 Journal of Cancer Therapy, 2013, 4, 485-491 http://dx.doi.org/10.4236/jct.2013.43A059 Published Online March 2013 (http://www.scirp.org/journal/jct) 485 Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling Soham Puvvada1, Lisa Rimsza2 1Department of Medicine, University of Arizona, Tucson, USA; 2Department of Pathology, University of Arizona, Tucson, USA. Email: sohampuvvada@email.arizona.edu Received January 23rd, 2013; revised February 27th, 2013; accepted March 6th, 2013 ABSTRACT This review discusses the impact of gene expression profiling and sequencing discoveries on new therapeutic strategies in Non-Hodgkin Lymphomas, particularly Diffuse Large B cell Lymphoma. Alterations in oncogenes, over-active sig- naling pathways down-stream of the B cell receptor, and epigenetic gene mutations will be described. We will also re- view new targeting strategies aimed at each of these aspects of cell biology encompassing BCL2, BTK, PKCβ, PI3K/ mTOR and HDAC inhibition. Specific new drugs in clinical trials and early trial results are included as well. Keywords: BCR Signaling; Epigenetics 1. Introduction Non Hodgkin Lymphomas (NHL) are the 7th leading cause of cancer death in the United States [1]. Aggre- ssive B subtypes of NHL include Diffuse Large B cell Lymphoma (DLBCL), Mantle Cell Lymphoma (MCL), and Burkitts Lymphoma (BL). Of these, DLBCL is the most common with approximately 30,000 new cases each year and accounting for nearly 40% of all Non Ho- dgkin Lymphomas [1]. Although the majority of cases are curable with traditional anthracycline based regimens, up to 40% of patients fail to achieve a remission or even- tually relapse. This variability in response highlights the underlying biological heterogeneity of DLBCL. In the last decade, several new technologies have been deve- loped for investigating lymphoma and other tumors. In combination with known molecular techniques, the appli- cation of gene expression profiling (GEP), and next ge- neration sequencing have created a paradigm shift in our understanding of DLBCL. In a seminal publication, using a customized Lym- phochip array, new molecular subtypes of DLBCL were identified: Germinal Center B cell (GCB) type, and the activated B cell (ABC) type resembling activated peri- pheral blood B cells [2]. Subsequently, Primary Medi- astinal (PMBL) was identified as a DLBCL type with a better prognosis [3]. These three molecular subtypes have different pathogenetic mechanisms with corresponding prognostic implications despite similar clinical features and risk stratifications by International Prognostic Index (IPI) [4,5]. It was noted that the PMBL group had favor- able clinical features and prognosis while the ABC sub- type had the worst prognosis and response to anthracy- cline based chemotherapy: CHOP (Cyclophosphamide, Hydroxydaunorubicin/Adriamycin, Oncovin/Vincristine, Prednisone) [3,6]. This has been further validated by se- veral groups of investigators that used GEP to demon- strate the clinical significance of the GCB versus ABC distinction in R-CHOP (Rituxan-CHOP) treated patients [5-7]. Further GEP and sequencing data have led to the development of several new therapeutic strategies that at- tempt to target the different subtypes of DLBCL. 2. Targeting Oncogenes MYC and BCL2 are well characterized oncogenes in Lymphoma, which impact proliferation and evasion of apoptosis respectively. MYC translocations are detected in B-cell lymphoma, unclassifiable with features inter- mediate between DLBCL and BL as well as 10% of DL- BCL. These usually involve non-Immunoglobulin trans- location partners [8]. Many of these lymphomas are “Double Hit” containing MYC translocations and trans- locations or other abnormalities of BCL2 and/or BCL6. BCL2 is translocated with the Immunoglobulin Heavy chain gene in approximately 15% of DLBCL; Most of these cases are of the GCB subtype (35%) [9]. Develop- ing effective therapeutic strategies that target MYC has Copyright © 2013 SciRes. JCT  Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling 486 been elusive. Recently, it has been recognized that acetyl lysine bromodomains on known MYC coactivator pro- teins regulate its transcription [10]. Therefore, targeting bromodomains