 J. Biomedical Science and Engineering, 2013, 6, 236-248 JBiSE http://dx.doi.org/10.4236/jbise.2013.62A029 Published Online February 2013 (http://www.scirp.org/journal/jbise/) A novel over-sampling method and its application to miRNA prediction Xuan Tho Dang1*, Osamu Hirose2, Thammakorn Saethang1, Vu Anh Tran1, Lan Anh T. Nguyen1, Tu Kien T. Le1, Mamoru Kubo2, Yoichi Yamada2, Kenji Satou2 1Graduate School of Natural Science and Technology, Kanazawa University, Kanazawa, Japan 2Institute of Science and Engineering, Kanazawa University, Kanazawa, Japan Email: *thodx@hnue.edu.vn Received 15 December 2012; revised 14 January 2013; accepted 20 January 2013 ABSTRACT MicroRNAs (miRNAs) are short (~22 nt) non-coding RNAs that play an indispensable role in gene regula- tion of many biological processes. Most of current computational, comparative, and non-comparative methods commonly classify human precursor micro- RNA (pre-miRNA) hairpins from both genome pseudo hairpins and other non-coding RNAs (ncRNAs). Al- though there were a few approaches achieving prom- ising results in applying class imbalance learning methods, this issue has still not so lved completely and successfully yet by the existing methods because of imbalanced class distribution in the datasets. For example, SMOTE is a famous and general over-sam- pling method addressing this problem, however in some cases it cannot improve or sometimes reduces classification performance. Therefore, we developed a novel over-sampling method named incre-mental- SMOTE to distinguish human pre-miRNA hairpins from both genome pseudo hairpins and other ncRNAs. Experimental results on pre-miRNA datasets from Batuwita et al. showed that our method achieved bet- ter Sensitivity and G-mean than the control (no over- sampling), SMOTE, and several successsors of modi- fied SMOTE including safe-level-SMOTE and bor- der-line-SMOTE. In addition, we also applied the novel method to five imbalanced benchmark datasets from UCI Machine Learning Repository and ach- ieved improvements in Sensitivity and G-mea n. These results suggest that our method outperforms SMOTE and several successors of it in various biomedical classification problems including miRNA classifica- tion. Keywords: Imbalanced Dataset; Over-Sampling; SMOTE; miRNA Classification 1. INTRODUCTION MicroRNAs (miRNAs) are short (~22nt) non-coding RNAs (ncRNAs) that play an indispensable role in gene regulation of many biological processes. The tiny miRNAs can target numerous mRNAs to induce mRNA degrada- tion or translational repression or both, they could regulate 20% - 30% of human genes [1]. The miRNAs are tran- scribed as long primary miRNAs (pri-miRNAs) which are processed into 60 - 70 nt precursor miRNAs (pre- miRNAs) by Drosha-DGCR8. The pre-miRNA is trans- ferred from Nucleus to Cytoplasm by Exp5-RanGTP and then split by Dicer-TRBP into miRNA duplex (~22 nt) [1]. The first miRNAs were characterized in early 1990s [2] but research on miRNAs has not revealed multiple roles in gene regulation (transcript degradation, translational suppression or transcriptional and translational activation) yet. Until the early 2000s, miRNAs were recognized as essential components in most biological processes [3-6]. Subsequently, miRNAs have become a hot topic and a large number of miRNAs, particularly 21,264 different miRNAs have been identified in various species so far according to the release 19 of miRBase [7]. However, the identification of miRNAs from a genome by existing experiment techniques is so difficult, expensive, and re- quires a large amount of time. Therefore, computational methods with two main approaches based on compara- tive and non-comparative methods play important role to detect new miRNAs. The rationale idea of the first ap- proach—comparative methods is that miRNA genes are conserved in closely related genomes in hairpin second- dary structures. Several comparative methods are pre- sented such as RNAmicro [8], MiRscan [9], miRseeker [10], MIRcheck [11], and MiRFinder [12]. These con- servation-dependent comparative methods are successful to predict hundreds of miRNAs with high sensitivity in closely related species. However, they are unable to identify novel miRNAs without close homologies due to lack of current data or unreliability of alignment algo- rithms [13], especially due to possibly rapid evolution of *Corresponding author. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 237 miRNAs. Particularly, the report from Berezikov et al. [14] has emphasized that non-conserved miRNAs in hu- man genome missed by comparative methods are rela- tively large and even have not still been recognized yet. Meanwhile, the second approach—non-comparative methods are promising for recognizing additional miRNAs which are non-conserved miRNAs. The main idea of these methods is based on hairpin secondary structures of pre-miRNAs. Sewer et al. [15] used clus- tering approach to predict novel miRNAs in the same cluster with known miRNAs. Xue et al. [16] proposed the classification of real and pseudo pre-miRNA hairpins based on 32 features of structure-sequence triplet. Clote et al. [17] and Hertel et al. [8] also identified that hairpin secondary structures are popular in many types of ncRNAs and a huge number of pseudo hairpins and hair- pin structures can be found among secondary structures of other ncRNAs. In addition, machine learning ap- proaches including random forest prediction model [18, 19]; hybrid of genetic algorithm and support vector ma- chine (GA-SVM) [20]; feature selection strategies based on SVM and boosting method [21]; hidden Markov model (HMM) [22] have also been used. Therefore, both genome pseudo hairpins, other ncRNAs and machine learning approaches should be applied. In addition, miRNA prediction problem should be also considered as imbalance class distribution. Batu- wita et al. [23] proposed an effective classifier system, namely microPred which