Food and Nutrition Sciences
Vol. 4 No. 9A (2013) , Article ID: 37116 , 10 pages DOI:10.4236/fns.2013.49A1028
Regulatory Role of Free Fatty Acids (FFAs)—Palmitoylation and Myristoylation
![]()
Center for Food Safety and Applied Nutrition, Division of Toxicology, Office of Applied Research and Safety Assessment, Food and Drug Administration, Laurel, USA.
Email: chung.kim@fda.hhs.gov
Copyright © 2013 Chung S. Kim, Ivan A. Ross. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 16th, 2013; revised June 16th, 2013; accepted June 24th, 2013
Keywords: Free Fatty Acids; Palmitoylation; Myristoylation; Microbial and Chemical Toxicity; Innate Immune Response
ABSTRACT
Multicellular organisms use chemical messengers to transmit signals among organelles and to other cells. Relatively small hydrophobic molecules such as lipids are excellent candidates for this signaling purpose. In most proteins, palmitic acid and other saturated and some unsaturated fatty acids are esterified to the free thiol of cysteines and to the N-amide terminal. This palmitoylation process enhances the surface hydrophobicity and membrane affinity of protein substrates and plays important roles in modulating proteins’ trafficking, stability, and sorting etc. Protein palmitoylation has been involved in numerous cellular processes, including signaling, apoptosis, and neuronal transmission. The palmitoylation process is involved in multiple diseases such as Huntington’s disease, various cardiovascular and T-cell mediated immune disorders, as well as cancer. Protein palmitoylation through the thioester (S-acylation) is unique in that it is the only reversible lipid modification. Our study on lipopolysaccharide (LPS) and deoxynivalenol (DON) treatment to rats provides some insights to the complex role of protein palmitoylation in chemical and microbial toxicity. In contrast, myrisoylated proteins contain the 14-carbon fatty acid myristate attached via amide linkage to the N-terminal glycine residue of protein, and occur cotranslationally. The bacterial outer membrane enzyme lipid A palmitoyltransferase (PagP) confers resistance to host immune defenses by transferring a palmitate chain from a phospholipid to the lipid A component of LPS. PagP is sensitive to cationic antimicrobial peptides (CAMP) which are included among the products of the Toll-like receptor 4 (TLR4) signal transduction pathway. This modification of lipid A with a palmitate appears to both and protects the pathogenic bacteria from host immune defenses and attenuates the activation of those same defenses through the TLR4 signal transduction pathway.
1. Introduction
Saturated FFAs play important roles for various biological functions such as the production of hormones, cellular membrane signaling, and the stabilization processes in the body. These are the 18-carbon stearic acid, the 16-carbon palmitic acid, and the 14-carbon myristic acid. When these important saturated fatty acids are not readily available, certain growth factors in the cells and organs will not be properly aligned. This is because the various receptors, such as G-protein receptors, need to be coupled with lipids in order to provide localization of function [1,2]. A biochemical process called palmitoylation, in which the body uses palmitic acid in stabilization processes, is very important to the body by regulating G protein-coupled receptor signaling [3,4]. Myristic acid, also called tetradecanoic acid, is a saturated fatty acid. It is commonly added co-transitionally to the nitrogen terminus of glycine in receptor-associated kinases to confer the membrane localization of the enzyme. Myristic acid has a sufficiently high hydrophobicity to become incurporated into the fatty acyl core of the phospholipid bilayer of the plasma membrane. In this way, myristic acid acts as a lipid anchor in biomembranes [5]. Myristic acid is known to be a very important fatty acid which the body uses to stabilize many different proteins, including proteins that are used in the immune system and also those that fight tumors [6]. This function is called myristoylation; it occurs when myristic acid is attached to the protein in a specific position where it functions usefully [7-10]. For example, the body has the ability to suppress production of tumors from lung cancer cells if a certain genetically determined suppressor gene is available. This gene codes for Fus-1 which is a protein that has been modified with covalent addition of the saturated fatty acid myristic acid [6]. Myristoleic acid is an omega-5- fatty acid that is biosynthesized from myristic acid by the enzyme delta-9-desaturase. This uncommon (n-9) fatty acid has been described in the retina; acylating an NH2 terminus of a protein related to signal transduction in photoreceptors [11]. Furthermore, the presence of this fatty acid was shown to be of diagnostic value in patients with defects of long-chain fatty acid oxidation [12], and is known to be cytotoxic to tumor cells. It induces apoptosis and necrosis in human prostate cancer LNCaP cells [13].
Multicellular organisms use chemical messengers to transmit signals among organelles and to other cells. Relatively small hydrophobic molecules such as lipids are excellent candidates for this signaling purpose. The two most common modifications, myristoylation and palmitoylation, differ with respect to the type and chemical nature of fatty acid attachment to the polypeptide backbone.
1.1. Myristoylation
After the initiating methionine is removed, the 14-carbon fatty acid myristate is attached via amide linkage to the N-terminal glycine residue. The reaction occurs cotranslationally and is catalyzed by the soluble enzyme Nmyristoyl transferase (NMT). NMT exhibits strict specificity for an N-terminal glycine and mutation of this glycine to alanme abrogates myristoylation [14].
FFAs are known to be markers of cellular membrane degradation through lipid peroxidation and are substrates for the production of reactive oxygen species (ROS) [15]. Since the generation of ROS during the metabolism of arsenic is thought to be involved in arsenic toxicosis, understanding the deleterious effects caused by ROS that attack the vital molecules like DNA, has become important [16].
