American Journal of Plant Sciences
Vol.5 No.13(2014), Article
ID:47467,9
pages
DOI:10.4236/ajps.2014.513221
An Efficient Protocol for Total RNA Isolation from Healthy and Stressed Tissues of Mulberry (Morus sp.) and Other Species
Figure 1. Denaturing agarose gel stained with ethidium bromide showing total RNA isolated from leaf tissues of mulberry using different RNA isolation methods (Logemann et al. (1987), Gasic et al. (2004), Salzman et al. (1999), TRIzol® (Invitrogen), RNeasy®(Qiagen) and modified protocol). Starting material and the volume of the DEPC-treated water used to dissolve RNA were kept constant and 2 μl total RNA was loaded for comparison.
(Figure 1). The reaction components included RNase inhibitors, chelator (EDTA), and protein denaturants (SDS and β-mercaptoethanol). We used PVPP instead of PVP in the standardized protocol. Addition of PVPP in nucleic acid extraction protocols was shown to be very effective for preventing tissue browning [5] [18] . Further, pH of the solution was maintained in the acidic range to allow efficient and preferable partitioning of RNA in the aqueous phase leaving DNA in the phenolic phase [19] .
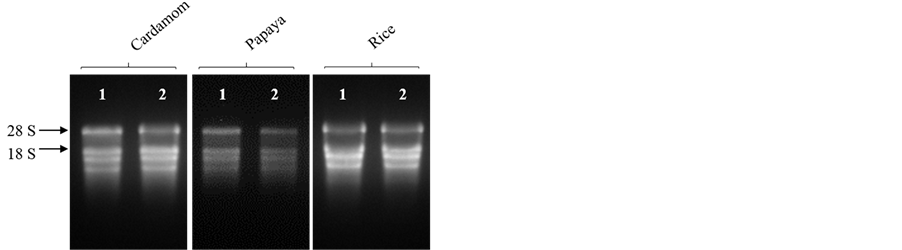
The modified protocol yielded a white, water soluble RNA precipitate from different tissues of mulberry. Denaturing agarose gel stained with ethidium bromide (EtBr) indicated intact RNA as there was clear distinct bands of 28 and 18S rRNA (Figure 1). About 520.00 ± 2.60 μg total RNA per gram of fresh leaf tissue was harvested from mulberry and the ratios of 260/280 and 260/230 were 2.02 ± 0.02 and 2.06 ± 0.02, respectively suggesting low quantities of interfering compounds such as polysaccharides, polyphenols and proteins (Table 2). The method developed was further used to extract RNA from different plant tissues (root, stem, inflorescence and immature fruits) of mulberry. As shown in the Figure 2, and Table 3, good quality RNA was extracted from these tissues, signifying that the proposed protocol could be useful for different tissue types. Similarly, the proposed protocol yielded satisfactory results for RNA extraction from plant species, such as cardamom, papaya and rice. The total RNA isolated from leaf tissues contained intact 28 and 18S rRNA signatures with high quantity (Figure 3, Table 4). We believe that this protocol could be useful for extracting RNA from many other species having metabolites that act as nucleic acids interfering molecules.
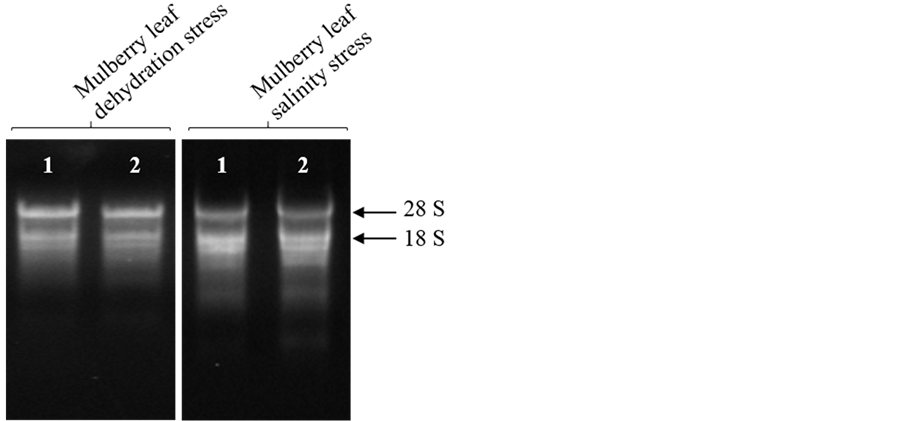
Many studies on stress biology require extraction of high quality RNA and many protocols failed to yield quality RNA in sufficient quantities [20] [21] . However, using this standardised protocol, we could isolate quality RNA in sufficient quantities from mulberry leaf tissues subjected to dehydration and salinity stresses. We isolated high quality RNA from stressed leaf tissues (Figure 4), and the average yield of total RNA was 348.83 ± 12.20 and 293.78 ± 7.20 μg/g of fresh weight under dehydration and salinity stresses, respectively (Table 4).