used a complete pseudo hairpin dataset and ncRNAs. In their research, they focused on handling with class imbalance problem in datasets where samples from the majority class (9248 = 8494 pseudo hairpins + 754 other ncRNAs) significantly outnumber the minority class (691 pre-miRNAs). Xiao et al. [24] presented several network parameters based on two-di- mensional network of pre-miRNA secondary structure such as bracketed, tree, dual graph, etc. Their dataset contained 3928 positive samples (animal pre-miRNAs) and 8897 negative samples (8487 pseudo hairpins and 410 ncRNAs). Therefore, this dataset is suffered from imbalance problem, that is, the negative dataset outnum- bers the positive dataset. The main problem of class im- balance distribution is that normal learners are often bi- ased to the majority class, leading to good classification for the majority class samples while misclassification for many minority class ones. In order to solve these class imbalance learning problems, some solutions have been developed, including two main types: the external meth- ods at data processing level and the internal methods at algorithm level. One of remarkable solutions is SMOTE which is a famous and general over-sampling method addressing this problem. For example, experimental re- sults of Batuwita et al. [23] suggested that the best clas- sifier has been developed by applying the SMOTE method. However, there are still some drawbacks of SMOTE, particularly in some cases it cannot improve or sometimes reduces classification performance. Therefore, in our research we developed a novel over-sampling method to achieve better classification performance than both of the control method (no over-sampling) and SMOTE in the classification of pre-miRNAs for human miRNA gene prediction. Moreover, in order to demon- strate the applicability of our methods, we also compare our methods with several successors of modified SMOTE including safe-level-SMOTE [25] and border- line-SMOTE [26]. The structure of this paper is organized as follows: Section 2 gives a brief introduction to SMOTE and some related works, then shows its drawback; Section 3 intro- duces three variations of our novel method, incremental- SMOTE, which is improved from SMOTE; Section 4 analyses the experiments and compares our novel me- thod with the control, SMOTE, safe-level-SMOTE, and borderline-SMOTE methods. Finally, conclusions are described in Section 5. 2. METHOD 2.1. SMOTE Chawla et al. developed a minority over-sampling tech- nique called SMOTE [27] in which the minority class samples are over-sampled by creating synthetic samples rather than by over-sampling with replacement. SMOTE provided a new approach to over-sampling and intro- duced a bias towards the minority class. The results in [27] showed that this approach could improve the per- formance of classifiers for the minority class. In a less application-specific manner, synthetic sam- ples are generated by operating in “feature space” rather than “data space”. The minority class is over-sampled by synthesizing new samples along the line joining the mi- nority samples and their nearest neighbors. Depending on the requirement of over-sampling amount, nearest neighbors are selected by chance. Synthetic samples are generated in the following way: firstly, compute the dif- ference of feature vector between each minority class sample and its randomly selected nearest neighbor. Then, multiply this difference by a random number between 0 and 1, and finally add it to the feature vector of the mi- nority sample. In this way, the synthetic minority sample is generated along the line segment between two specific features. This approach is effective in forcing the decision re- gion of the minority class to become more general as shown in Figure 1. Figure 1(a) presents a typical case of imbalanced data where samples from the majority class greatly outnumber the minority class. As a result, the majority class samples are well-classified whereas many Copyright © 2013 SciRes. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 Copyright © 2013 SciRes. 238 thetic samples are generated between two sets of minor- ity class samples named Source (S) and Destination (D). Figure 2(b) describes step by step the generation of syn- thetic samples by using SMOTE method: at the first step, a set of synthetic samples (X1) is generated, and at the next step, another new set (X2) is generated; the process is repeated and the sets of generated synthetic samples can be different in each step. samples from the minority class are easy to be misclassi- fied. Therefore, imbalanced dataset problem requires a new and more adaptive method such as SMOTE. Figure 1(b) describes synthetic samples generated by SMOTE to achieve more balanced distribution so that the classi- fier recognizes all samples correctly. A variation of SMOTE, namely borderline-SMOTE is proposed by Han et al. [26] as the improvement of SMOTE. The authors analyzed most of the classification algorithms and attempt to learn the borderline of each class as exactly as possible in the training process. Those samples far from the borderline may make a little con- tribution to classification. Therefore, their method was based on the same over-sampling technique to SMOTE, but the difference is it only over-sampled the borderline samples of minority class instead of over-sampling all samples of the class as in SMOTE. In Figure 2(b), we could realize the drawbacks of SMOTE method: S and D do not change in the process of generating synthetic samples; and synthetic minority class samples will not be paid any attention after they are generated. Therefore, in order to address these drawbacks and improve classification accuracy of the SMOTE me- thod, we focus on how to utilize generated synthetic sam- ples as members of Source and Destination sets for further generation. This idea will be presented in next section, a novel method namely