Ross et al. [17] have investigated the FFAs profile from the rats treated with sodium arsenite (NaAs O2), a single oral dose, in water, and in a lipid medium, to pregnant rats on gestational day (GD) 10, a time point at midorganogenesis. NaAsO2 was administered in deionized water (AsH2O) or in half and half dairy cream (AsHH) at a dose of 41 mg NaAsO2/kg body weight. Control animals were treated with either dairy cream (HH) or deionized water (H2O). The animals were sacrificed on GD 20.
The study reveals an elevation of FFAs in the maternal liver and brain, and the fetal brain. In the fetal brain, myristic and stearic acids concentrations were higher in animals treated with AsHH vs. AsH2O (Figure 1). This could be due to the aggregation of NaAs with HH which could delay its metabolism and excretion as compared to NaAs in H2O that can be readily cleared by the kidneys. The fact that NaAs crosses the blood-brain barrier as well as the placental barrier freely [18-20], the NaAs-HH complex could be retained longer in the brain as compared to NaAs and AsH2O which are washed out of the brain easily by the efflux system of the brain [21]. This prolonged presence of AsHH in the cells probably induces the process of myristoylation and palmitoylation in the cellular signaling pathways. The increase of oleic and arachidonic acid in the liver of AsH2O vs. H2O group, indicates a direct effect of NaAs on these fatty acids. The depletion of myristoleic acid in the maternal brain, as indicated in Figure 2, could be due to the active participation of myristoylation against the toxic effect of NaAs in the maternal brain. Myristoylation is an irreversible process and normally precedes the process of palmitoylation [14]. The palmitoylation process is, in contrast, reversible and palmitic acid can be substituted by fatty acids
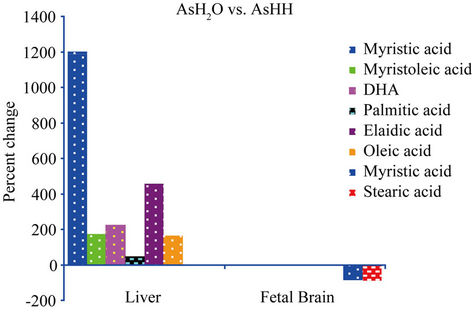
Figure 1. The formula used for the calculation of percent changes in Figure 1 is as follows: [{(AsH2O − AsHH)/AsHH} × 100].
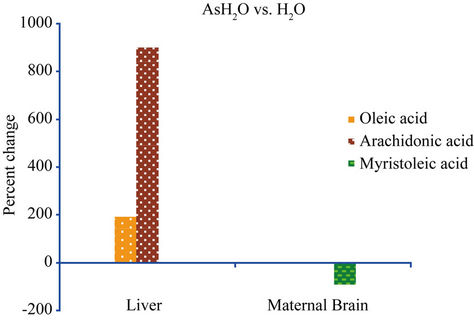
Figure 2. The formula used for the calculation of percent changes in Figure 2 is as follows: [{(AsH2O − H2O)/H2O} × 100].
such as stearic, oleic, and arachidonic acids for the palmitoylation process to regulate the cellular functions [22]. As shown in the fetal brain (Figure 3), the enhancement of myristic, stearic, and arachidonic acids in the AsHH group could be a protective effect due to the slow clearance of the AsHH from the fetal brain. This, in turn, could be explained by differences in the sensitivity of membrane functions for signal pathways during the developmental stages of the fetal brain, as compared to the maternal brain. Taken together, findings of different levels of myristic and myristoleic acids in the maternal and fetal brains and the other organs indicate that the antitumoric activity against NaAsO2 treatment is more pronounced in the brain than in the kidney and liver.
1.2. Palmitoylation
In contrast, palmitoylated proteins contain the 16-carbon fatty acid palmitate attached via throester linkage to one or more cysteme residues. Palmitoylation is a posttranslational reaction that appears to be mediated by a membrane-bound palmitoyl acyl transferase. Unlike myristoylation, whrch is generally a relatively stable modification, palmitoylatron can be reversed by the action of thioesterases.
In most proteins, palmitic acid and some other fatty acids are esterified to the free thiol of cysteines and to the N-amide terminal as shown in Figure 4. This palmitoylation process enhances the surface hydrophobicity and membrane affinity of protein substrates and play important roles in modulating proteins’ trafficking, stability and sorting. Since this linkage between the palmitate and protein is readily cleaved, cycles of palmitoylation and depalmitoylation occur in a regulated manner for many proteins [23].
Protein palmitoylation has been involved in numerous cellular processes, including signaling, apoptosis, and neuronal transmission [24]. The palmitoylation process is also involved in multiple diseases such as Huntington’s disease, various cardiovascular and T-cell mediated immune disorders, as well as cancer. Protein palmitoylation through the thioester (S-acylation) is unique in that it is the only reversible lipid modification.
• Palmitoylation is involved in the process of protein trafficking between organelles and in the segregation or clustering of proteins in membrane compartments [25-27].
• Palmitoylation increases the hydrophobicity of proteins to promote protein-membrane association [28, 29].
• Modification of proteins to control protein-protein interaction [30-32], lipid raft targeting [33] and intracellular trafficking [34,35].
• The palmitoylation process is involved in multiple diseases such as Huntington’s disease, various cardiovascular and T-cell mediated immune disorders, as well as cancer [36].