Table
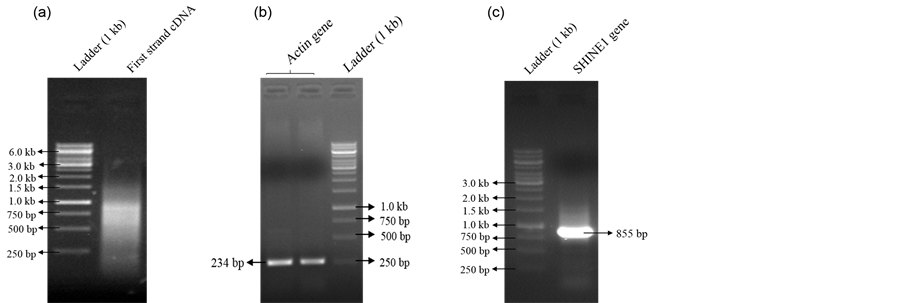
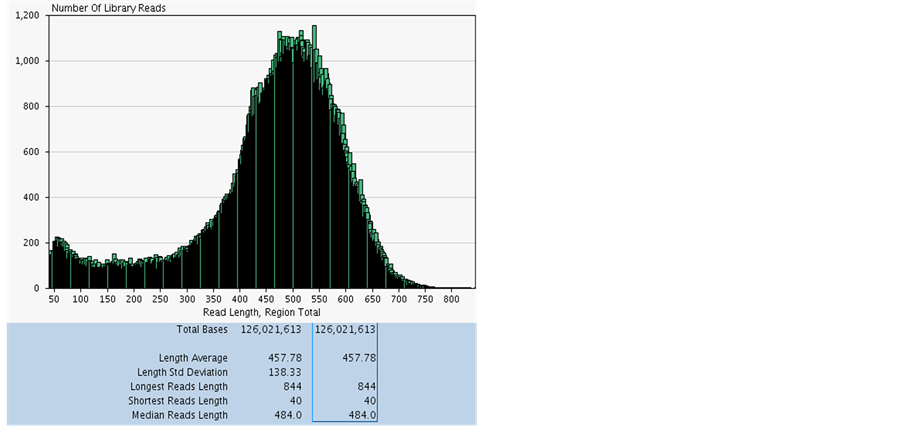
Radha Sivarajan Sajeevan1,2, Manchanahally Byrappa Shivanna2, Karaba N. Nataraja1* 1Department of Crop Physiology, University of Agricultural Sciences, GKVK, Bangalore, India 2Department of Studies in Applied Botany, Kuvempu University, Shankaraghatta, Shimoga District, India Email: *nataraja_karaba@yahoo.com Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Received 1 May 2014; revised 30 May 2014; accepted 21 June 2014 ABSTRACT Extraction of quality RNA for molecular biology applications from perennial woody plants like mulberry is complicated due to the presence of high polysaccharides, polyphenols and other secondary metabolites. Since the existing methods failed to yield quality RNA in sufficient quantity from leaf and root tissues of mulberry, in this study, we modified the CTAB-based protocol. The standardised protocol yielded high quantity (520.00 μg/g fresh weight of leaf tissue) of quality RNA and the RNA extracted was suitable for all downstream applications such as cDNA synthesis, PCR and whole transcriptome analysis. The method developed was also found to be useful for isolating good quality and quantity total RNA from desiccated and salinity stressed leaf tissues of mulberry. The protocol was also applied successfully to isolate total RNA from leaf tissues of other species such as cardamom, papaya and rice. Keywords:Mulberry, Cardamom, Papaya, Rice, Dehydration, Salinity, RNA Isolation 1. Introduction Mulberry (Morus sp.), a perennial tree with high economic importance in sericulture industry is the sole food plant for the silkworm, Bombyx mori L. Fast growth and ability to adapt to adverse environmental conditions make the tree a good source to prospect novel genes and pathways. Isolating quality RNA from mulberry is prerequisite for gene discovery and functional validation studies. Although the existing plant RNA isolation protocols could be used to extract RNA from mulberry tissues, high-quality RNA extraction is problematic due to the presence of polysaccharides, polyphenolics and other unknown compounds that bind or co-precipitate with the RNA. Leaves of mulberry contain latex, large amount of secondary metabolites, and many uncharacterised biomolecules [1] that interfere with nucleic acid extractions. Mulberry leaf extract contains 44.82% polyphenols and have comparatively high values of phenolic compounds which ranged from 8.33 to 11.79 mmol/100 g of dry weight [2] [3] . When oxidized, phenolic compounds give rise to quinones [4] , which in turn bind to nucleic acids [5] , making them unsuitable for downstream applications. Moreover, isolating quality RNA from tissues exposed to different types of environmental stresses such as desiccation and salinity for gene expression analysis is difficult due to increased accumulation of reactive oxygen species, secondary metabolites and other compounds that easily degrades nucleic acids [6] . The general protocols described by Logemann et al. (1987) [7] , Salzman et al. (1999) [5] , Gasic et al. (2004) [8] , and/or commercially available reagent-based methods (TRIzol®, Invitrogen, USA) or RNA isolation kits (RNeasy®, Qiagen, Germany) are not useful in mulberry because of the poor RNA yield. In this study, we used the basic protocol described by Gasic et al. (2004) [8] for tissues rich in polyphenols and polysaccharides to develop an efficient and