incremental- SMOTE. Another modification of SMOTE, safe-level-SMOTE is also presented by Bunkhumpornpat et al. [25]. Instead of randomly synthesizing the minority samples along the line joining a minority sample and its selected nearest neighbours, this method ignored nearby majority samples. The safe-level-SMOTE method carefully generated syn- thetic samples along the same line with different weight level, called safe level. The safe level was computed by using nearest neighbour minority samples. Moreover, although X is generated by S and D, S is the decisive factor in generating X. In contrast, D is only a randomly selected nearest neighbor to be used in com- bination with S in this generation. Therefore, the change of S will lead to the change of X as shown below in in- cremental-SMOTE2 and incremental-SMOTE3. Based on the analysis, we present three new methods, incremental-SMOTE1, incremental-SMOTE2, and incre- mental-SMOTE3. Although SMOTE and several successors of modified SMOTE including borderline-SMOTE and safe-level- SMOTE are famous and general over-sampling methods addressing the imbalanced class distribution problems, in some cases they cannot improve or sometimes reduce classification accuracy. 2.3. Incremental-SMOTE1 The idea of incremental-SMOTE1 is simple: the Des- tination set is expanded incrementally while keeping the Source set to be unchanged in all steps. Figure 3 illus- trates more details about this idea. In step 1, using SMOTE, a set of synthetic samples X1 is generated from two sets—Source (S) and Destination (D) sets as men- tioned above. In step 2, the Destination set is expanded by merging X1 into it. Similarly in step 3, the Destina- tion set is expanded again by merging X2 into it. The process is repeated in further steps. 2.2. Drawback of SMOTE As discussed above, one particular synthetic sample is generated by using SMOTE as shown in Figure 2(a). Blue sample x is a synthetic sample generated along the line joining a minority class sample s and its randomly selected nearest neighbor d. Generally, the set of syn- (a) (b) Figure 1. Advantages of SMOTE. Black, red, and blue dots indicate majority class samples, minority class samples, and synthetic minority class samples, respectively. The discrimina- tion hyperplane is the brown line. (a) The original dataset with an erroneous classifier biased by the imbalanced dataset; (b) Synthesize some new minority class samples by applying SMOTE with a perfect classifier. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 239 (a) (b) Figure 2. Synthetic samples generated normally by SMOTE. Both red and blue dots indicate minority class samples. The latter indicate synthetic minority class samples generated by using SMOTE. (a) Particularly, one synthetic minority class sample is generated along the line of two minority class samples by using SMOTE; (b) Generally, a set of synthetic sam- ples is generated from two sets named Source (S) and Destination (D). Figure 3. The idea of incremental-SMOTE1. Pseudo-code of incremental-SMOTE1 is as follows: Algorithm incremental-SMOTE1 (T, N). Input: The number of minority class samples T; Am- ount of incremental-SMOTE1 N (%); Output: (N × T/100) synthetic minority class samples 0) Initialize and assign two new sets with the same size as the number of minority class samples. Source = T; Destination = T; 1) Generate the new set X of T synthetic samples by using SMOTE. X = new synthetic samples generated at this step; 2) Merge X into the Destination set. Destination = Destination + X; 3) Repeat from Step 1 N/100 times. 2.4. Incremental-SMOTE2 Incremental-SMOTE2 is based on a reversal idea: the Destination set now is kept to be the same in every step, and the Source set is expanded incrementally. It is shown clearly in Figure 4. Pseudo-code of incremental-SMOTE2 is as follows: Algorithm incremental-SMOTE2 (T, N). Input: The number of minority class samples T; Amount of incremental-SMOTE2 N (%); Output: (N × T/100) synthetic minority class samples 0. Initialize and assign two new sets with the same size as the number of minority class samples. Source = T; Destination = T; 1) Generate the new set X of T synthetic samples by using SMOTE. X = new synthetic samples generated at this step; 2) Merge X into the Source set. Source = Source + X; 3) Repeat from Step 1 until (N × T/100) synthetic mi- nority class samples are generated. 2.5. Incremental-SMOTE3 The idea of incremental-SMOTE3 is the combination of incremental-SMOTE1 and incremental-SMOTE2: both Source and Destination sets are expanded. Figure 5 shows more details for this idea. Pseudo-code of incremental-SMOTE3 is as follows: Algorithm incremental-SMOTE3 (T, N). Input: The number of minority class samples T; Amount of SMOTE N (%); Output: (N * T/100) synthetic minority class samples 0) Initialize and assign two new sets with the same size as the number of minority class samples. Copyright © 2013 SciRes. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 240 Figure 4. The idea of incremental-SMOTE2. Figure 5. Thes idea of incremental-SMOTE3. Source = T; Destination = T; 1) Generate the new set X of T synthetic samples by using SMOTE. X = new synthetic samples generated at this step; 2) Merge X into the Source and Destination sets. Source = Source + X; Destination = Destination + X; 3) Repeat from Step 1 until (N × T/100) synthetic mi- nority class samples are generated. 