenular influence on dorsal raphe neu- rons. Brain Research Bulletin, 41(1), 47-52. [51] Smith, Y., Bevan, M.D., Shink, E. and Bolam, J.P. (1998) Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience, 86(2), 353-387. [52] Sartorius, A. and Henn, F.A. (2007) Deep brain stimula- tion of the lateral habenula in treatment resistent major depression. Medical Hypotheses, 69(6), 1305-1308. [53] Oroz, M.C.R., Obeso, J.A., Lang, A.E., Houeto, J.L., Pol- lak, P., Rehncrona, S., Kulisevsky, J., Albanese, A., Volk- mann, J., Hariz, M.I., Quinn, N.P., Speelman, J.D., Guridi, J., Zamarbide, I., Gironell, A., Molet, J., Sedano, B. P., Pidoux, B., Bonnet, A.M., Agid, Y., Xie, J., Benabid, A.L., Lozano, A.M., Cyr, J.S., Romito, L., Contarino, M.F., Scerrati, M., Fraix, V. and Blercom, N.V. (2005) Bilateral deep brain stimulation in Parkinson’s disease: A multicen- tre study with 4 years follow-up. Brain, 128, 2240-2249. [54] Servello, D., Porta, M., Sassi, M., Brambilla, A., Robert- son, M.M., (2008) Deep brain stimulation in 18 patients with severe Gilles de la Tourette syndrome refractory to treatment: The surgery and stimulation. Journal of Neu- rology, Neurosurgery and Psychiatry, 79(2), 136-142. [55] Greenberg, B.D., Gabriels, L.A., Jr, D.A.M., Rezai, A.R., Friehs, G.M., Okun, M.S., Shapira, N.A., Foote, K.D., Cosyns, P.R., Kubu, C.S., Malloy, P.F., Salloway, S.P., Giftakis, J.E., Rise, M.T., Machado, A.G., Baker, K.B., Stypulkowski, P.H., Goodman, W.K., Rasmussen, S.A. and Nuttin, B.J. (2010) Deep brain stimulation of the ven- tral internal capsule/ventral striatum for obsessive-compulsive disorder: worldwide experience. Molecular Psychiatry, 15(1), 64-79. [56] Gubellini, P., Salin, P., Goff, L.K.L. and Baunez, C. (2009) Deep brain stimulation in neurological diseases and ex- perimental models: From molecule to complex behaviour. Progress in Neurobiology, 89(1), 79-123. |