reproducible RNA isolation method for mulberry plant tissues. The modified protocol mainly focussed on removal of polysaccharides and polyphenolics and other secondary metabolites by using an insoluble, inert material polyvinylpolypyrrolidone (PVPP). The protocol standardized was used to isolate total RNA from leaf, stem, inflorescence, immature fruit and root tissues of mulberry. The protocol was also found to be useful for isolating RNA from leaf tissue exposed to desiccation and salinity stress. Further, the efficiency of the protocol was tested in different plant species such as cardamom (Elettaria cardamomum L.), papaya (Carica papaya L.) and rice (Oryza sativa L.) using healthy leaf samples. The protocol described in this study, allows rapid isolation of high-quality RNA from healthy and stressed plant tissues. 2. Material and Methods 2.1. Plant Materials Mulberry plants maintained at University of Agricultural Sciences, GKVK Campus, Bangalore were used for the study. Tissue samples were collected from root, stem, inflorescence and immature fruits of mulberry. To create desiccation stress the mulberry plants grown in pots were exposed to 40% soil field capacity (FC) [9] and leaf tissues were harvested for RNA extraction. To induce salinity stress, 10 cm long twigs were cut from the top of the healthy plants and exposed to 250 mM NaCl under laboratory conditions for 16 - 18 h by dipping the cut ends in the solution. Healthy leaf samples were collected from cardamom, papaya and rice plants maintained in the green house, and used for RNA extraction. All the tissues were washed with sterile diethyl pyrocarbon (DEPC, Sigma, USA) treated water, blotted on tissue paper, quickly frozen in liquid nitrogen before transportation to the laboratory and stored at −80˚C. 2.2. Solutions and Reagents General care: Glassware used for reagent preparation was baked overnight at 180˚C; mortar and pestles and all plastic ware was either new disposable or treated with 0.1% (v/v) DEPC treated water and autoclaved before use; gel-running apparatus was treated with 5% hydrogen peroxide (H2O2) or 80% ethanol for 30 min and dried; all solutions used in the RNA extractions, except those containing Tris, were treated with 0.1% (v/v) DEPC to destroy potentially damaging RNases. Powder free nitrile gloves were used throughout to avoid potential contamination. The reagents and chemical used are given below. 1) 4% PVPP (w/v) (Sigma P-6755) 2) Phenol (water saturated):chloroform:isoamyl alcohol (25:24:1, v/v/v) 3) Chloroform:isoamyl alcohol (24:1, v/v) 4) 6 M and 2 M LiCl 5) 3 M sodium acetate (NaOAc, adjusted to pH 5.2) 6) Ice cold 70% ethanol (v/v) 7) Ice cold absolute ethanol 8) RNase free sterile water (0.1% DEPC—treated autoclaved water) Extraction buffer: 100 mM Tris-HCl (adjusted to pH 7.0), 500 mM NaCl, 10 mM Ethylene diamine tetra acetic acid (EDTA, adjusted to pH 8), 2% (w/v) Sodium Dodecyl Sulphate (SDS) and 4% (v/v) β-mercaptoethanol (freshly added before use). 2.3. RNA Extraction Procedure Frozen plant tissue (1 g) was ground to fine powder in a mortar and pestle using liquid nitrogen along with PVPP (4%) and homogenised in 10 ml of extraction buffer containing 400 ml of β-mercaptoethanol. 