2.6. Classifier For binary class classification, Support Vector Machine (SVM) is widely used to build a classifier discriminating the classes [28]. SVM is based on simple ideas origi- nated in statistical learning theory [29] which has high generalization capability, optimizes global classification solution and could be successfully applied in bioinfor- matics. In addition, in order to more generally evaluate the performance of our method, two different classifica- tion methods other than SVM were used, namely k- Nearest Neighbour (k-NN) and Random Forest (RF). Implementation of SVM, k-NN, and RF in kernlab [30], class [31], and random Forest [32]package avail- able at the Comprehensive R Archive Network (CRAN) was used, respectively. In our research, we used Radial Basis kernel (Gaussian kernel) of kernlab for SVM. Kernlab is an extensible package for kernel-based ma- chine learning methods in R and includes various kernels such as Linear kernel, Radial Basis kernel (Gaussian kernel), Polynomial kernel, etc. Moreover, all hyper- parameters for k-NN and RF, as well as other hyper-pa- rameters for SVM such as cost, class weights, etc. were also set to be default values. 2.7. Evaluation Measures A confusion matrix of binary class classification is shown in Ta bl e 1. In the field of binary classification in imbalanced data, most of the studies consider the class label of the minority class as positive. Thus, the class labels positive and negative are given to the samples in minority and majority classes, respectively. In Tab le 1, the first column presents the actual class label of the samples, and the first row is their predicted outcome class label. TP and TN denote the number of positive and negative samples that are classified correctly, while FN and FP denote the number of misclassified positive and negative samples, respectively. If the dataset is extremely imbalanced, for example, with an imbalance ratio of 99 to 1, even when the classi- fier classifies all the samples as negative, the accuracy of classification is still high up to 99%. As a result, accu- racy is not used to evaluate the performance of classifier for imbalance datasets, and more reasonable evaluation metrics should be presented [33,34]. In medical science, bioinformatics, and machine lear- ning communities [23,24,33,34], the sensitivity (SE) and the specificity (SP) are two metrics used to evaluate the performance of classifiers. Sensitivity measures the pro- portion of actual positives which are correctly identified as such, while specificity can be defined as the propor- tion of negatives which are correctly identified. Kubat et al. [35] proposed the Geometric mean metric defined as follows. G-mean =SE SP There are many researches applying this metric for evaluating classifiers as commonly used in imbalanced Table 1. A confusion matrix for binary class classification. Predicted Positive Predicted Negative Observed Positive TP FN Observed NegativeFP TN Copyright © 2013 SciRes. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 Copyright © 2013 SciRes. 241 OPEN ACCESS class distribution [23,34-37]. Therefore, we use this met- ric to measure the performances of the classifiers in our research. 3. RESULTS 3.1. miRNA Dataset The miRNA datasets selected in this research were downloaded from the website of microPred classifier system [23] and miRNA network-level [24]. MicroPred consists of three kinds of non-redundant human se- quences: 691 pre-miRNAs, 8494 pseudo hairpins, and 754 other ncRNAs (9248 hairpins). The first type is posi- tive, and the others are negative. Meanwhile, miRNA network-level dataset contains 3928 positive samples (animal pre-miRNAs) and 8897 negative samples (8487 pseudo hairpins and 410 ncRNAs). Class imbalance ratio of the positive to negative dataset of microPred and miRNA network-level was 1:13 and 1:2, respectively. It means that these datasets have imbalanced class distribu- tion with majority class samples outnumbering minority ones. In microPred dataset, 48 features were used to rep- resent each sample while there were only 21 features used in miRNA network-level dataset. This was shown clearly by Xiao et al. [24] who presented 24 network features but three of them (Girth, M_coreness, and Tran- sitivity) were meaningless because all samples are the same value in both classes. Subsequently, the rest 21 features were meaningful and thus used in miRNA net- work-level dataset. 3.2. Classification Imbalance Learning Results The experiments were executed to compare five methods: control method (no over-sampling), SMOTE, safe-level- SMOTE, borderline-SMOTE, and incremental-SMOTE. SVM, k-NN, and RF were used as the classifiers. The classification performance of the methods was estimated by 10-fold cross-validation. For each test, nine-tenth of the complete dataset was used as a training set. Then, in case of SMOTE, safe-level-SMOTE, borderline-SMOTE, and incremental-SMOTE, minority samples in the train- ing set over-sampled with the value of k is set to 5 like as SMOTE. After the training by an SVM, k-NN, or RF model using the (possibly over-sampled) training set, the model was tested against the remaining one-tenth of the dataset (i.e. test set). This process was repeated for all 10-fold with different combination of training and test sets. The values for the criteria of performance, sensitiv- ity, specificity, and G-mean were calculated by averaging 20 independent runs of 10-fold cross-validation and summarized in Table 2. Furthermore, two-sample t-test with equal variance was conducted to assess whether the averages of G-mean by different methods are signifi- cantly different. Table 2. Classification performance for microPred and miRNA network-level datasets expressed in percent. SVM k-NN RF Dataset Method SE SP G-mean SE SP G-meanSE SP G-mean No over-sampling 97.82 99.98 98.89 58.54 99.82 76.44 99.46 99.97 99.72 SMOTE 98.68 99.96 99.32 81.40 98.06 89.34 99.70 99.97 99.84 increSMOTE1 99.10 99.95 99.52 84.74 97.26 90.78 99.78 99.97 99.88 increSMOTE2 98.94 99.95 99.44 90.98 94.52 92.74 99.80 99.97 99.88 increSMOTE3 98.93 99.95 99.44 89.49 94.66 92.04 99.79 99.97 99.88 Safe level-SMOTE 98.72 99.96 99.34 81.27 97.53 89.03 99.72 99.97 99.84 Borderline-SMOTE1 98.65 99.96 99.30 79.39 97.73 88.08 99.64 99.97 99.80 microPred Borderline-SMOTE2 98.80 99.93 99.36 83.93 96.58 90.03 99.97 99.94 99.96 No over-sampling 80.43 96.51 88.10 79.85 94.92 87.06 82.58 95.92 89.00 SMOTE 88.08 91.32 89.69 84.77 