1.3. The Role of Palmitoylation in Microbial and Chemical Toxicity: Signal Pathways, Protein Binding and Trafficking
Lipopolysaccharide (LPS)
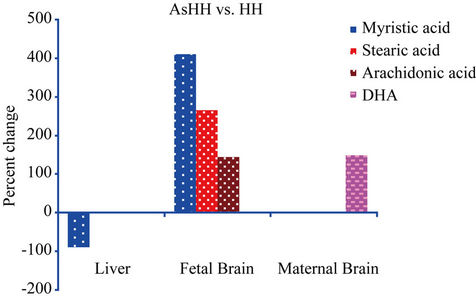
Figure 3. The formula used for the calculation of percent changes in Figure 3 is as follows: [{(AsHH − HH)/HH} × 100].
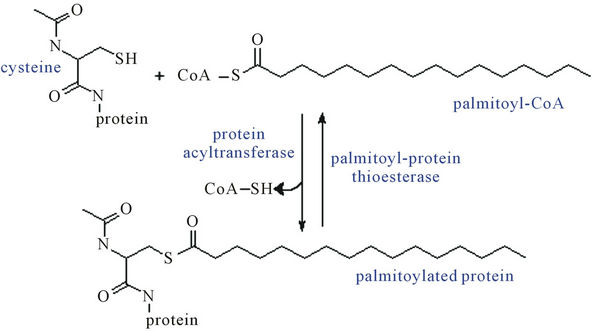
Figure 4. Protein S-palmitoylation—the thioester linkage of long-chain fatty acids to cysteine residues in proteins.
An essential virulence property of bacterial pathogens is the ability to sense the environment within the host tissues and to coordinate the expression of the virulence factors that promote the bacterial survival and replication strategies.
• The infected host also senses the presence of invading bacteria and responds by activation of the innate immune system. In Gram-negative bacteria, Lipid A is the region of LPS (Figure 5), also known as endotoxin, which is responsible for the immunostimulatory activity of LPS.
• The inflammatory response is necessary to eliminate most infections and, at the same time, responsible for some of the main pathophysiological symptoms associated with persistent infections. Bacterial pathogens can coexist with their hosts in part because they modify the structure of lipid A to attenuate the inflammatory response and evade immune recognition (Figure 6) [37].
• Palmitoylated lipid A can both protect pathogenic bacteria from host immune defenses and attenuate the activation of those same defenses through the Tolllike receptor 4 (TLR4) signal transduction pathway (Figure 7).
Deoxynivalenol (DON)
Several DON contamination episodes have caused the FDA to establish regulatory measures needed to control DON in foods [38].
• Exposure to sublethal levels of trichothecenes can stimulate or suppress immune parameters such as lymphocyte proliferation, host resistance, cell-mediated immunity, and humoral immune function in a variety of animal and cell culture models depending on dose, exposure frequency, and timing of exposure [39].
• LPS-induced nitric oxide (NO) production by RAW264 cells was dose-dependently inhibited by DON [40].
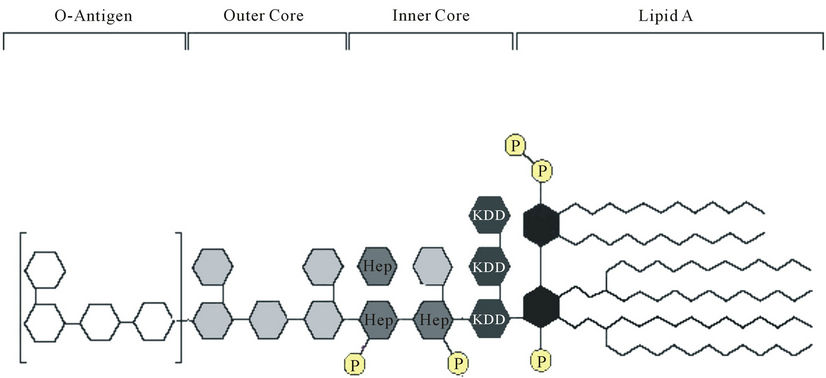
Figure 5. Lipopolysaccharide—the main component of the outer leaflet of the bacteria outer membrane is the immunodominant antigen of most Gram-negative pathogen.
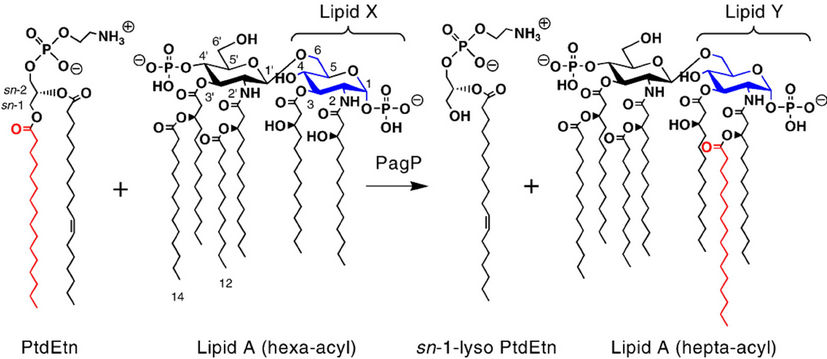
Figure 6. A palmitate chain from a phospholipid is incorporated into lipid A by an outer membrane enzyme PagP.