is an attractive option. JQ1 is a novel small molecule bromodomain inhibitor that downre- gulates MYC transcription and transcription of MYC dependent genes.When JQ1 was tested in multiple mye- loma cell lines, it decreased proliferation and caused cell cycle arrest [10]. JQ1 has been shown to decrease MYC and IL7 receptor in high-risk Acute Lymphoblastic Leu- kemia cell lines [11]. G-quadruplexes are secondary DNA structures resulting from DNA folding. It has recently been shown that MYC can form a G-Quadruplex. Quar- floxin is a fluroquinolone derivative. It has been chemi- cally modified so that it interacts with and stabilizes G- quadruplexes [12]. It is currently in phase I clinical trials in advanced solid tumors and lymphoma [13]. Oblimersen, an antisense nucleotide, was one of the first agents targeting BCL2 in development. It was tested in clinical trials in several solid tumors and hematologic malignancies. Early phase clinical data were promising in CLL but phase III data have been unconvincing, and its future development remains uncertain [14]. Obatoclax was developed as a small molecule with affinity to BCL- 2, BCL-XL, and MCL-1. Interestingly, obatoclax did not cause thrombocytopenia which has served as an in vivo marker of pharmacodynamics of BCL-XL inhibition [15] Obatoclax has shown modest clinical activity in a phase I combination with fludarabine and rituximab in relapsed CLL with partial response(PR) of 54% with no complete remissions [16]. In Small cell lung cancer, while early phase data indicated responses, a randomized phase II clinical trial in combination with chemotherapy did not show a statistically significant benefit in overall response rate (ORR) [17]. ABT-263 and ABT-199 are the next generation of BCL2 inhibitors. ABT263 has been very active in clinical trials with good response rates but th- rombocytopenia has been a dose limiting toxicity. Pre- clinical data have shown reproducible thrombocytopenia caused by mitochondrial membrane permeabilization in platelets thought to be mediated by BCLXL, and leading to apoptosis and clearance by liver and spleen macro- phages [18]. This is also suggested by relative resistance of younger platelets to ABT 263 that have higher levels of circulating BCLXL. This has been mitigated by the development of ABT 199. A key feature of this drug is that it lacks the high affinity binding to BCL-XL. This decreases the amount of thrombocytopenia allowing for higher dosing and correspondingly better response rates [19]. It is currently undergoing phase I trials in CLL and NHL. Additional agents in preclinical development in- clude Sabutoclax which is a novel pan pro survival BCL2 family protein inhibitor currently in preclinical develop- ment [20]. These agents are summarized in Table 1. 2.1. Targeting B Cell Receptor Signaling Recently in both Chroniclymphocytic leukemia (CLL) and particularly the ABC-DLBCL subtype, Chronic ac- tive B cell receptor (BCR) signaling has been recently identified as an important pathway. While it was known that CARD11 mutations are present in 10% of ABC- DLBCL, the oncogenic drivers associated with wildtype CARD11 were unknown. Through RNAi screen data, BCR signaling and Bruton’s Tyrosine Kinase (BTK) were identified as key components of survival in ABC- DLBCL [24]. Germline mutations in BTK are associated with X-linked Agammaglobulinemia. It has a variable phenotype characterized by recurrent bacterial infections in affected males in the first two years of life [25]. More than 600 different mutations in BTK have been reported, and two thirds of mutations are premature stop codons, splice defects, or frameshift mutations that interfere with translational processing of the BTK transcript [26]. BCR signaling is mediated by CD79A and CD79B. When an antigen occupies BCR, Srcfamily kinases pho- sphorylate tyrosines in the ITAM motifs of CD79A and CD79B [27]. This activates spleen tyrosine kinase (SYK) by which in turn initiates a signaling cascade that in- volves BTK, phospholipase Cγ, and protein kinase C β (PKCβ). BTK forms a complex with B-cell linker (BL- NK) and Phospholipase Cγ leading to the activation of multiple pathways including Nuclear factor-kappaB (NF- κB), Phosphatidylinositol 