10 ml of phenol (water saturated): chloroform: isoamyl alcohol mixture was added and mixed thoroughly. The suspension appeared milky white in color (temperature more than 4˚C and/or oxidation condition, colour would change to brown). The mixture was transferred to a 50 ml oak ridge centrifuge tube and centrifuge at 15,000 rpm for 15 min. The top colourless aqueous phase containing nucleic acids, was transferred to a fresh oak ridge centrifuge tube and equal volume of chloroform: isoamyl alcohol mixture was added, and mixed thoroughly by inverting the tube for 15 to 20 times. The mixture was then centrifuged at 15,000 rpm for 10 min at 4˚C, and the aqueous phase was transferred to a fresh oak ridge centrifuge tube. 6 M LiCl was added to a final concentration of 3 M and incubated in −80˚C for 5 h or −20˚C for 10 h. The solution was then centrifuged at 15,000 rpm for 30 min at 4˚C to collect RNA as precipitate. The precipitate was resuspended in 4 to 6 ml of 2M LiCl and centrifuged at 15000 rpm for 20 min at 4˚C. The supernatant was discarded and the RNA pellet was washed with 70% ice cold ethanol. The pellet was then air dried and alcohol free pellet was dissolved in 250 to 500 ml of DEPC treated sterile water. For checking the quality, 2 µl of the total RNA was loaded on formaldehyde denaturing agarose gel (1%, w/v) [10] . 2.4. Optional Steps for Getting High Quality RNA without DNA Contamination To the RNA solution, DNase (2 U/µg DNA) was added with 10X DNase buffer to a final concentration of 1X and incubated at 37˚C for 1 h. An equal volume of chloroform: isoamyl alcohol mixture was added and mixed thoroughly by inverting the tube for 10 to 15 times and centrifuged at 10,000 rpm for 10 min at 4˚C. The top aqueous phase was transferred carefully to a fresh 1.5 ml RNase free sterile eppendorf tube and 1 volume of 3 M sodium acetate (pH 5.2) and 2.5 volumes of 100% ethanol were added, mixed by inversion and incubated over night at −20˚C. The content was then centrifuged at 12,000 rpm for 30 min at 4˚C. The pellet was collected and washed twice with 70% (v/v) alcohol and air dried to evaporate alcohol. The RNA pellet was subsequently dissolved in sterile water (100 to 250 µl) and used after checking the quality. 2.5. Estimation of Quantity and Quality of RNA The quality of total RNA isolated was assessed spectrophotmetrically using UV-VIS spectrophotometer (Shimanzo, Japan) by estimating the ratio of absorbance at 260/280 and 260/230 nm [10] . RNA quantity was estimated using the formula, 1 O.D. at 260 nm = 40 ng of RNA [10] . Integrity of the RNA samples were checked in a denaturing agarose (1% w/v) gel, stained with ethidium bromide, and visualized under UV light [10] . 2.6. Synthesis of cDNA and PCR About 3 µg of total RNA was heat denatured at 65˚C for 5 min and reverse transcribed by MMLV reverse transcriptase (100 U; MBI, Fermentas Life Sciences, USA) in 20 µl reaction mixture containing 40 pmol of OligodT (22 mer) primer. For complimentary DNA (cDNA) synthesis, the reaction mixture was incubated at 42˚C for 1 h [10] . The enzyme was heat inactivated at 65˚C for 15 min and the cDNA synthesised was either used immediately, or stored at 4˚C until further use. The cDNA synthesised was separated on agarose gel [10] to check the quality. Polymerase chain reaction (PCR) was performed using the cDNA as template. The PCR was carried out to amplify housekeeping gene actin using specific primers (forward 5’ TCCATAATGAAGTGTGATGT 3’ and reverse 5’ GGACCTGACTCGTCATACTC 3’), and 1 µl of reverse transcribed product in 20 µl reaction mixture. The reaction mixure contained 1 X Taq buffer, 1.5 mM dNTPs, and 1 U Taq polymerase (MBI, Fermentas Life Sciences, USA). The reaction conditions were 94˚C for 1 min followed by 30 cycles of 94˚C for 20 s, 52˚C for 30 s, and 72˚C for 20 s and a single final elongation step of 72˚C for 10 min. Similarly, a transcription factor (TF) gene, SHINE1 (SHN1) was amplified using the cDNA as template with the primers (forward 5’ ATGGTACAGTCAAAGAAGTTCAGAGG 3’ and reverse 5’ CTAGTGTCAATTACACCACAATTAC 3’) under standardised PCR conditions. The annealing temperature used to amplify the TF gene SHN1 was 64˚C and analysed by separating the PCR product on agarose gel [10] . 2.7. Whole Transcriptome Sequencing From the total RNA, messenger RNA (mRNA) was isolated using mRNA isolation kit (Oligotex mRNA Mini kit Qiagen, CA, USA). The mRNA enriched fraction was converted to 454 barcoded cDNA library as reported [11] . The process of library preparation, emulsion-based clonal amplification and sequencing on the 454 Genome Sequencer FLX Titanium system were performed in the Biodiagnostic, Inc. USA according to the manufacturer’s instructions (454 Life Sciences, USA). 2.8. Data Analysis Yield data (µg/g FW), absorbance ratios (A260/A280 and A260/A230 ratios) were analysed by analysis of variance (ANOVA, SPSS software package for Windows, release 15.0; SPSS Inc., Chicago, IL, USA) and significant difference between means was compared at the 0.05 probability level using Duncan’s Multiple Range Test (DMRT; P = 0.05). 3. Results and Discussion Morus species tissues contain high amount of latex, and rich in polyphenolics, polysaccharides and other secondary metabolites that rapidly oxidize during sample grinding [2] [3] . Although there are many protocols standardized for isolating RNA from various plant species and tissues with varying amount of cellular composition [5] [7] [8] [12] [13] , the methods were not suitable for all types of tissues and plant species. The existing methods could not yield high quality RNA in sufficient quantities in mulberry probably because of high amount of interfering compounds such as polyphenolics and polysaccharides (Figure 1). The difficulty in RNA extraction from different species and also from the same species grown under different environments have been earlier reported by many researchers [14] -[17] . We modified the protocol described by Gasic et al. (2004) [8] with SDS, PVPP, phenol: chloroform: isoamyl alcohol instead of CTAB, PVP and chloroform: isoamyl alcohol as mentioned in Table 1. The modified protocol yielded good quality and quantity RNA from mulberry leaf tissues Table 2. Quantification of total RNA extracted using different protocols from leaf tissues of mulberry. The results are the average of three replications ± standard error (SE). Different superscript lowercase letters indicate significant difference at p ≤ 0.05, FW: fresh weight. Table 3. Quantification of total RNA extracted from different tissues of mulberry. The results are the average of three replications ± standard error (SE). Different superscript lowercase letters indicate significant difference at p ≤ 0.05, FW: fresh weight. Table 4. Quantification of total RNA extracted from healthy leaf tissues of cardamom, papaya, rice, and dehydrated and salinity stressed leaf tissues of mulberry. The results are the average of three replications ± standard error (SE). Different superscript lowercase letters indicate significant difference at p ≤ 0.05, FW: fresh weight. Figure 2. Denaturing agarose gel stained