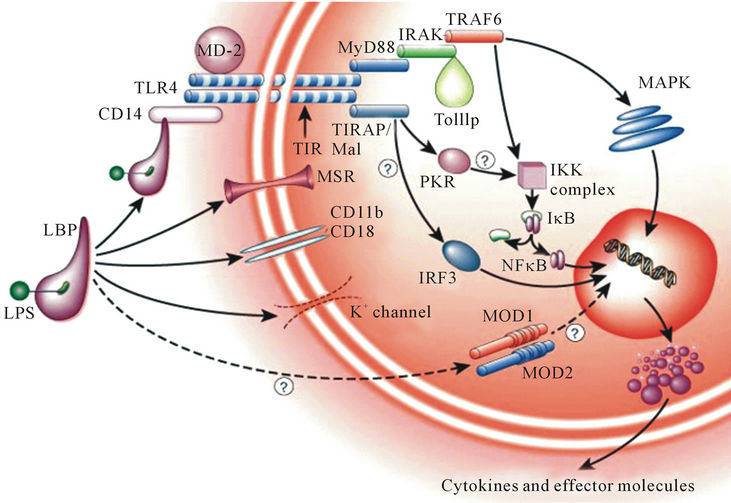
Figure 7. A group of proteins that comprise the TLR4 family of receptors detects invading pathogen and mount a rapid defensive response.
• DON has also suppressed LPS-induced inducible NO synthase (iNOS) promoter activity and the expression of iNOS protein in a similar concentration range to that of the inhibitory effect on NO production [41].
• DON induced IgA nephropathy in mice by upregulating IL-6 expression, which is suppressed by the consumption of some of the polyunsaturated fatty acids [42].
To examine the role of palmitoylation in animals treated with lipopolysaccharide and deoxynivalenol individually and concurrently at doses that alone would not cause overt toxicity, the following study has been carried out and presented at the Society of Toxicology Annual Meetings [43,44].
2. Methods
Male Harlan Hsd:Sprague-Dawley rats (virus and antibody-free), 9 weeks of age, were acclimated to their environment for approximately one week. The animals were housed individually in polycarbonate cages in a temperature controlled environment (24˚C - 26˚C), and a relative humidity of 40% - 70%. A light-dark cycle was maintained with lights on at 7:00 AM and off at 7:00 PM. After a period of acclimation, the rats were randomly assigned to the different treatment groups of eight animals each. The animals were fed ground Purina rodent chow 5002 (Purina Mills, Inc., Richmond, IN) and water from an in-house water system ad libitum throughout the study. The rats were divided into 4 groups. Group 1 was administered an IP dose of 10 mg DON/kg; group 2: 83 µg LPS/kg; group 3: 10 mg DON and 83 µg LPS/kg; group 4: 1 mL saline/kg. Animals were sacrificed at 3, 24, and 72 hr after dosing. The liver and brain were immediately harvested and frozen in liquid nitrogen. The specimens were then stored at −80˚C until the fatty acids were extracted. Extraction of free fatty acids and analysis by gas chromatography were described in Ross et al. [17].
3. Statistical Analysis
All response parameters were tested using the ShapiroWilk Test to determine whether the response parameter follows a normal distribution. This information was used to determine whether a parametric or non-parametric analysis was used for subsequent analysis. Time differences for each level of DON and LPS (that is, presence or absence) were tested by a one-way Analysis of Variance (ANOVA). If the ANOVA was statistically significant (p < 0.05), a protested LSD t-test was used for pairwise comparison of 3, 24 and 72 hours. For each time period and level of DON, a t-test was used to compare presence and absence of LPS. For each time period and level of LPS, a t-test was used to compare presence and absence of DON. Time differences for each level of DON and LPS were analyzed by the nonparametric Kruskal-Wallis test. If this test was significant (p < 0.05), the Mann-Whitney test was used for pair wise comparison of 3, 24 and 72 hours. For each time period and level of DON, the Mann-Whitney test was used to compare presence and absence of LPS. For each time period and level of LPS, the Mann-Whitney test was used to compare presence and absence of DON.
4. Results
The individual and concurrent administration of LPS and DON had no effect on the fatty acids of the brain and the glutathione levels of the brain and liver (data not shown). In the liver of animals treated with DON, there was a significant decrease in palmitic acid at 24 hours (Figure 8(b)) compared to 3 hr (Figure 8(a)) and an increase at 72 hr (Figure 8(c)) compared to 24 hr (Figure 8(b)) after dosing. This profile is an indication of palmitic acid utilization between 3 and 24 hours and its restoration by 72 hr, when the mild toxic effect was neutralized. In the liver of animals treated with LPS, elaidic acid was significantly decreased at 24 hr (Figure 8(e)) and at 72 hr Figure 8(f)) when compared to 3 hr (Figure 8(d)). Stearic acid was significantly decreased at 72 hr (Figure 8(f)) compared to 3 hr (Figure 8(d)) after dosing. The minimally toxic doses of LPS and DON concurrent administration produced no changes in the fatty acids of the liver (Figures 8(g)-(i)).