90.23 87.46 87.28 91.82 89.52 increSMOTE1 88.57 90.99 89.77 84.69 90.74 87.67 86.24 93.02 89.57 increSMOTE2 89.62 89.31 89.47 87.58 85.71 86.64 87.01 92.12 89.53 increSMOTE3 89.67 89.24 89.45 87.47 85.27 86.36 86.38 92.78 89.52 Safelevel-SMOTE 88.22 91.20 89.70 84.97 89.84 87.37 85.66 93.51 89.50 Borderline-SMOTE1 86.58 93.14 89.80 83.35 91.25 87.21 85.47 93.59 89.44 miRNA network level Borderline-SMOTE2 86.63 93.01 89.76 86.09 85.50 85.80 85.09 93.92 89.39  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 242 Table 3. The description of the imbalanced datasets from UCI. Name Examples Attributes Imbalance ratio Breast-w 683 10 1:1.90 Haberman 306 3 1:2.78 Blood 748 4 1:3.20 Breast-p 198 32 1:3.21 Yeast 1484 8 1:28.10 Ionosphere 351 34 1:1.79 Glass 214 9 1:6.38 Satimage 6435 36 1:9.28 Experimental results on the microPred dataset showed that our method achieved better G-mean than control method, SMOTE, safe-level-SMOTE, and borderline- SMOTE on two of three classifiers. For example, with using SVM, although the sensitivity and G-mean in- creased for control method (97.82% and 98.89%), SMOTE (98.68% and 99.32%), safe-level-SMOTE (98.72% and 99.34%), borderline-SMOTE1 (98.65% and 99.30%), and borderline-SMOTE2 (98.80% and 99.36%), they also increased by (99.10% and 99.52%), (98.94% and 99.44%), and (98.93% and 99.44%) for incremental- SMOTE1, incremental-SMOTE2, and incremental- SMOTE3, respectively. However, it is different in the criterion of the specificity: in comparison with control method (99.98%), the specificity was decreased by 0.02% for SMOTE, safe-level-SMOTE, and borderline- SMOTE1 (99.96%, 99.96% and 99.96%); 0.03% for in- cremental-SMOTE1, incremental-SMOTE2, and incre- mental-SMOTE3 (99.95%, 99.95%, 99.95%, and 99.95%); and 0.05% for borderline-SMOTE2 (99.93%). The assessment by t-test on the microPred dataset suggested that SMOTE, safe-level-SMOTE, borderline- SMOTE1, and borderline-SMOTE2 significantly out- performs the control method (with p-value 2.2E−16, 2.2E−16, 4.7E−15, and 2.2E−16, respectively) and this is also similar with incremental-SMOTE1, incremental- SMOTE2, and incremental-SMOTE3 in comparison with the control method (with p-values 7.24E−14, 4.5E−13, and 6.2E−10, respectively). Furthermore, it is easily rec- ognized that three methods used in our research also re- markably outperform SMOTE, safelevel-SMOTE, bor- derline-SMOTE1, and borderline-SMOTE2 with p-values (5.4E−5, 3.7E−3, and 2.1E−2); (1.9E−4, 1.2E−2, and 4.0E−2); (3.4E−5, 2.4E−3, and 1.3E−2); and (5.6E−4, 3.2E−2, and 8.0E−2), respectively. In addition, the experimental results and the assess- ment by t-test on the miRNA network-level dataset also suggested that our method significantly achieved better G-mean than control method, SMOTE, and safe-level- SMOTE by using all three classifiers, and significantly outperformed borderline-SMOTE on two of three classi- fiers (for more details, see Tables 2 and 5). 3.3. Benchmark Datasets To demonstrate the applicability of our methods, we also performed experiments using eight real-world imbal- anced benchmark datasets from UCI Machine Learning Repository [38]: Radar data (ionosphere), Breast Cancer Wisconsin (breast-w), Haberman’s Survival (haberman), Blood Transfusion Service Center (blood), Wisconsin Prognostic Breast Cancer (breast-p), Glass Identification (glass), Landsat Satellite (satimage), and Yeast (yeast) with different class imbalance ratio as shown in Table 3. For highly imbalanced problems, the classes “head- lamps”, “damp grey soil”, and “ME2” of glass, satimage, and yeast datasets, respectively, were converted into mi- nority class and the remaining classes of each dataset became majority class. Except ionosphere, glass, and satimage, these datasets contain biomedical data. The experiments were executed under the settings al- most the same as above. The values for the performance criteria, sensitivity, specificity, and G-mean were calcu- lated by averaging 20 independent runs of 10-fold cross- validation and summarized in Ta bl e 4 . The results also suggested that our method achieved better G-mean than the control method, SMOTE, safe-level-SMOTE, and borderline-SMOTE methods. Furthermore, the assess- ment by two-sample t-test showed that SMOTE, safe- level-SMOTE, and borderline-SMOTE significantly outperforms the control and our methods remarkably outperform the control, SMOTE, safe-level-SMOTE, and borderline-SMOTE with p-values smaller than 0.05 in most cases (for more details, see Table 5). In addition, we calculated the correlation between the proportion of negative samples in the dataset (i.e. degree of imbalance) and improvement by the methods each with different classifiers as shown in Ta ble 6. In case of SMOTE, the improvement from no-oversampling was calculated. However, in case of other methods, im- provement from SMOTE was calculated. The results suggested that improvements by SMOTE, incremental- SMOTE2, and incremental-SMOTE3 have positive cor- relation to the degree of imbalance. In contrast, other methods have opposite characteristics. Among them, the improvements by SMOTE have shown relatively stronger correlation. These characteristics were observed more clearly when we used RF. Actually, only the combina- tions of RF with SMOTE, safelevel-SMOTE, and incre- mental-SMOTE2 showed the correlation values with p- values less than 0.05. 4. CONCLUSION In this paper, we addressed a problem in human miRNA gene recognition, and showed that it requires a better Copyright © 2013 SciRes. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 243 Table 4. The comparison of Sensitivity (SE), Specificity (SP) and G-mean expressed in percent. SVM k-NN RF Dataset Method SE SP G-meanSE SP G-mean SE SP G-mean No over-sampling 98.44 94.13 96.26 95.71 97.10 96.40 96.35 97.02 96.68 SMOTE 98.71 95.12 96.90 98.32 96.41 97.36 97.14 96.56 96.85 Safelevel-SMOTE 98.86 95.49 97.16 99.25 95.38 97.30 96.99 96.56 96.78 Borderline-SMOTE1 99.07 94.00 96.50 98.40 95.82 97.10 96.78 96.66 96.72 Borderline-SMOTE2 99.44 94.07 96.72 98.98 95.26 97.10 97.03 96.47 96.75 increSMOTE1 99.44 95.48 97.44 99.25 96.05 97.64 97.43 96.56 96.99 increSMOTE2 99.48 95.43 97.43 99.38 95.80 97.57 97.55 96.43 96.99 Breast-w increSMOTE3 99.54 95.41 97.46 99.23 95.90 97.55 97.51 96.48 97.00 No over-sampling 18.77 92.87 41.71 28.09 87.87 49.65 23.52 91.13 46.26 SMOTE 53.21 65.49 58.99 55.43 65.38 60.15 41.48 77.04 56.49 Safelevel-SMOTE 66.79 56.18 61.22 66.05 54.36 59.89 44.57 75.71 58.06 Borderline-SMOTE1 66.98 55.18 60.76 61.67 56.31 58.91 40.19 75.16 54.93 Borderline-SMOTE2 69.07 54.67 61.41 69.14 49.73 58.61 41.17 74.13 55.20 increSMOTE1 68.40 57.47 62.67 62.78 60.67 61.69 45.80 77.98 59.73 increSMOTE2 68.02 57.36 62.44 63.83 59.96 61.81 50.37 76.00 61.84 Haberman increSMOTE3 66.48 57.78 61.95 62.53 60.76 61.61 51.73 70.67 60.43 No over-sampling 30.65 94.21 53.71 27.56 91.03 50.07 29.24 90.04 51.27 SMOTE 74.04 60.35 66.84 51.88 74.70 62.24 50.28 72.47 60.35 Safelevel-SMOTE 65.76 68.61 67.08 57.67 68.39 62.79 54.07 71.11 62.00 Borderline-SMOTE1 61.91 71.97 66.65 55.70 70.23 62.53 45.79 74.71 58.47 Borderline-SMOTE2 50.11 84.89 65.22 63.96 62.87 63.39 55.31 64.75 59.81 increSMOTE1 73.85 61.98 67.65 70.39 57.96 63.87 55.70 68.05 61.55 increSMOTE2 73.17 62.71 67.74 58.03 69.37 63.43 63.96 63.42 63.68 Blood increSMOTE3 73.43 62.54 67.76 61.49 65.69 63.54 58.37 66.00 62.06 No over-sampling 10.11 99.47 31.36 20.85 87.88 42.69 19.68 98.54 43.87 SMOTE 53.30 73.81 62.68 59.47 57.58 58.45 43.30 83.68 60.16 Safelevel-SMOTE 52.98 74.93 62.98 61.17 56.62 58.80 41.91 84.54 59.45 Borderline-SMOTE1 48.40 79.74 62.09 63.94 54.07 58.75 40.11 83.64 57.87 Borderline-SMOTE2 50.64 73.64 61.00 68.40 50.63 58.81 50.43 70.13 59.38 increSMOTE1 57.98 71.49 64.35 68.30 53.84 60.59 46.70 82.12 61.86 increSMOTE2 59.89 69.27 64.39 68.72 53.44 60.58 48.72 81.66 63.05 Breast-p increSMOTE3 56.17 73.41 64.19 66.91 55.63 60.95 47.13 81.59 62.00 Copyright © 2013 SciRes. OPEN ACCESS 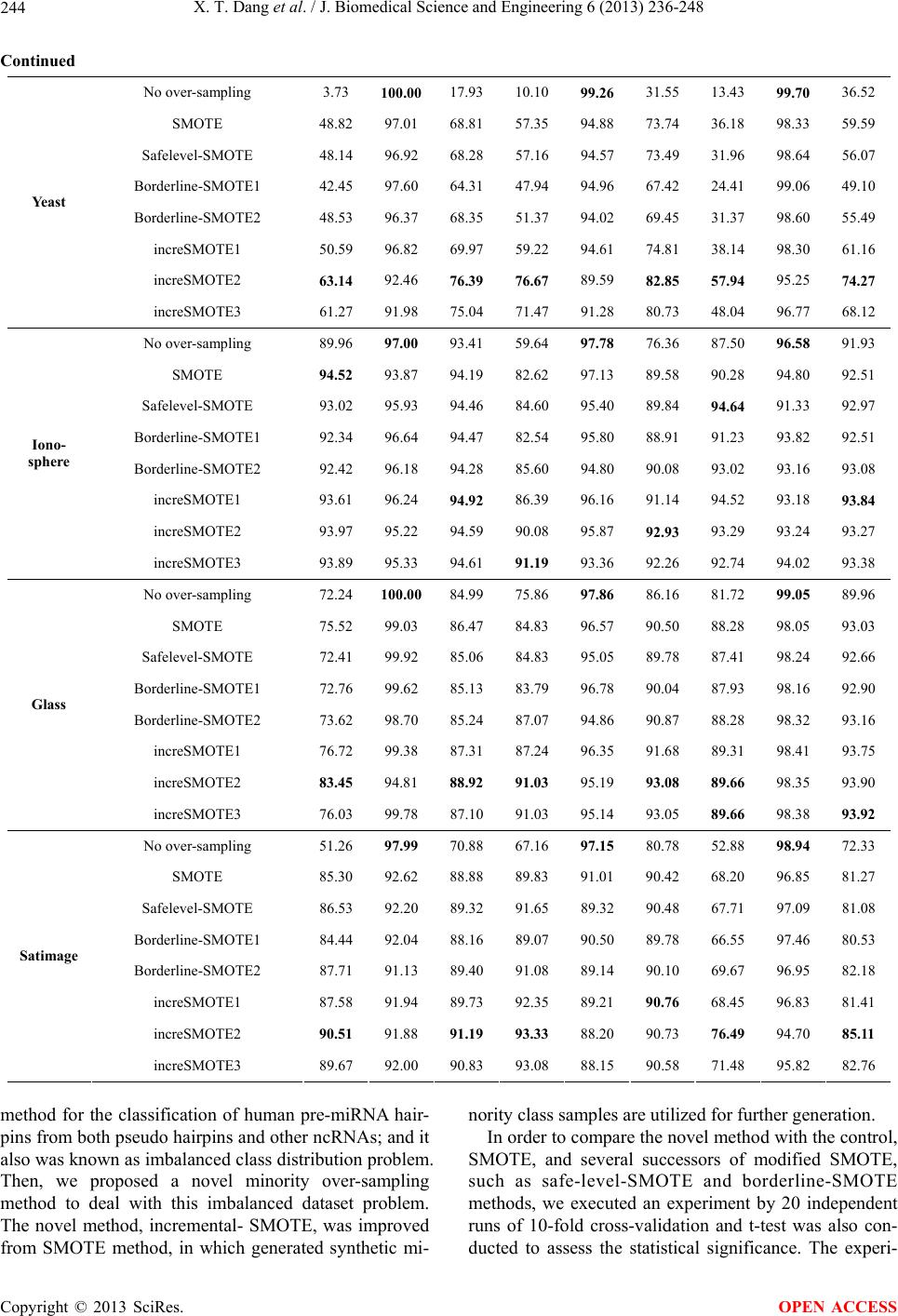 X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 244 Continued No over-sampling 3.73 100.00 17.93 10.10 99.26 31.55 13.43 99.70 36.52 SMOTE 48.82 97.01 68.81 57.35 94.88 73.74 36.18 98.33 59.59 Safelevel-SMOTE 48.14 96.92 68.28 57.16 94.57 73.49 31.96 98.64 56.07 Borderline-SMOTE1 42.45 97.60 64.31 47.94 94.96 67.42 24.41 99.06 49.10 Borderline-SMOTE2 48.53 96.37 68.35 51.37 94.02 69.45 31.37 98.60 55.49 increSMOTE1 50.59 96.82 69.97 59.22 94.61 74.81 38.14 98.30 61.16 increSMOTE2 63.14 92.46 76.39 76.67 89.59 82.85 57.94 95.25 74.27 Yeast increSMOTE3 61.27 91.98 75.04 71.47 91.28 80.73 48.04 96.77 68.12 No over-sampling 89.96 97.00 93.41 59.64 97.78 76.36 87.50 96.58 91.93 SMOTE 94.52 93.87 94.19 82.62 97.13 89.58 90.28 94.80 92.51 Safelevel-SMOTE 93.02 95.93 94.46 84.60 95.40 89.84 94.64 91.33 92.97 Borderline-SMOTE1 92.34 96.64 94.47 82.54 95.80 88.91 91.23 93.82 92.51 Borderline-SMOTE2 92.42 96.18 94.28 85.60 94.80 90.08 93.02 93.16 93.08 increSMOTE1 93.61 96.24 94.92 86.39 96.16 91.14 94.52 93.18 93.84 increSMOTE2 93.97 95.22 94.59 90.08 95.87 92.93 93.29 93.24 93.27 Iono- sphere increSMOTE3 93.89 95.33 94.61 91.19 93.36 92.26 92.74 94.02 93.38 No over-sampling 72.24 100.00 84.99 75.86 97.86 86.16 81.72 99.05 89.96 SMOTE 75.52 99.03 86.47 84.83 96.57 90.50 88.28 98.05 93.03 Safelevel-SMOTE 72.41 99.92 85.06 84.83 95.05 89.78 87.41 98.24 92.66 Borderline-SMOTE1 72.76 99.62 85.13 83.79 96.78 90.04 87.93 98.16 92.90 Borderline-SMOTE2 73.62 98.70 85.24 87.07 94.86 90.87 88.28 98.32 93.16 increSMOTE1 76.72 99.38 87.31 87.24 96.35 91.68 89.31 98.41 93.75 increSMOTE2 83.45 94.81 88.92 91.03 95.19 93.08 89.66 98.35 93.90 Glass increSMOTE3 76.03 99.78 87.10 91.03 95.14 93.05 89.66 98.38 93.92 No over-sampling 51.26 97.99 70.88 67.16 97.15 80.78 52.88 98.94 72.33 SMOTE 85.30 92.62 88.88 89.83 91.01 90.42 68.20 96.85 81.27 Safelevel-SMOTE 86.53 92.20 89.32 91.65 89.32 90.48 67.71 97.09 81.08 Borderline-SMOTE1 84.44 92.04 88.16 89.07 90.50 89.78 66.55 97.46 80.53 Borderline-SMOTE2 87.71 91.13 89.40 91.08 89.14 90.10 69.67 96.95 82.18 increSMOTE1 87.58 91.94 89.73 92.35 89.21 90.76 68.45 96.83 81.41 increSMOTE2 90.51 91.88 91.19 93.33 88.20 90.73 76.49 94.70 85.11 Satimage increSMOTE3 89.67 92.00 90.83 93.08 88.15 90.58 71.48 95.82 82.76 method for the classification of human pre-miRNA hair- pins from both pseudo hairpins and other ncRNAs; and it also was known as imbalanced class distribution problem. Then, we proposed a novel minority over-sampling method to deal with this imbalanced dataset problem. The novel method, incremental- SMOTE, was improved from SMOTE method, in which generated synthetic mi- nority class samples are utilized for further generation. In order to compare the novel method with the control, SMOTE, and several successors of modified SMOTE, such as safe-level-SMOTE and borderline-SMOTE methods, we executed an experiment by 20 independent runs of 10-fold cross-validation and t-test was also con- ducted to assess the statistical significance. The experi- Copyright © 2013 SciRes. OPEN ACCESS 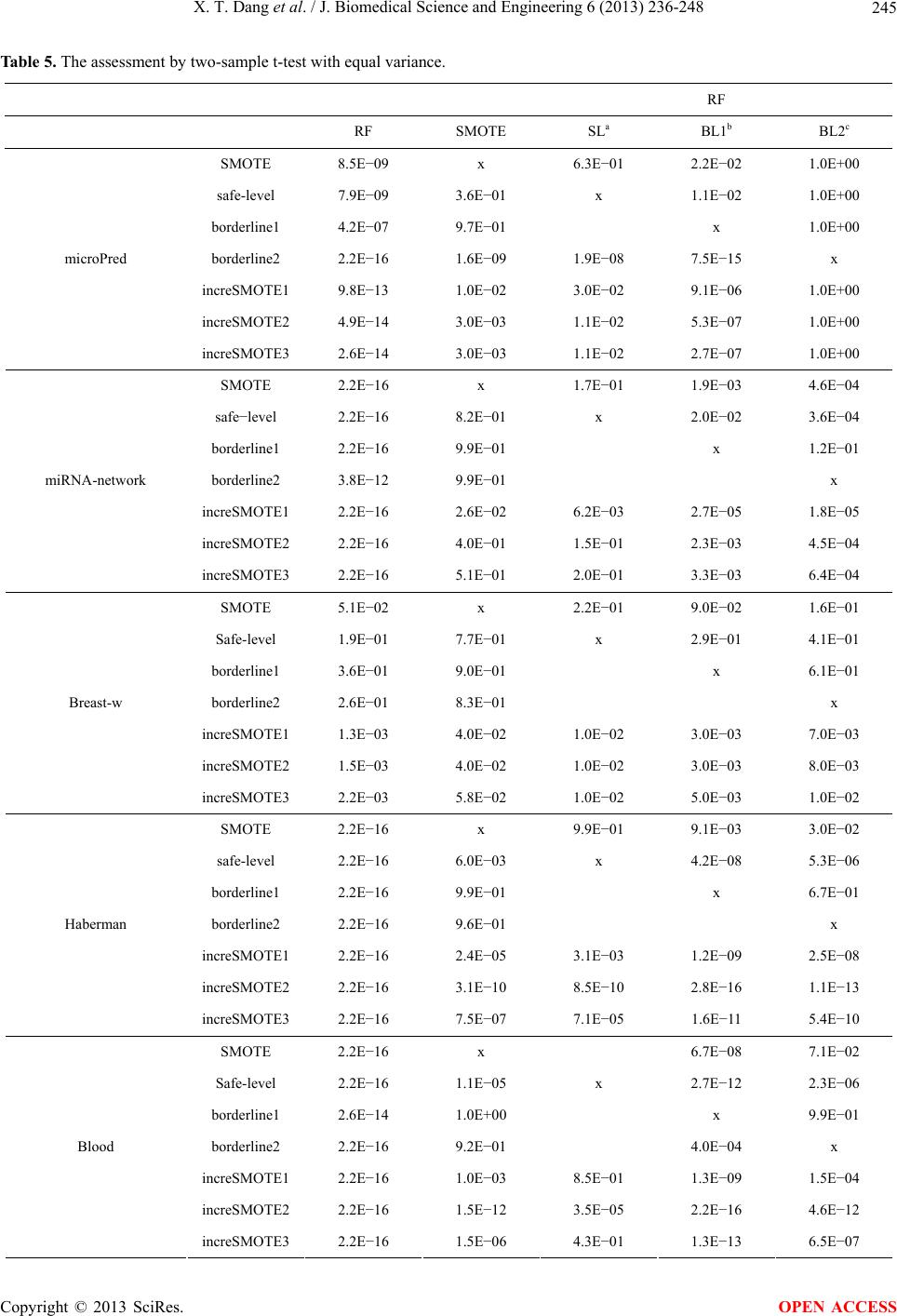 X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 245 Table 5. The assessment by two-sample t-test with equal variance. RF RF SMOTE SLa BL1b BL2c SMOTE 8.5E−09 x 6.3E−01 2.2E−02 1.0E+00 safe-level 7.9E−09 3.6E−01 x 1.1E−02 1.0E+00 borderline1 4.2E−07 9.7E−01 x 1.0E+00 borderline2 2.2E−16 1.6E−09 1.9E−08 7.5E−15 x increSMOTE1 9.8E−13 1.0E−02 3.0E−02 9.1E−06 1.0E+00 increSMOTE2 4.9E−14 3.0E−03 1.1E−02 5.3E−07 1.0E+00 microPred increSMOTE3 2.6E−14 3.0E−03 1.1E−02 2.7E−07 1.0E+00 SMOTE 2.2E−16 x 1.7E−01 1.9E−03 4.6E−04 safe−level 2.2E−16 8.2E−01 x 2.0E−02 3.6E−04 borderline1 2.2E−16 9.9E−01 x 1.2E−01 borderline2 3.8E−12 9.9E−01 x increSMOTE1 2.2E−16 2.6E−02 6.2E−03 2.7E−05 1.8E−05 increSMOTE2 2.2E−16 4.0E−01 1.5E−01 2.3E−03 4.5E−04 miRNA-network increSMOTE3 2.2E−16 5.1E−01 