with ethidium bromide showing total RNA isolated from different parts of mulberry tissues using the modified protocol. Total RNA (2 μl) was separated on agarose (1% w/v) gel. Lane 1 and 2 are replications. The components in RNA extraction mixture and also the protocol influence the downstream application in molecular biology experiments. Reverse transcription yielded sufficient amount of quality cDNA (Figure 5(a)) and the cDNA was also found to be suitable for target gene amplification. We amplified 234 bp housekeeping actin gene (Figure 5(b)) and also 855 bp of stress related TF, SHN1 (Figure 5(c)) using the mulberry cDNA. The identity of all amplified products was confirmed by sequencing (data not shown). In all reactions, total RNA was used as a template to confirm the PCR product was not a result of DNA contamination. Further, we also tested the suitability of total RNA isolated for whole transcriptome analysis using mulberry leaf tissue. The transcriptome analysis carried out from the total RNA isolated from drought stressed mulberry leaf tissues using Figure 3. Denaturing agarose gel stained with ethidium bromide showing total RNA isolated from leaf tissues of cardamom, papaya and rice plants using the modified protocol. Total RNA (2 μl) was separated on agarose (1% w/v) gel. Lane 1 and 2 are replications. Figure 4. Denaturing agarose gel stained with ethidium bromide showing total RNA isolated from dehydrated and salinity stressed leaf tissues of mulberry using the modified protocol. Total RNA (2 μl) was separated on agarose (1% w/v) gel. Lane 1 and 2 are replications. Figure 5. Agarose gel stained with ethidium bromide showing (a) first strand cDNA after RT reaction; (b) and (c) RT-PCR for actin and SHN1 genes, respectively. The arrows indicate the actin and SHN1 genes products; DNA ladder (1 kb; MBI, Fermentas Life Sciences, USA). Figure 6. Summary statistics of 454-FLX Titanium sequencing using the total RNA isolated from drought stressed leaf tissues of mulberry plant. X-axis is no. of library reads and Y-axis is read length. The shortest read length was 40 bp and longest was 844 bp. The average length of transcript sequence was about 458 bp and the sequence length ranged from 180 bp to 5600 bp. the protocol reported in this study generated quality transcriptome data. We used 454 Genome Sequencer FLX Titanium system to generate over 126,021,613 bases with 500 bps of average length of sequence (Figure 6). Taken together, the data suggest the RNA protocol developed did not have any bad effect on the downstream molecular biology applications. 4. Conclusion The protocol described here is simple, inexpensive and effective for isolating total RNA from tissues with varying levels of polysaccharides, polyphenolics and secondary metabolites. The protocol was also found to be suitable for isolating intact RNA from plant tissues exposed to different types of stresses, without any effect on downstream molecular biology applications. Acknowledgments We gratefully acknowledge the Department of Biotechnology (DBT), Government of India, New Delhi, (BT/PR 13457/PBD/19/214/2010) for providing financial support to carry out this work. References NOTES *Corresponding author.



 1. Comparison of components of extraction buffers between two RNA isolation protocols.
1. Comparison of components of extraction buffers between two RNA isolation protocols.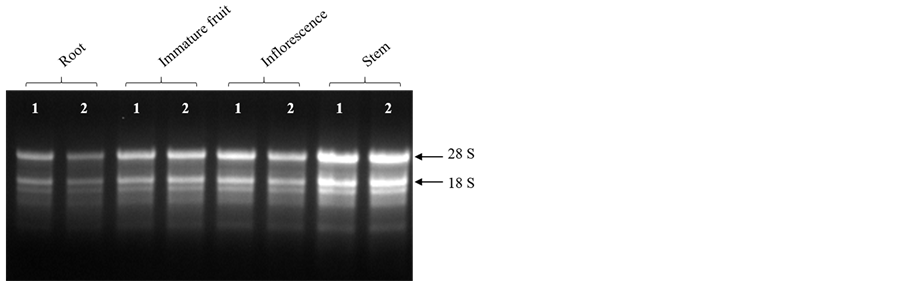

Journal Menu >>