5. Discussion
The administration of LPS (83 µg/kg BW) activated the innate immune response of the Sprague-Dawley rat and induced a classical but reversible (48 - 72 hr) sickness syndrome response with clear evidence of inflammation [45]. Liver histopathology of this study revealed early mild hepatotoxicity following IP dose of 10 mg/kg DON [46]. The fatty acid profile of this study indicates utilization of palmitic acid in the liver of animals treated with DON (Figure 8(b)) and the utilization of stearic and elaidic acids in the liver of animals treated with LPS (Figure 8(f)). This is indication of the involvement of these fatty acids in the palmitoylation process. The decrease in palmitic acid at 24 hr with DON treatment is indication of the utilization of palmitic acid by the early exposure of the cells to the toxin. After this mild toxic effect was counteracted, the level of palmitic acid was restored. The decrease of stearic (C18:0) and elaidic acids (C18:1 t) in the liver of animals treated with LPS could be due to substitution of these fatty acids for palmitic acid. LPS administration intraperitoneally induced hepatic vascular cell adhesion molecule (VCAM) mRNA as early as 0.5 hour after dosing [47]. Therefore, it is possible that palmitic acid (C16:0) was depleted and restored before our first sampling time of 3 hour after dosing, and by this time stearic and elaidic acids were substituted for palmitic acid. There were no changes in the free fatty acids of animals treated with LPS and DON concurrently. This lack of fatty acid activity may be due to the competition of DON and LPS for binding sites, and also to the combination of activities induced by DON
 (a)
(a) (b)
(b) (c)
(c) (d)
(d) (e)
(e) (f)
(f) (g)
(g) (h)
(h) (i)
(i)
Figure 8. (a) The effect of DON on the fatty acids of the liver 3 hours after dosing. Values are means ± SEM of 8 animals; (b) Effect of DON on fatty acids of the liver 24 hours after dosing. Values are means ± SEM of 8 animals. *Palmitic acid significantly decreased from 3 after dosing (p < 0.02); (c) Effect of DON on fatty acids of the liver 72 hours after dosing. Values are means ± SEM of 7 animals. *Palmitic acid significantly increased from 24 hours after dosing (p < 0.01); (d) Effect of LPS on fatty acids of the liver 3 hours after dosing. Values are means ± SEM of 8 animals; (e) Effect of LPS on fatty acid of the liver 24 hours after dosing. Values are means ± SEM of 8 animals. *Elaidic acid significantly decreased from 3 hours after dosing (p < 0.01); (f) Effect of LPS on fatty acids of the liver 72 hours after dosing. Values are means ± SEM of 7 animals. *Elaidic acid significantly decreased from 3 hours after dosing (p < 0.01). **Stearic acid significantly decreased from 3 hours after dosing (p < 0.03); (g) Effect of LPS and DON concurrent administration on fatty acids of the liver 3 hours after dosing. Values are means SEM of 8 animals; (h) Effect of LPS and DON concurrent administration on fatty acids of the liver 24 hours after dosing. Values are means SEM of 7 animals; (i) Effect of LPS and DON concurrent administration on fatty acids of the liver 72 hours after dosing. Values are means SEM of 8 animals.
and LPS. For example, the LPS binding protein CD14 recognizes LPS and aids in the loading of LPS onto the LPS receptor complex [48]. It has been observed by Wache et al., 2009 [49] that DON decreased the cell surface expression of CD14 in a dose-dependent manner. Also, macrophages stimulated by LPS produce large amounts of NO [50] and DON has been shown to suppressed LPS-induced NO production by the mouse macrophage cell line, RAW264, in a concentration-dependent manner. Significant inhibitory effect was also produced at a concentration as low as 500 ng/mL. DON also suppressed LPS-induced iNOS promoter activity and the expression of iNOS protein in a similar concentration range to that of the inhibition effect of NO production [41]. NO production by iNOS contributes to host defence and pathophysiological changes in inflammation, including sepsis [51]. Low concentrations of NO produced by iNOS are beneficial for the antimicrobial activity of macrophages against pathogens [52], and excessive production of NO and its derivatives can provoke pathogenesis by septic shock and autoimmune disorders [53]. Palmitoylation of iNOS is necessary for the intracellular transit towards subcellular domains where NO synthesis is required [36]. NO production by palmitoyl-specific radioprotective domain (P-SRD) has been shown to inhibit the inflammatory responses caused by endotoxin [40] and the inhibition was probably due to the competition of the P-SRD for LPS binding site on the macrophage cells. Since iNOS is S-acylated with palmitic acid, the inhibition of LPS-induced NO production by DON has consequentially diminished the utilization of palmitic acid that is required for palmitoylation in NO synthesis.
6. Conclusion
This study provides a model to explore the involvement of the palmitoylation process in the interaction of LPS with environmental toxins, such as DON, that can adversely affect the body. The data provide insights on the role of fatty acids in bacterial pathogenesis and some understanding of the diversity of bacterial survival strategies and the important role that NO plays for host protection against invading bacteria.