2.0E−01 3.3E−03 6.4E−04 SMOTE 5.1E−02 x 2.2E−01 9.0E−02 1.6E−01 Safe-level 1.9E−01 7.7E−01 x 2.9E−01 4.1E−01 borderline1 3.6E−01 9.0E−01 x 6.1E−01 borderline2 2.6E−01 8.3E−01 x increSMOTE1 1.3E−03 4.0E−02 1.0E−02 3.0E−03 7.0E−03 increSMOTE2 1.5E−03 4.0E−02 1.0E−02 3.0E−03 8.0E−03 Breast-w increSMOTE3 2.2E−03 5.8E−02 1.0E−02 5.0E−03 1.0E−02 SMOTE 2.2E−16 x 9.9E−01 9.1E−03 3.0E−02 safe-level 2.2E−16 6.0E−03 x 4.2E−08 5.3E−06 borderline1 2.2E−16 9.9E−01 x 6.7E−01 borderline2 2.2E−16 9.6E−01 x increSMOTE1 2.2E−16 2.4E−05 3.1E−03 1.2E−09 2.5E−08 increSMOTE2 2.2E−16 3.1E−10 8.5E−10 2.8E−16 1.1E−13 Haberman increSMOTE3 2.2E−16 7.5E−07 7.1E−05 1.6E−11 5.4E−10 SMOTE 2.2E−16 x 6.7E−08 7.1E−02 Safe-level 2.2E−16 1.1E−05 x 2.7E−12 2.3E−06 borderline1 2.6E−14 1.0E+00 x 9.9E−01 borderline2 2.2E−16 9.2E−01 4.0E−04 x increSMOTE1 2.2E−16 1.0E−03 8.5E−01 1.3E−09 1.5E−04 increSMOTE2 2.2E−16 1.5E−12 3.5E−05 2.2E−16 4.6E−12 Blood increSMOTE3 2.2E−16 1.5E−06 4.3E−01 1.3E−13 6.5E−07 Copyright © 2013 SciRes. OPEN ACCESS 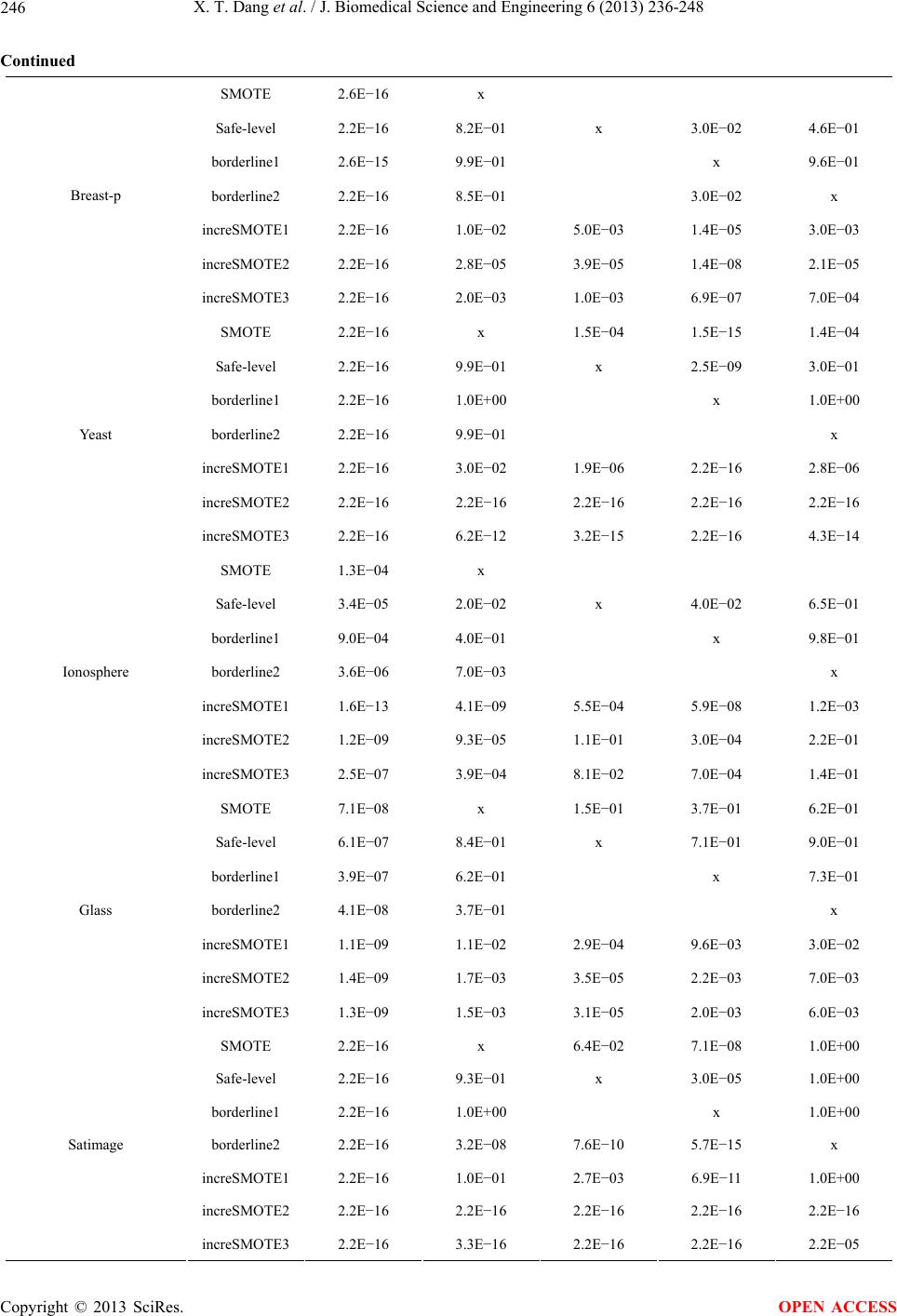 X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 246 Continued SMOTE 2.6E−16 x Safe-level 2.2E−16 8.2E−01 x 3.0E−02 4.6E−01 borderline1 2.6E−15 9.9E−01 x 9.6E−01 borderline2 2.2E−16 8.5E−01 3.0E−02 x increSMOTE1 2.2E−16 1.0E−02 5.0E−03 1.4E−05 3.0E−03 increSMOTE2 2.2E−16 2.8E−05 3.9E−05 1.4E−08 2.1E−05 Breast-p increSMOTE3 2.2E−16 2.0E−03 1.0E−03 6.9E−07 7.0E−04 SMOTE 2.2E−16 x 1.5E−04 1.5E−15 1.4E−04 Safe-level 2.2E−16 9.9E−01 x 2.5E−09 3.0E−01 borderline1 2.2E−16 1.0E+00 x 1.0E+00 borderline2 2.2E−16 9.9E−01 x increSMOTE1 2.2E−16 3.0E−02 1.9E−06 2.2E−16 2.8E−06 increSMOTE2 2.2E−16 2.2E−16 2.2E−16 2.2E−16 2.2E−16 Yeast increSMOTE3 2.2E−16 6.2E−12 3.2E−15 2.2E−16 4.3E−14 SMOTE 1.3E−04 x Safe-level 3.4E−05 2.0E−02 x 4.0E−02 6.5E−01 borderline1 9.0E−04 4.0E−01 x 9.8E−01 borderline2 3.6E−06 7.0E−03 x increSMOTE1 1.6E−13 4.1E−09 5.5E−04 5.9E−08 1.2E−03 increSMOTE2 1.2E−09 9.3E−05 1.1E−01 3.0E−04 2.2E−01 Ionosphere increSMOTE3 2.5E−07 3.9E−04 8.1E−02 7.0E−04 1.4E−01 SMOTE 7.1E−08 x 1.5E−01 3.7E−01 6.2E−01 Safe-level 6.1E−07 8.4E−01 x 7.1E−01 9.0E−01 borderline1 3.9E−07 6.2E−01 x 7.3E−01 borderline2 4.1E−08 3.7E−01 x increSMOTE1 1.1E−09 1.1E−02 2.9E−04 9.6E−03 3.0E−02 increSMOTE2 1.4E−09 1.7E−03 3.5E−05 2.2E−03 7.0E−03 Glass increSMOTE3 1.3E−09 1.5E−03 3.1E−05 2.0E−03 6.0E−03 SMOTE 2.2E−16 x 6.4E−02 7.1E−08 1.0E+00 Safe-level 2.2E−16 9.3E−01 x 3.0E−05 1.0E+00 borderline1 2.2E−16 1.0E+00 x 1.0E+00 borderline2 2.2E−16 3.2E−08 7.6E−10 5.7E−15 x increSMOTE1 2.2E−16 1.0E−01 2.7E−03 6.9E−11 1.0E+00 increSMOTE2 2.2E−16 2.2E−16 2.2E−16 2.2E−16 2.2E−16 Satimage increSMOTE3 2.2E−16 3.3E−16 2.2E−16 2.2E−16 2.2E−05 Copyright © 2013 SciRes. OPEN ACCESS  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 Copyright © 2013 SciRes. 247 Ta bl e 6. The correlation between degree of imbalance and im- provement by the methods each with different classifiers. SVM k-NN RF SMOTE 0.70 0.42 0.94 Safelevel-SMOTE −0.59 −0.31 −0.72 Borderline-SMOTE1 −0.54 −0.49 −0.65 Borderline-SMOTE2 0.01 −0.39 −0.35 increSMOTE1 −0.66 −0.50 −0.48 increSMOTE2 0.66 0.23 0.83 increSMOTE3 0.36 0.21 0.59 OPEN ACCESS mental results showed that our method achieved better G-mean and Sensitivity than both of the control, SMOTE, safe-level-SMOTE, and borderline-SMOTE methods with the p-value less than 0.05 in most cases. These re- sults suggest that our method outperforms SMOTE and several successors of modified SMOTE in various bio- medical classification problems, including human miRNA gene prediction. Although incremental-SMOTE achieved better per- formances in various biomedical classification problems, the advantages and disadvantages of three variations of incremental-SMOTE are still unclear in real applications. Moreover, there are still several topics left to be consid- ered further such as: the combination of our novel me- thod with feature selection methods, application of other novel under-sampling methods, extraction of a new and appropriate set of features from pre-miRNA hairpins dataset, and so on. In future work, we will find the solu- tion to these problems. 5. ACKNOWLEDGEMENTS The authors wish to thank Batuwita et al. and Xiao et al. for providing us with microPred and miRNA network-level datasets, respectively. REFERENCES [1] Kim, V.N. and Nam, J.-W. (2006) Genomics of mi- croRNA. Trends in Genetic, 22, 165-173. doi:10.1016/j.tig.2006.01.003 [2] Lee, R.C., Feinbaum, R.L. and Ambros, V. (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843- 854. doi:10.1016/0092-8674(93)90529-Y [3] Harfe, B.D., Mcmanus, M.T., Mansfield, J.H., Hornstein, E. and Tabin, C.J. (2005) The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the ver- tebrate limb. Proceedings of the National Academy of Sciences, 102, 10898-10903. doi:10.1073/pnas.0504834102 [4] Wilfred, B.R., Wang, W. and Nelson, P.T. (2007) Ener- gizing miRNA research: A review of the role of miRNAs in lipid metabolism, with a prediction that miR-103/107 regulates human metabolic pathways. Molecular Genetics and Metabolism, 91:209-217. doi:10.1016/j.ymgme.2007.03.011 [5] Lodish, H.F., Chen, C. and Bartel, D.P. (2004) Micro- RNAs modulate hematopoietic lineage differentiation. Science, 303, 83-86. doi:10.1126/science.1091903 [6] Lim, L.P., Lau, N.C., Garrett-engele, P. and Grimson, A. (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature, 292, 288-292. [7] Kozomara, A. and Griffiths-Jones, S. (2011) miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Rese arch, 39, D152-D157. doi:10.1093/nar/gkq1027 [8] Hertel, J. and Stadler, P.F. (2006) Hairpins in a Haystack: Recognizing microRNA precursors in comparative ge- nomics data. Bioinformatics, 22, e197-e202. doi:10.1093/bioinformatics/btl257 [9] Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B. and Bartel, D.P. (2003) Vertebrate microRNA genes. Science, 299, 1540. doi:10.1126/science.1080372 [10] Lai, E.C., Tomancak, P., Williams, R.W. and