3-kinase (PI3K), Extracellular signal-regulated kinase (ERK), Mitogen activated protein kinase (MAP) and Nuclear factor of activated T cells (NFAT) [27]. This process is distinct from tonic BCR that promotes cell survival in mature B lymphocytes in mouse models; in mouse cells, when the BCR was condi- tionally ablated, all mature B cells died over a course of 2 weeks [28]. Implication of BTK in chronic active BCR signaling, and discovery of targeted BTK inhibitors has led to impressive clinical data. Recently, in an interim analysis of a multicenter, Open-Label, Phase 2 study of PCI32765 (targeted inhibitor to BTK) in ABC-DLBCL, the ORR in relapsed refractory DLBCL was 40%. 60% responses occurred in ABC-DLBCL tumors with CD79B mutations but responses were also seen in 37% wildtype CD79B suggesting that responses to PCI-32765 do not require a BCR mutation [29]. PKCβ is a serine threonine kinase expressed by normal and malignant B lymphocytes. It is required for BCR survival signals including activation of NF-κB; it phos- phorylates CARMA1 via IKK/TAK1. Enzastaurin is an acyclic bisindolylmaleimide that was initially developed as an adenosine triphosphate-competitive, selective inhi- bitor of PKCβ [30]. The compound also modulates the PI3K/AKT pathway in selected tumor models. In pre- clinical studies, Enzastaurin induced apoptosis and inhi- Copyright © 2013 SciRes. JCT  Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling Copyright © 2013 SciRes. JCT 487 Table 1. Targeting oncogenes in lymphoma. Process Target Small Molecule Inhibitor Development/ Response Focal Adverse Event Reference JQ1:BET Bromodomain Inhibitor Preclinical: Multiple myeloma All N/A [10,11] Proliferation MYC Quarfloxcin Ongoing Phase I N/A [13] Oblimersen Phase I NHL: 1/21 CR Thrombocytopenia [21] Obatoclax Phase I CLL in combination with Fludarabine/ Rituximab: 54% PR. Reversible mental status changes. [22] ABT 263 Phase 1: Relapsed Refractory Lymphoid Malignancies: 10/46 PR Relapsed Refractory CLL: 31% PR Thrombocytopenia Myelosuppression [19, 22] Apoptosis BCL2 ABT 199 Ongoing first in human Phase CLL/NHL N/A [23] Sabutoclax Preclinical: CML patient samples N/A [20] bited the proliferation of DLBCL cell lines and xeno- grafts at low micro molar doses [30]. In a randomized phase II trial, Enzastaurin in combination with Rituxan- CHOP (R-CHOP) was associated with an increased pro- gression free survival (PFS) and complete response (CR) rates [31]. It is currently in phase III trials: PRELUDE in DLBCL [32]. Fostamatinib is a SYK inhibitor that is orally bioavailable. In preclinical experiments, target in- hibition of SYK abrogated BCR signaling and induced apoptosis. When SYK was inhibited by R406 and R788 (Fostamatinib), it was associated with tumor regression [33]. This led to a phase I/II trial of Fostamatinib in DL- BCL. At a phase II dose of 200mg twice daily, the drug was well tolerated but ORR of 23.5% was noted with one only 1 CR [34]. The future of this drug in DLBCL re- mains uncertain. Another, downstream pathway activated by BCR is the PI3K/AKT (Protein Kinase B)/mTOR (Mammalian tar- get of Rapamycin) [35]. It is clinically significant in lym- phoma and CLL.mTOR is serine-threonine kinase that is present in two distinct complexes: mTORC1 and mTORC2. mTORC2 phosphorylates AKT, SGK1(serum and glucocorticoid-regulated kinase), PKC, and mTORC1 mediates cell growth and proliferation via the eIF4E (Eu- karyotic translation initiation factor 4E) binding proteins [36]. Everolimus is a rapamycin analog approved by the FDA for Renal cell carcinoma. Everolimusinduced a G1 arrest in DLBCL cell lines. Further, increased cytotoxic- ity was observed in rituximab sensitive cell lines [37]. It was evaluated in a phase II trial of 77 patients NHL and median age of 70. The predominant NHL subtypes en- rolled on the trial included DLBCL, MCL, and Grade III Follicular lymphoma (FL). The ORR was 30% with ma- jority being PRs and three CRs. Grade III toxicities in- cluded myelosuppression, hyperglycemia, and respiratory Infections [38]. P110α, p110β, p110δ and p110γ mediate the activation of the PI3K pathway via cell surface receptors. Of these, the p110δ isoform is highly expressed inhematopoetic cells, and