REFERENCES
- C. Wilcox, J. S. Hu and E. N. Olson, “Acylation of Proteins with Myristic Acid Occurs Cotranslationally,” Science, Vol. 238, No. 4831, 1987, pp. 1275-1278. doi:10.1126/science.3685978
- I. Izawa, M. Nishizawa, Y. Hayashi and M. Inagaki, “Palmitoylation of ERBN Is Required for Its Plasma Membrane Localization,” Genes Cells, Vol. 13, No. 7, 2008, pp. 691-701. doi:10.1111/j.1365-2443.2008.01198.x
- Z. Xie, W. T. Ho and J. H. Exton, “Requirements and Effects of Palmitoylation of Rat PLD1,” The Journal of Biological Chemistry, Vol. 276, No. 12, 2001, pp. 9383- 9391. doi:10.1074/jbc.M009425200
- N. D. Holliday and H. M. Cox, “Control of Signaling Efficacy by Palmitoylation of the Rat Y1 Receptor,” British Journal of Pharmacology, Vol. 139, No. 3, 2003, pp. 501-512. doi:10.1038/sj.bjp.0705276
- A. Harishchandran and R. Nagaraj, “Interaction of a Pseudosubstrate Peptide of Protein Kinase C and Its Myristoylated from with Lipid Vesicles: Only the Myristoylated Form Translocates into the Lipid Bilayer,” Biochimica et Biophysica Acta, Vol. 1713, No. 2, 2005, pp. 73-82. doi:10.1016/j.bbamem.2005.05.008
- F. Uno, J. Sasaki, M. Nishizaki, G. Carboni, K. Xu, E. N. Atkinson, M. Kondo, J. D. Minna , J. A. Roth and L. Ji, “Myristoylation of the Fus1 Protein Is Required for Tumor Suppression in Human Lung Cancer Cells,” Cancer Research, Vol. 64, No. 9, 2004, pp. 2969-2976. doi:10.1158/0008-5472.CAN-03-3702
- A. Aitken, P. Cohen, S. Santikarn, D. H. Williams, A. G. Calder, A. Smith and C. B. Klee, “Identification of the NH2-Terminal Blocking Group of Calcineurin B as Myristic Acid,” FEBS Letters, Vol. 150, No. 2, 1982, pp. 314-318. doi:10.1016/0014-5793(82)80759-X
- S. A. Carr, K. Biemann, S. Shoji, D. C. Parmalee and K. Titani, “N-Tetradecanoyl Is the NH2-Terminal Blocking Group of the Catalytic Submits of Cyclic AMP-Dependent Protein Kinase from Bovine Cardiac Muscles,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 79, No. 20, 1982, pp. 6128- 6131. doi:10.1073/pnas.79.20.6128
- E. N. Olson and G. Spizz, “Fatty Acylation of Cellular Proteins. Temporal and Subcellular Differences between Palmitate and Myristate Acylation,” The Journal of Biological Chemistry, Vol. 261, No. 5, 1986, pp. 2458-2466.
- E. N. Olson, “Modification of Proteins with Covalent Lipids,” Progress in Lipid Research, Vol. 27, No. 3, 1988, pp. 177-197. doi:10.1016/0163-7827(88)90012-4
- A. M. Dizhoor, L. H. Ericsson, R. S. Johnson, S. Kumar, E. Olshevskaya, S. Zozulya, T. A. Neubert, L. Stryer, J. B. Hurley and K. A. Walsh, “The NH2 Terminus of Retinal Recoverin Is Acylated by a Small Family of Fatty Acids,” The Journal of Biological Chemistry, Vol. 267, No. 23, 1992, pp. 16033-16036.
- W. Onkenhout, V. Venizelos, P. F. van der Poel. M. P. van den Heuvel and B. J. Poorthuis, “Identification and Quantification of Intermediates of Unsaturated Fatty Acid Metabolism in Plasma of Patients with Fatty Acid Oxidation Disorders,” Clinical Chemistry, Vol. 41, No. 10, 1995, pp. 1467-1474.
- K. Iguchi, N. Okumura, S. Usui, H. Sajiki, K. Hirota and K. Hirano, “Myristoleic Acid, a Cytotoxic Component in the Extract From Serenoa Repens, Induces Apoptosis and Necrosis in Human Prostatic LNCaP Cells,” Prostate, Vol. 47, No. 1, 2001, pp. 59-65. doi:10.1002/pros.1047
- A. Wolven, W. van’t Hof and M. D. Resh, “Analysis of Myristoylated and Palmitoylated Src Family Proteins,” Methods in Molecular Biology, Vol. 84, 1998, pp. 261- 266.
- S. Tardivel, A. Gousset-Dupont, V. Robert, M. L. Pourci, A. Grynberg and B. Lacour, “Protective Effects of EPA and Deleterious Effects of DHA on eNOS Activity in Ea hy 926 Culture with Lysophosphatidylcholine,” Lipids, Vol. 44, No. 3, 2009, pp. 225-235. doi:10.1007/s11745-009-3284-8
- K. Ramanathan, M. Anusuyadevi, S. Shila and C. Panneerselvam, “Ascorbic Acid and Alpha-Tocopherol as Potent Modulators of Apoptosis on Arsenic Induced Toxicity in Rats,” Toxicology Letters, Vol. 156, No. 2, 2005, pp. 297-306. doi:10.1016/j.toxlet.2004.12.003
- I. A. Ross, T. Boyle, W. D. Johnson, R. L. Sprando, M. W. O’Donnell, D. Ruggles and C. S. Kim, “Free Fatty Acids Profile of the Fetal Brain and the Plasma, Liver, Brain, and Kidneys of Pregnant Rats Treated with Sodium Arsenite at Mid-organogenesis,” Toxicology and Industrial Health, Vol. 26, No. 10, 2010, pp. 657-666. doi:10.1177/0748233710375952
- J. Dong and S. Y. Su, “The Association between Arsenic and Children’s Intelligence: A Meta-Analysis,” Biological Trace Element Research, Vol. 129, No. 1-3, 2008, pp. 88-93.
- J. Kang, Y. Jin, Y. Cheng and K. Wu, “Effects of Arsenic in Drinking Water on Children’s Intelligence,” Wei Sheng Yan Jiu, Vol. 36, No. 3, 2007, pp. 347-349.
- G. Samanta, D. Das, B. K. Mandal, T. R. Chowdhury, D. Chakraborti, A. Pal and S. Ahamed, “Arsenic in the Breast Milk of Lactating Women in Arsenic-Affected Areas of West Bengal, India and It Effects on Infants,” Journal of Environmental Science and Health, Part A. Toxic/Hazardous Substances and Environmental Engineerin, Vol. 42, No. 12, 2007, pp. 1815-1825.