Rubin, G.M. (2003) Computational identification of Drosophila mi- croRNA genes. Genome Biology, 4, R42. doi:10.1186/gb-2003-4-7-r42 [11] Jones-Rhoades, M.W. and Bartel, D.P. (2004) Computa- tional identification of plant microRNAs and their targets, including a stress-induced miRNA. Molecular Cell, 14, 787-799. doi:10.1016/j.molcel.2004.05.027 [12] Bonnet, E., Wuyts, J., Rouzé, P. and Van de Peer, Y. (2004) Detection of 91 potential conserved plant mi- croRNAs in Arabidopsis thaliana and Oryza sativa identi- fies important target genes. Proceedings of the National Academy of Sciences, 101, 11511-11516. doi:10.1073/pnas.0404025101 [13] Ng, K.L.S. and Mishra, S.K. (2007) De novo SVM clas- sification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bio- informatics, 23, 1321-1330. doi:10.1093/bioinformatics/btm026 [14] Berezikov, E., Guryev, V., Van de Belt, J., Wienholds, E., Plasterk, R.H.A. and Cuppen, E. (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell, 120, 21-24. doi:10.1016/j.cell.2004.12.031 [15] Sewer, A., Paul, N., Landgraf, P., Aravin, A., Pfeffer, S., Brownstein, M.J., Tuschl, T., Van Nimwegen, E. and Zavolan, M. (2005) Identification of clustered micro- RNAs using an ab initio prediction method. BMC Bioin- formatics, 6, 267. doi:10.1186/1471-2105-6-267 [16] Xue, C., Li, F., He, T., Liu, G-.P., Li, Y. and Zhang, X. (2005) Classification of real and pseudo microRNA pre- cursors using local structure-sequence features and sup- port vector machine. BMC Bioinformatics, 6, 310. doi:10.1186/1471-2105-6-310 [17] Clote, P., Ferré, F., Kranakis, E. and Krizanc, D. (2005)  X. T. Dang et al. / J. Biomedical Science and Engineering 6 (2013) 236-248 248 Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA, 11, 578- 591. doi:10.1261/rna.7220505 [18] Jiang, P., Wu, H., Wang, W., Ma, W., Sun, X. and Lu, Z. (2007) MiPred: Classification of real and pseudo micro- RNA precursors using random forest prediction model with combined features. Nucleic Acids Re search, 35, 339- 344. doi:10.1093/nar/gkm368 [19] Tang, X., Xiao, J., Li, Y., Wen, Z., Fang, Z. and Li, M. (2012) Systematic analysis revealed better performance of random forest algorithm coupled with complex net- work features in predicting microRNA precursors. Che- mometrics and Intelligent Laboratory Systems, 118, 317- 323. doi:10.1016/j.chemolab.2012.05.001 [20] Wang, Y., Chen, X., Jiang, W., Li, L., Li, W., Yang, L., Liao, M., Lian, B., Lv, Y., Wang, S., Wang, S. and Li, X. (2011) Predicting human microRNA precursors based on an optimized feature subset generated by GA-SVM. Ge- nomics, 98, 73-78. doi:10.1016/j.ygeno.2011.04.011 [21] Zhang, Y., Yang, Y., Zhang, H., Jiang, X., Xu, B., Xue, Y., Cao, Y., Zhai, Q., Zhai, Y., Xu, M., Cooke, H.J. and Shi, Q. (2011) Prediction of novel pre-microRNAs with high accuracy through boosting and SVM. Bioinformatics, 27, 1436-1437. doi:10.1093/bioinformatics/btr148 [22] Kadri, S., Hinman, V. and Benos, P.V. (2009) HHMMiR: Efficient de novo prediction of microRNAs using hierar- chical hidden Markov models. BMC Bioinformatics, 10, S35. doi:10.1186/1471-2105-10-S1-S35 [23] Batuwita, R. and Palade, V. (2009) microPred: Effective classification of pre-miRNAs for human miRNA gene prediction. Bioinformatics, 25, 989-995. doi:10.1093/bioinformatics/btp107 [24] Xiao, J., Tang, X., Li, Y., Fang, Z., Ma, D., He, Y. and Li, M. (2011) Identification of microRNA precursors based on random forest with network-level representation me- thod of stem-loop structure. BMC Bioinformatics, 12, 165. doi:10.1186/1471-2105-12-165 [25] Bunkhumpornpat, C., Sinapiromsaran, K. and Lursinsap, C. (2009) Safe-Level-SMOTE: Safe-level-synthetic mi- nority over-sampling technique. Lecture Notes in Com- puter Science, 5476, 475-482. doi:10.1007/978-3-642-01307-2_43 [26] Han, H., Wang, W. and Mao, B. (2005) Borderline- SMOTE: A new over-sampling method in imbalanced data sets learning. Lecture Notes in Computer Science, 3644, 878-887. doi:10.1007/11538059_91 [27] Chawla, N.V., Bowyer, K.W. and Hall, L.O. (2002) SMOTE : Synthetic minority over-sampling technique. Journal of Artificial Intelligence Research, 16, 321-357. [28] Burges, C.J.C. (1998) A tutorial on support vector ma- chines for pattern recognition. Data Mining and Knowl- edge Discovery, 2, 121-167. doi:10.1023/A:1009715923555 [29] Vapnik, V.N. (1999) An overview of statistical learning theory. IEEE Transactions on Neural networks, 10, 988- 999. doi:10.1109/72.788640 [30] Karatzoglou, A. and Smola, A. (2004) kernlab—An S4 package for kernel methods in R. Journal of Statistical Software, 11, 1-20. [31] Venables, W.N. and Ripley, B.D. (2002) Modern applied statistics with S. 4th Edition. Springer, New York. doi:10.1007/978-0-387-21706-2 [32] Liaw, A. and Wiener, M. (2002) Classification and re- gression by random forest. R News, 2, 18-22. [33] Akbani, R., Kwek, S. and Japkowicz, N. (2004) Applying support vector machines to imbalanced datasets. Lecture Notes in Computer Science, 3201, 39-50. doi:10.1007/978-3-540-30115-8_7 [34] Anand, A., Pugalenthi, G., Fogel, G.B. and Suganthan, P.N. (2010) An approach for classification of highly im- balanced data using weighting and undersampling. Amino Acids, 39, 1385-1391. doi:10.1007/s00726-010-0595-2 [35] Kubat, M. and Matwin, S. (8-12 July 1997) Addressing the curse of imbalanced training sets: One-sided selection. Proceedings of the 14th International Conference in Ma- chine Learning, Nashville, 179-186. [36] Han, K. (2011) Effective sample selection for classifica- tion of pre-miRNAs. Genetics and Molecular Research, 10, 506-518. doi:10.4238/vol10-1gmr1054 [37] Xuan, P., Guo, M., Liu, X., Huang, Y., Li, W. and Huang, Y. (2011) PlantMiRNAPred: Efficient classification of real and pseudo plant pre-miRNAs. Bioinformatics, 27, 1368-1376. doi:10.1093/bioinformatics/btr153 [38] Frank, A. and Asuncion, A. (2010) UCI machine learning repository. University of California, Irvine. Copyright © 2013 SciRes. OPEN ACCESS
|