required for cells to respond to BCR [39]. CAL-101/GS1101 was identified as a potent and selec- tive inhibitor of p110δ. It blocked constitutive PI3K sig- naling resulting in decreased phosphorylation of AKT and induction of apoptosis [40]. Finally, it has been shown that activated BCR also results in tyrosine phos- phorylation of JAK-STAT via LYN. An in vitro phos- phorylation assay demonstrated that LYN directly phos- phorylates STAT3 [41]. Ruxolitinib is an oral selective inhibitor of JAK1 and JAK2 that is FDA approved for the treatment of Myelofibrosis. It is currently in a mul- ticenter phase II study in DLBCL [42]. These agents are summarized in Table 2. 2.2. Targeting Epigenetics Post translational modification of histones via epigenetic mechanisms is critical for maintaining integrity of chro- matin and gene expression. Recently, several studies have implicated epigenetic mechanisms in the development of DLBCL. In a recent study that initially sequenced ge- nomic DNA of 14 NHL and paired normal tissues, 717  Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling 488 Table 2. Targeting the bcr signaling pathway in lymphoma. Drug Target Disease Reference PCI-32765/Ibrutinib BTK ABC-DLBCL [29] Enzaustaurin PKC DLBCL [30-32] Fostamatinib SYK DLBCL [34] Everolimus mTOR DLBCL [38] GS1101 P110 CLL, Indolent NHL [40] Ruxolitinib JAK-STAT DLBCL [42] coding single nucleotide variants were identified affect- ing 651 genes. These 14 NHL cases were then reana- lyzed with another 113 samples including 83 DLBCL with RNA sequencing; the tumor and matched normal DNA from these cases were re-sequenced to confirm 109 genes with multiple somatic mutations. It was remark- able that genes with roles for histone modification were frequent targets for somatic mutations in DLBCL [43]. MLL2 (Myeloid/Lymphoid or mixed lineage leukemia gene) showed the most significant number of nonsense single nucleotide variants including 37% nonsense muta- tions, 46% read frame altering indels, 8% point muta- tions at splice sites, and 9% non-synonymous amino acid substitutions. MLL2 was mutated in 59% DLBCL cell lines, 32% patient samples and in none of the paired nor- mal tissues. It is one of the six human H3K4 specific methyl transferases. Tri methylated H3K4 (H3K4me3) is epigenetically associated with promoters of actively tran- scribed genes. These mutations are likely inactivating since both alleles of the genes were affected by the mu- tations resulting in loss of MLL2 activity. MLL2 muta- tions were distributed amongst both ABC and GCB-DL- BCL subtypes [43]. Histone acetylation is moderated by Histone acetyl- transferases (HATs); it isan epigenetic mechanism for keeping chromatin in an open, transcription ready state. Histone Deacetylases (HDAC) keep the chromatin in ad- eactelyated repressive state. MEF2 (Mycocyte enhancer factor-2) proteins are a family of transcription factors that can act as transcriptional coactivators or corepressors of HAT and HDAC. Under normal intracellular calcium levels, MEF2 is bound by type IIa HDACs [44]. In- creased cytoplasmic calcium displaces the bound HDAC allowing for competitive binding of the HATs CREB- binding protein (CREBBP) and E1A binding protein p300 (EP300). This enables transcription of MEF2 target genes by acetylation of lysine residues on Histone H3 (H3K27). Somatic mutations of MEF2 that involved non synonymous single nucleotide variantsare unique to FL and GCB-DLBCL [43,45,46]. Mutations in HATs prin- cipally CREBBP are one of the most frequent mutations in DLBCL found in >30% of cases [45]. Thus, HDAC inhibition is a promising clinical strategy in DLBCL. Several HDAC inhibitors are in clinical trials for DL- BCL. Panobinostat is currently undergoing phase II trials as a single agent or in combination with rituximab or Everolimus in DLBCL [47,48]. Vorinostat was well tol- erated as a single agent in DLBCL but had suboptimal responses. It is currently being evaluated in combination with cytotoxic therapy in multiple trials [49]. The next generation HDAC inhibitors in early clinical develop- ment include Abexinostat, Bellinostat, and Rocilinostat (Figure 1). In preclinical studies with CLL patient sam- ples, Abexinostat resulted in cell death