- K. Hiroyuki, H. Suzuki, M. Naito, T. Tsuruo and Y. Sugiyama, “Characterization of Efflux Transport of Organic Anions in a Mouse Brain Capillary Endothelial Cell Line,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 285, No. 3, 1998, pp. 1260-1265.
- A. D. Munday and J. A. Lopez, “Posttranslational Protein Palmitoylation: Promoting Platelet Purpose Arteriosclerosis,” Thrombosis, and Vascular Biology, Vol. 27, No. 7, 2007, pp. 1496-1499. doi:10.1161/ATVBAHA.106.136226
- M. D. Resh, “Fatty Acylation of Proteins: New Insights into Membrane Targeting of Myristoylated and Palmitoylated Proteins,” Biochimica et Biophysica Acta, Vol. 1451, No. 1, 1999, pp. 1-16. doi:10.1016/S0167-4889(99)00075-0
- S. I. Patterson, “Posttranslational Protein S-Palmitoylation and the Compartmentalization of Signaling Molecules in Neurons,” Biological Research, Vol. 35, No. 2, 2002, pp. 139-150. doi:10.4067/S0716-97602002000200005
- A. Mor and M. R. Philips, “Compartmentalized Ras/ MAPK Signaling,” Annual Review of Immunology, Vol. 24, No. 1, 2006, pp. 771-800. doi:10.1146/annurev.immunol.24.021605.090723
- S. J. Plowman and J. F. Hancock, “Ras Signaling from Plasma Membrane and Endomembrane Microdomains. Biochim,” Biochimica et Biophysica Acta, Vol. 1746, No. 3, 2005, pp. 274-283. doi:10.1016/j.bbamcr.2005.06.004
- L. P. Wright and M. R. Philips, “Thematic Review Series Lipid Posttranslational Modifications, CAAX Modification and Membrane Targeting of Ras,” The Journal of Lipid Research, Vol. 47, No. 5, 2006, pp. 883-891. doi:10.1194/jlr.R600004-JLR200
- M. Bijlmakers and M. Marsh, “The On-Off Story of Protein Palmitoylation,” Trends in Cell Biology, Vol. 13, No. 1, 2003, pp. 32-42. doi:10.1016/S0962-8924(02)00008-9
- K. Huang and A. D. El-Husseini, “Modulation of Neuronal Protein Trafficking and Function by Palmitoylation,” Current Opinion in Neurobiology, Vol. 15, No. 5, 2005, pp. 527-535. doi:10.1016/j.conb.2005.08.001
- K. L. Clark, A. Oelke, M. E. Johnson, K. D. Eilert, P. C. Simpson and S. C. Todd, “CD81 Associates with I4-3-3 in a Redox-regulated Palmitoylation-Dependent Manner,” The Journal of Biological Chemistry, Vol. 279, No. 19, 2004, pp. 19401-19406. doi:10.1074/jbc.M312626200
- X. Yang, O. V. Kovalenko, W. Tang, C. Claas, C. S. Stipp and M. E. Hemler, “Palmitoylation Supports Assembly and Function of Integrintetraspanin Complexes,” The Journal of Cell Biology, Vol. 167, No. 6, 2004, pp. 1231-1240. doi:10.1083/jcb.200404100
- B. Zhou, L. Liu, M. Reddivari and X. A. Zhang, “The Palmitoylation of Metastasis Suppressor KAII/CD82 Is Important for Its Motility-and Invasiveness-Inhibitory Activity,” Cancer Research, Vol. 64, No. 20, 2004, pp. 7455-7463. doi:10.1158/0008-5472.CAN-04-1574
- S. W. Wong, M. J. Kwon, A. M. Choi, H. P. Kim, K. Nakahira and D. H. Hwang, “Fatty Acids Modulate TollLike Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-Dependent Manner,” The Journal of Biological Chemistry, Vol. 284, No. 40, 2009, pp. 27384-27392.
- E. V. Kalinina and L. D. Fricker, “Palmitoylation of Carboxypeptidase D. Implications for Intracellular Trafficking,” The Journal of Biological Chemistry, Vol. 278, No. 11, 2003, pp. 9244-9249. doi:10.1074/jbc.M209379200
- I. Navarro-Lerida, M. M. Corvi, A. A. Barrientos, F. Gavilanes, L. G. Berthiaume and I. Rodriguez-Crespo, “Palmitoylation of Inducible Nitric Oxide Synthase at Cys-3 is required for Proper Intracellular Traffic and Nitric Oxide Synthesis,” The Journal of Biological Chemistry, Vol. 279, No. 53, 2004, pp. 55682-55689. doi:10.1074/jbc.M406621200
- J. M. Draper and C. D. Smith, “Palmitoyl Acyltransferase Assays and Inhibitors (Review),” Molecular Membrane Biology, Vol. 26, No. 1, 2009, pp. 5-13. doi:10.1080/09687680802683839
- R. E. Bishop, “Microreview: The Lipid A Pamitoyltransferase PagP: Molecular Mechanisms and Role in Bacterial Pathogenesis,” Molecular Microbiology, Vol. 57, No. 4, 2005, pp. 900-912.