with physiologi- cally achievable IC50. It was also found to be synergistic with Bortezomib. Increase in Histone H3 acetylation and P21 protein, a known cyclin dependent kinase inhibitor up regulated by histone acetylation were also noted [50]. Abexinostatwas well tolerated in patients with relapsed FL, and MCL. While the median PFS in MCL was 4 months, at a median follow up of 10.3 months the PFS was 86% in FL patients [51]. A co-operative group trial evaluated the efficacy of Bellinostat in relapsed refrac- tory DLBCL patients with up to 5 prior chemotherapy regimens. In this trial, 2 PRs were observed at 5 and 13 months after registration [52]. Rocilinostat is currently in preclinical development, and it is synergistic with Bor- tezomib in both DLBCL and Multiple Myeloma cell lines [53,54]. BCL6 is an oncogene also involved with HAT repres- sion and is frequently altered in DLBCL through trans- locations and other mechanisms involving the 3q27 locus. The protein is typically expressed in the GCB subtype of DLBCL and is a potent therapeutic target in DLBCL. A recombinant 120 amino acid peptide RI-BPI containing the SMRTdomain inhibited transcriptional repressor ac- tivity of BCL6 and had potent in vitro lymphoma effects [55]. It was also noted that BCL6 repressed the expres- sion of EP300 and its cofactor HLA-B associated tran- script 3(BAT3). RI-BPI induced expression of p300 and BAT3, resulting in acetylation of p300 targets including p53 and Heat shock protein (hsp) 90. Induction of p300 and BAT3 was required for the antilymphoma effects of RI-BPI, since specific blockade of either protein rescued human DLBCL cell lines from the BCL6 inhibitor (Fig- ure 1) [56]. RI-BPI has recently shown promising pre- clinical synergy with small molecule inhibitors of BCL2, NEDD8 activating enzyme and Bortezomib [57]. EZH2 is the catalytic subunit of the PRC2 (Polycomb repressive complex 2) that enables methylation of histone H2 on lysine 27 (H3K27), decreasing gene expression and inhibiting transcription of dependent genes. EZH2 overexpression correlates with poor prognosis in Prostate adenocarcinoma and Renal cell carcinoma. Somatic mu- tations have been recently identified in the SET domain of EZH2 occurring in FL and up to 22% GCB-DLBCL. A mutation that results in the replacement of tyrosine to Copyright © 2013 SciRes. JCT 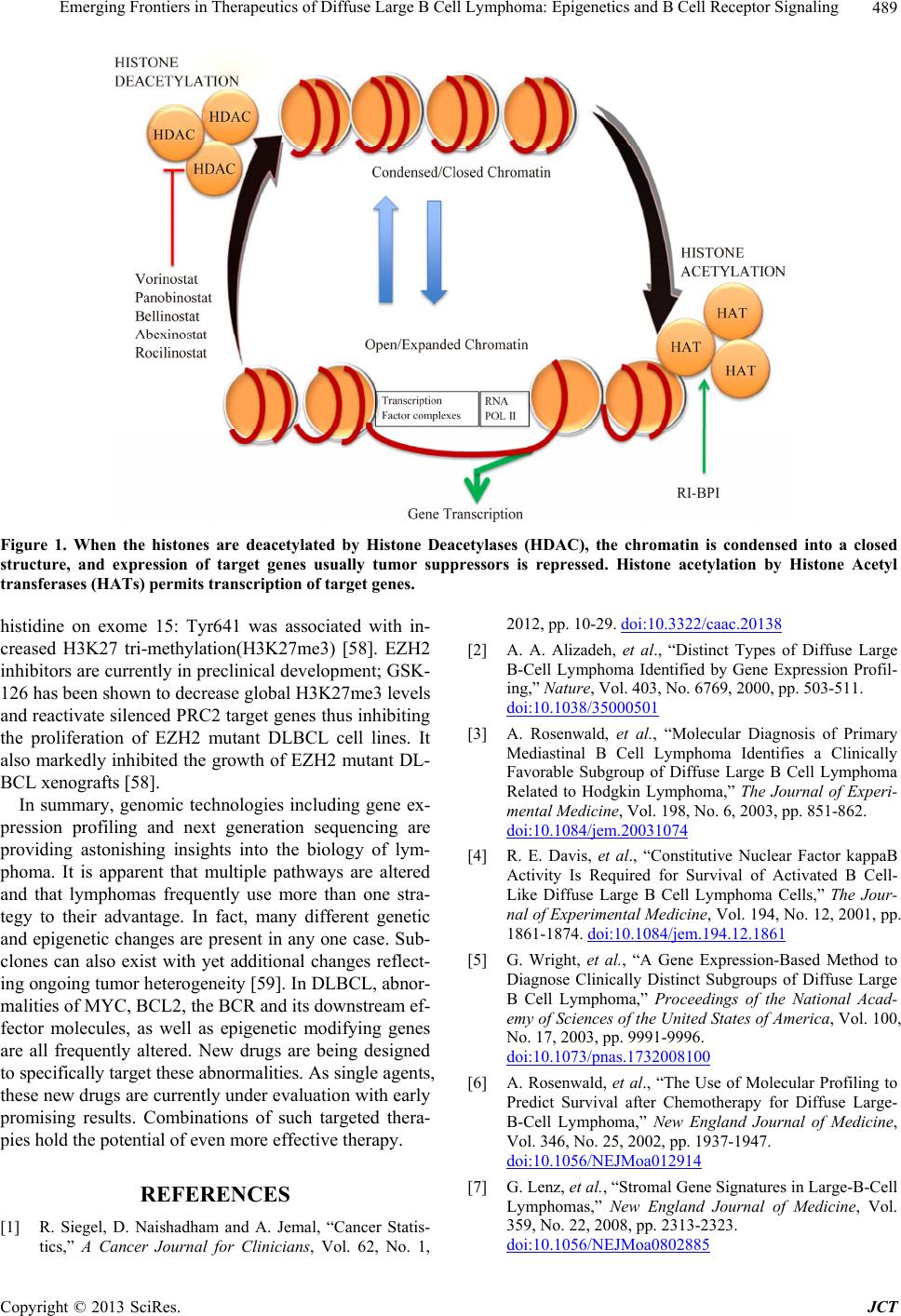 Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling Copyright © 2013 SciRes. JCT 489 Figure 1. When the histones are deacetylated by Histone Deacetylases (HDAC), the chromatin is condensed into a closed structure, and expression of target genes usually tumor suppressors is repressed. Histone acetylation by Histone Acetyl transferases (HATs) permits transcription of target genes. histidine on exome 15: Tyr641 was associated with in- creased H3K27 tri-methylation(H3K27me3) [58]. EZH2 inhibitors are currently in preclinical development; GSK- 126 has been shown to decrease global H3K27me3 levels and reactivate silenced PRC2 target genes thus inhibiting the proliferation of EZH2 mutant DLBCL cell lines. It also markedly inhibited the growth of EZH2 mutant DL- BCL xenografts [58]. In summary, genomic technologies including gene ex- pression profiling and next generation sequencing are providing astonishing insights into the biology of lym- phoma. It is apparent that multiple pathways are altered and that lymphomas frequently use more than one stra- tegy to their advantage. In fact, many different genetic and epigenetic changes are present in any one case. Sub- clones can also exist with yet additional changes reflect- ing ongoing tumor heterogeneity [59]. In DLBCL, abnor- malities of MYC, BCL2, the BCR and its downstream ef- fector molecules, as well as epigenetic modifying genes are all frequently altered. New drugs are being designed to specifically target these abnormalities. As single agents, these new drugs are currently under evaluation with early promising results. Combinations of such targeted thera- pies hold the potential of even more effective therapy. REFERENCES [1] R. Siegel, D. Naishadham and A. Jemal, “Cancer Statis- tics,” A Cancer Journal for Clinicians, Vol. 62, No. 1, 2012, pp. 10-29. doi:10.3322/caac.20138 [2] A. A. Alizadeh, et al., “Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profil- ing,” Nature, Vol. 403, No. 6769, 2000, pp. 503-511. doi:10.1038/35000501 [3] A. Rosenwald, et al., “Molecular Diagnosis of Primary Mediastinal B Cell Lymphoma Identifies a Clinically Favorable Subgroup of Diffuse Large B Cell Lymphoma Related to Hodgkin Lymphoma,” The Journal of Experi- mental Medicine, Vol. 198, No. 6, 2003, pp. 851-862. doi:10.1084/jem.20031074 [4] R. E. Davis, et al., “Constitutive Nuclear Factor kappaB Activity Is Required for Survival of Activated B Cell- Like Diffuse Large B Cell Lymphoma Cells,” The Jour- nal of Experimental Medicine, Vol. 194, No. 12, 2001, pp. 1861-1874. doi:10.1084/jem.194.12.1861 [5] G. Wright, et al., “A Gene Expression-Based Method to Diagnose Clinically Distinct Subgroups of Diffuse Large B Cell Lymphoma,” Proceedings of the National Acad- emy of Sciences of the United States of America, Vol. 100, No. 17, 2003, pp. 9991-9996. doi:10.1073/pnas.1732008100 [6] A. Rosenwald, et al., “The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large- B-Cell Lymphoma,” New England Journal of Medicine, Vol. 346, No. 25, 2002, pp. 1937-1947. doi:10.1056/NEJMoa012914 [7] G. Lenz, et al., “Stromal Gene Signatures in Large-B-Cell Lymphomas,” New England Journal of Medicine, Vol. 359, No. 22, 2008, pp. 2313-2323. doi:10.1056/NEJMoa0802885  Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling 490 [8] K. J. Savage, et al., “MYC Gene Rearrangements Are Associated with a Poor Prognosis in Diffuse Large B-Cell Lymphoma Patients Treated with R-CHOP Chemother- apy,” Blood, Vol. 114, No. 17, 2009, pp. 3533-3537. doi:10.1182/blood-2009-05-220095 [9] J. Iqbal, et al., “BCL2 Translocation Defines a Unique Tumor Subset within the Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma,” American Journal of Pathology, Vol. 165, No. 1, 2004, pp. 159-166. doi:10.1016/S0002-9440(10)63284-1 [10] J. E. Delmore, et al., “BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc,” Cell, Vol. 146, No. 6, 2011, pp. 904-917. doi:10.1016/j.cell.2011.08.017 [11] C. J. O. Nadja Kopp, L. Bird, R. Paranal, J. Qi, T. Bow- man, S. J. Rodig, A. L. Kung, J. E. Bradner and D. Wein- stock, “BET Bromodomain Inhibition Targets Both c- Myc and IL7R in Acute Lymphoblastic Leukemia,” Blood, Vol. 120, No. 21, 2012, Abstract No: 672. [12] S. Kendrick and L. H. Hurley, “The Role of G-Quadru- plex/i-Motif Secondary Structures as Cis-Acting Regula- tory Elements,” Pure and Applied Chemistry, Vol. 82, No. 8, 2010, pp. 1609-1621. doi:10.1351/PAC-CON-09-09-29 [13] NCT00955786, “Dose-Escalation Study of CX-3543 in Patients with Advanced Solid Tumors or Lymphomas,” National Institutes of Health, 2012. [14] M. S. Davids and J. A. Burger, “Cell Trafficking in Chro- nic Lymphocytic Leukemia,” Open Journal of Hematolog, Vol. 3, No. S1, 2012. [15] M. Konopleva, et al., “Mechanisms of Antileukemic Ac- tivity of the Novel Bcl-2 Homology Domain-3 Mimetic GX15-070 (Obatoclax),” Cancer Research, Vol. 68, No. 9, 2008, pp. 3413-3420. doi:10.1158/0008-5472.CAN-07-1919 [16] J. R. Brown, “Obatoclax in Combination with Fludara- bine and Rituximab (FR) Is Well Tolerated and Shows Promising Clinical Activity in Relapsed CLL/SLL,” Blood, Vol. 118, No. 21, 2012, Abstract No: 2865. [17] P. K. Paik, et al., “A Phase I Study of Obatoclax Mesy- late, a Bcl-2 Antagonist, Plus Topotecan in Solid Tumor Malignancies,” Cancer Chemotherapy and Pharmacology, Vol. 66, No. 6, 2010, pp. 1079-1085. doi:10.1007/s00280-010-1265-5 [18] K. D. Mason, et al., “Programmed Anuclear Cell Death Delimits Platelet Life Span,” Cell, Vol. 128, No. 6, 2007, pp. 1173-1186. doi:10.1016/j.cell.2007.01.037 [19] W. H. Wilson, et al., “Navitoclax, a Targeted High-Affi- nity Inhibitor of BCL-2, in Lymphoid Malignancies: A Phase 1 Dose-Escalation Study of Safety, Pharmacoki- netics, Pharmacodynamics, and Antitumour Activity,” The Lancet Oncology, Vol. 11, No. 12, 2010, pp. 1149-1159. doi:10.1016/S1470-2045(10)70261-8 [20] S. H. Leu, “Sabutoclax, a Novel Pan BCL2 Family In- hibitor, Sensitizes Dormant Blast Crisis Chronic Myeloid Leukemia Stem Cells to Dasatinib,” Blood, 2012. [21] J. S. Waters, et al., “Phase I Clinical and Pharmacokinetic