- US Food and Drug Administration, “Guidance for Industry and FDA: Advisory levels for Deoxynivalenol (DON) in Finished Wheat Products for Human Consumption and Grains and Grain By-Products used for Animal Feed,” 2010. http://www.fda.gov/Food
- J. J. Pestka and A. T. Smolinski, “Deoxynivalenol: Toxicology and Potential Effects on Humans,” Journal of Toxicology and Environmental Health. Part B, Critical Reviews, Vol. 8, No. 1, 2005, pp. 39-69. doi:10.1080/10937400590889458
- V. K. Singh, T. M. Seed and K. Kumar, “N-Palmitoylation of the Radioprotective Domain of Interleukin-I Affords Inhibition of LPS-Induced Nitric Oxide Generation,” Immunopharmacology and Immunotoxicology, Vol. 26, No. 2, 2004, pp. 193-202. doi:10.1081/IPH-120037714
- K. Sugiyama, M. Muroi, K. Tanamoto, M. Nishijima and M. Sugita-Konishi, “Deoxynivalenol and Nivalenol Inhibit Lipopolysaccharide-Induced Nitric Oxide Production by Mouse Macrophage Cells,” Toxicology Letters, Vol. 192, No. 2, 2010, pp. 150-154. doi:10.1016/j.toxlet.2009.10.020
- Q. Jia, H. R. Zhou, Y. Shi and J. J. Pestka,” Docosahexaenoic Acid Consumption Inhibits Deoxynivalenol-Induced CREB/ATF1 Activation and IL-6 Gene Transcription in Mouse Macrophages,” The Journal of Nutrition, Vol. 136, No. 2, 2006, pp. 366-372.
- I. A. Ross, T. Boyle, W. D. Johnson, L. H. Garthoff, S. M. Ahn, M. W. O’Donnell and C. S. Kim, “The Individual and Interactive Effects of Lipopolysaccharide and Deoxynivalenol on Free Fatty Acids in the Liver and Brain of Rats,” Society of Toxicology Annual Meeting, 2011.
- I. A. Ross and C. S. Kim, “The Role of Palmitoylation in Chemical and Microbial Toxicity: Signal Pathways, Protein Binding and Trafficking,” Society of Toxicology Annual Meeting, 2013.
- P. L. Wiesenfeld, L. H. Garthoff, T. J. Sobotka, J. K. Suagee and C. N. Barton, “Acute Oral Toxicity of Colchicines in Rats: Effects of Gender, Vehicle Matrix and Pre-Exposure to Lipopolysaccharide,” Journal of Applied Toxicology, Vol. 27, No. 5, 2007, pp. 421-433. doi:10.1002/jat.1198
- S. Sahu, L. H. Garthoff, M. G. Robl, S. J. Chirtel, D. I. Ruggles, T. J. Flynn and T. J. Sobotka, “Rat Liver Clone-9 Cells in Culture as a Model for Screening Hepatotoxic Potential of Food-Related Products: Hepatotoxicity of Deoxynivalenol,” Journal of Applied Toxicology, Vol. 28, No. 6, 2008, pp. 765-772. doi:10.1002/jat.1337
- H. Huang, L. Tongzheng, J. L. Rose, R. L. Stevens and D. G. Hoyt, “Sensitivity of Mice to Lipopolysaccharide Is Increased by a High Saturated Fat and Cholesterol Diet,” Journal of Inflammation, Vol. 4, No. 22, 2007, pp. 1-11.
- S. Doll, J. A. Schrickx, H. Valenta, S. Danicke and J. Fink-Gremmels, “Interactions of Deoxynivalenol and Lipopolysaccharides on Cytotoxicity, Protein Synthesis and Metabolism of DON on Porcine Hepatocytes and Kupffer Cell Enriched Hepatocyte Cultures,” Toxicology Letters, Vol. 189, No. 2, 2009, pp. 121-129. doi:10.1016/j.toxlet.2009.05.011
- Y. J. Wache, L. Hbabi-Haddioui, L. Guzylack-Piriou and H. Belkhelfa, “The Mycotoxin Deoxynivalenol Inhibits the Cell Surface Expression of Activation Markers in Human Macrophages,” Toxicology, Vol. 262, No. 3, 2009, pp. 239-244. doi:10.1016/j.tox.2009.06.014
- A. S. Baldwin, “The NF-Kappa B and I Kappa B Proteins: New Discoveries and Insights,” Annual Review of Immunology, Vol. 14, 1996, pp. 649-683. doi:10.1146/annurev.immunol.14.1.649
- P. Valance and J. Collier, “Biology and Clinical Relevance of Nitric Oxide,” British Medical Journal, Vol. 309, 1994, pp. 453-457. doi:10.1136/bmj.309.6952.453
- H. T. Cook and V. Cattell, “Role of Nitric Oxide in Immune-Mediated Diseases,” Clinical Science, Vol. 91, No. 4, 1996. pp. 375-384.
- T. Nguyen, D. Brunson, C. L. Crespi, B. W. Penman, J. S. Wishnok and S. R. Tannenbaum, “DNA Damage and Mutation in Human Cells Exposed to Nitric Oxide in Vitro,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, No. 7, 1992, pp. 3030-3034. doi:10.1073/pnas.89.7.3030
Abbreviations
FFAs, free fatty acids; LPS, lipopolysaccharide; DON, deoxynivalenol; ROS, reactive oxygen species; NaAsO2, sodium arsenite; AsH2O, NaAsO2 in deionized water; AsHH, NaAsO2 in half and half dairy cream; NO, nitric oxide; iNOs, inducible NO synthase; CAMP, cationic antimicrobial peptides; TLR4, Toll-like receptor 4.