Study of bcl-2 Antisense Oligonucleotide Therapy in Pa- tients with Non-Hodgkin’s Lymphoma,” Journal of Cli- nical Oncology, Vol. 18, No. 9, 2000, pp. 1812-1823. [22] M. S. Davids, and A. Letai, “Targeting the B-Cell Lym- phoma/Leukemia 2 Family in Cancer,” Journal of Clini- cal Oncology, Vol. 30, No. 5, 2012, pp. 3127-3135. doi:10.1200/JCO.2011.37.0981 [23] NCT01328626, “A Phase 1 Study Evaluating the Safety and Pharmacokinetics of ABT-199 in Subjects with Re- lapsed or Refractory Chronic Lymphocytic Leukemia and Non-Hodgkin Lymphoma,” 2012. [24] R. E. Davis, et al., “Chronic Active B-Cell-Receptor Sig- nalling in Diffuse Large B-Cell Lymphoma,” Nature, Vol. 463, No. 7277, 2010, pp. 88-92. doi:10.1038/nature08638 [25] M. E. Conley, et al., “Mutations in btk in Patients with Presumed X-Linked Agammaglobulinemia,” American Journal of Human Genetics, Vol. 62, No. 5, 1998, pp. 1034-1043. doi:10.1086/301828 [26] J. Valiaho, C. I. Smith and M. Vihinen, “BTKbase: The Mutation Database for X-Linked Agammaglobulinemia,” Human Mutation, Vol. 27, No. 12, 2006, pp. 1209-1217. doi:10.1002/humu.20410 [27] L. M. Staudt, “Oncogenic Activation of NF-kappaB,” Cold Spring Harbor Perspectives in Biology, Vol. 2, No. 6, 2010, Article ID: a000109. doi:10.1101/cshperspect.a000109 [28] M. Kraus, et al., “Survival of Resting Mature B Lym- phocytes Depends on BCR Signaling via the Igalpha/Beta Heterodimer,” Cell, Vol. 117, No. 6, 2004, pp. 787-800. doi:10.1016/j.cell.2004.05.014 [29] W. H. Wilson, “The Bruton’s Tyrosine Kinase (BTK) Inhibitor, Ibrutinib (PCI-32765) Has Preferential Activity in the ABC Subtype of Relapsed/Refractory de Novo Dif- fuse Large B Cell Lymphoma (DLBCL): Interim Results of a MultiCenter, Open-Label, Phase II Study, in Ameri- can Society of Hematology,” Blood, 2012. [30] M. J. Robertson, et al., “Phase II study of Enzastaurin, a Protein Kinase C Beta Inhibitor, in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma,” Journal of Clinical Oncology, Vol. 25, No. 13, 2007, pp. 1741- 1746. doi:10.1200/JCO.2006.09.3146 [31] J. D. E. A. Hainsworth, “Randomized Phase II Study of R-CHOP Plus Enzastaurin versus R-CHOP in the First Line Treatment of Patients iwth Intermediate and High Risk Diffuse Large B Cell Lymphoma: Preliminary Ana- lysis,” Journal of Clinical Oncology, Vol. 29, No. 8016, 2011. [32] NCT00332202, “Prelude: Study to Investigate the Pre- vention of Relapse in Lymphoma Using Daily Enzas- taurin,” National Institutes of Health: Clinical Trials, 2012. [33] R. M. Young, et al., “Mouse Models of Non-Hodgkin Lymphoma Reveal Syk as an Important Therapeutic Tar- get,” Blood, Vol. 113, No. 11, 2009, pp. 2508-2516. doi:10.1182/blood-2008-05-158618 [34] J. W. Friedberg, et al., “Inhibition of Syk with Fosta- matinib Disodium Has Significant Clinical Activity in Non-Hodgkin Lymphoma and Chronic Lymphocytic Leu- kemia,” Blood, Vol. 115, No. 13, 2010, pp. 2578-2585. doi:10.1182/blood-2009-08-236471 [35] A. Bernal, et al., “Survival of Leukemic B Cells Pro- Copyright © 2013 SciRes. JCT  Emerging Frontiers in Therapeutics of Diffuse Large B Cell Lymphoma: Epigenetics and B Cell Receptor Signaling Copyright © 2013 SciRes. JCT 491 moted by Engagement of the Antigen Receptor,” Blood, Vol. 98, No. 10, 2001, pp. 3050-3057. doi:10.1182/blood.V98.10.3050 [36] R. J. Dowling, et al., “Dissecting the Role of mTOR: Les- sons from mTOR Inhibitors,” Biochimica et Biophysica Acta, Vol. 1804, No. 3, 2010, pp. 433-439. doi:10.1016/j.bbapap.2009.12.001 [37] K. Wanner, et al., “Mammalian Target of Rapamycin Inhibition Induces Cell Cycle Arrest in Diffuse Large B Cell Lymphoma (DLBCL) Cells and Sensitises DLBCL Cells to Rituximab,” British Journal of Haematology, Vol. 134, No. 5, 2006, pp. 475-484. doi:10.1111/j.1365-2141.2006.06210.x [38] T. E. Witzig, et al., “A Phase II Trial of the Oral mTOR Inhibitor Everolimus in Relapsed Aggressive Lymphoma,” Leukemia, Vol. 25, No. 2, 2011, pp. 341-347. doi:10.1038/leu.2010.226 [39] S. T. Jou, et al., “Essential, Nonredundant Role for the Phosphoinositide 3-Kinase p110delta in Signaling by the B-Cell Receptor Complex,” Molecular and Cellular Bi- ology, Vol. 22, No. 24, 2002, pp. 8580-8591. doi:10.1128/MCB.22.24.8580-8591.2002 [40] B. J. Lannutti, et al., “CAL-101, a p110delta Selective Phosphatidylinositol-3-Kinase Inhibitor for the Treatment of B-Cell Malignancies, Inhibits PI3K Signaling and Cel- lular Viability,” Blood, Vol. 117, No. 2, 2011, pp. 591- 594. doi:10.1182/blood-2010-03-275305 [41] L. Wang, T. Kurosaki and S. J. Corey, “Engagement of the B-Cell Antigen Receptor Activates Stat through Lyn in a Jak-Independent Pathway,” Oncogene, Vol. 26, No. 20, 2007, pp. 2851-2859. doi:10.1038/sj.onc.1210092 [42] NCT01431209, “Ruxolitinib Phosphate (Oral JAK Inhibi- tor INCB18424) in Treating Patients with Relapsed or Refractory Diffuse Large B-Cell or Peripheral T-Cell Non- Hodgkin Lymphoma,” National Institutes of Healt, 2012. [43] R. D. Morin, et al., “Frequent Mutation of Histone- Modifying Genes in Non-Hodgkin Lymphoma,” Nature, Vol. 476, No. 7360, 2011, pp. 298-303. doi:10.1038/nature10351 [44] H. D. Youn and J. O. Liu, “Cabin1 represses MEF2-De- pendent Nur77 Expression and T Cell Apoptosis by Con- trolling Association of Histone Deacetylases and Acety- lases with MEF2,” Immunity, Vol. 13, No. 1, 2000, pp. 85-94. doi:10.1016/S1074-7613(00)00010-8 [45] L. Pasqualucci, et al., “Inactivating Mutations of Acetyl- transferase Genes in B-Cell Lymphoma,” Nature, Vol. 471, No. 7337, 2011, pp. 189-195. doi:10.1038/nature09730 [46] L. Pasqualucci, et al., “Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma,” Nature Genetics, Vol. 43, No. 9, 2011, pp. 830-837. doi:10.1038/ng.892 [47] NCT00967044, “Panobinostat (LBH589) Plus Everoli- mus (RAD001) in Patients with Relapsed and Refractory Lymphoma,” National Institutes of Health, 2012. [48] NCT01238692, “A Phase II Study of Oral Panobinostat (LBH589) and Rituximab to Treat Diffuse Large B Cell Lymphoma,” National Institutes of Health, 2012. [49] M. Crump, et al., “Phase II Trial of Oral Vorinostat (Sub- eroylanilide Hydroxamic Acid) in Relapsed Diffuse Large- B-Cell Lymphoma,” Annals of Oncology, Vol. 19, No. 5, 2008, pp. 964-969. doi:10.1093/annonc/mdn031 [50] S. Bhalla, et al., “PCI-24781 Induces Caspase and Reac- tive Oxygen Species-Dependent Apoptosis through NF- kappaB Mechanisms and Is Synergistic with Bortezomib in Lymphoma Cells,” Clinical Cancer Research, Vol. 15, No. 10, 2009, pp. 3354-3365. doi:10.1158/1078-0432.CCR-08-2365 [51] A. M. Evens, “A Phase II Multicenter Study of the His- tone Deacetylase Inhibitor (HDACi) Abexinostat (PCI- 24781) in Relapsed/Refractory Follicular Lymphoma (FL) and Mantle Cell Lymphoma (MCL), in ASH Annual Meeting Abstracts,” Blood, 2012. [52] D. O. Persky, “A Phase II Study of PXD101 (belinostat) in Relapsed and Refractory Aggressive B-Cell Lympho- mas (rel/ref ABCL); SWOG 0520 in ASCO,” Journal of Clinical Oncology, Vol. 30, 2012, Article ID: e18536. [53] R. Noopur, “Rocilinostat (ACY-1215), a Selective HDAC 6 Inhibitor, Alone and in Combination with Bortezomib in Multiple Myeloma: Preliminary Results from the First- in-Humans Phase I/II Study, in ASH Annual Meeting Abstracts,” Blood, 2012. [54] L. Santo, et al., “Preclinical Activity, Pharmacodynamic, and Pharmacokinetic Properties of a Selective HDAC6 Inhibitor, ACY-1215, in Combination with Bortezomib in Multiple Myeloma,” Blood, Vol. 119, No. 11, 2012, pp. 2579-2589. doi:10.1182/blood-2011-10-387365 [55] L. C. Cerchietti, et al., “BCL6 Repression of EP300 in Human Diffuse Large B Cell Lymphoma Cells Provides a Basis for Rational Combinatorial Therapy,” Journal of Clinical Investigation, Vol. 120, No. 12, 2010, pp. 4569- 4582. doi:10.1172/JCI42869 [56] L. C. Cerchietti, et al., “A Peptomimetic Inhibitor of BCL6 with Potent Antilymphoma Effects in Vitro and in Vivo,” Blood, Vol. 113, No. 15, 2009, pp. 3397-3405. doi:10.1182/blood-2008-07-168773 [57] T. E. A. Dupont, “Combinatorial Targeting of BCL6 and Anti-Apoptotic Proteins in Diffuse Large B-Cell Lym- phoma (DLBCL) and Follicular Lymphoma (FL),” Blood, 2012. [58] M. T. McCabe, et al., “EZH2 Inhibition as a Therapeutic Strategy for Lymphoma with EZH2-Activating Muta- tions,” Nature, Vol. 492, No. 7427, 2012, pp. 108-112. doi:10.1038/nature11606 [59] M. Gerlinger, et al., “Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequenc- ing,” New England Journal of Medicine, Vol. 366, No. 10, 2012, pp. 883-892. doi:10.1056/NEJMoa1113205
|